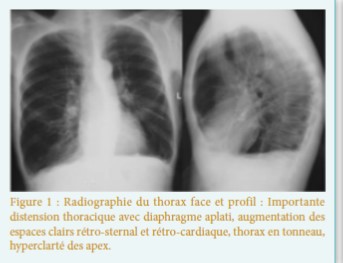
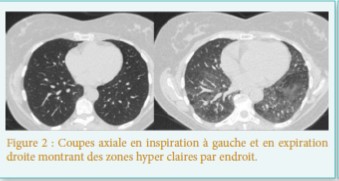
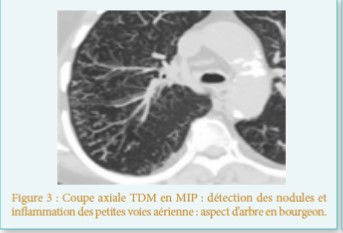
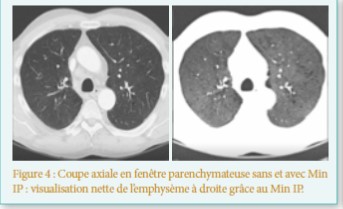
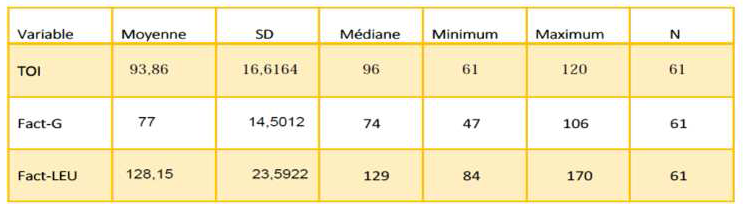
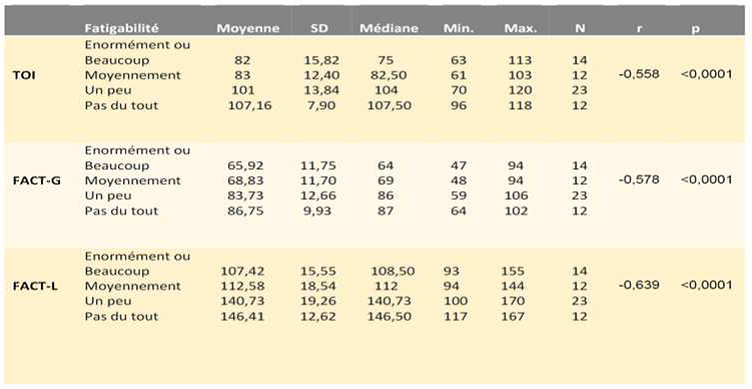
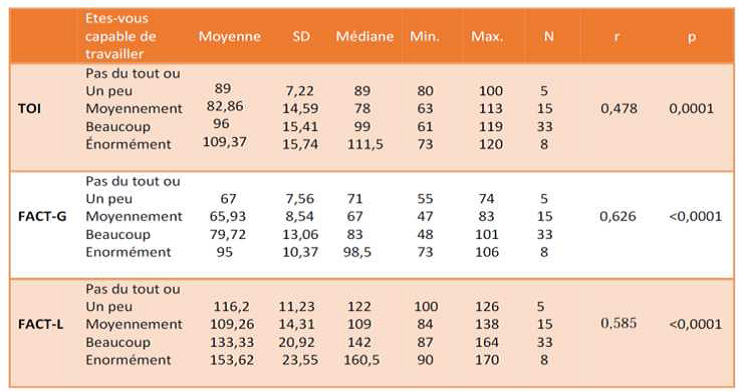
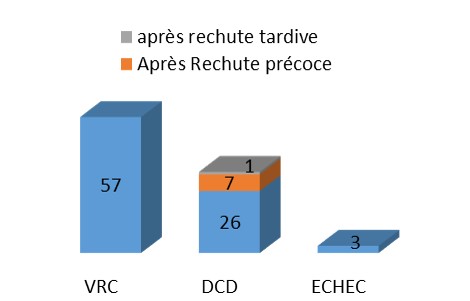
La maladie de Fabry[1] est une maladie héréditaire du métabolisme, de transmission liée au chromosome X, est due à un déficit total ou partiel d’une enzyme lysosomale, l’alpha-galactosidase-A. Elle aboutit dès le plus jeune âge à une accumulation progressive ubiquitaire de glycosphingolipides (principalement le globotriaosylcéramide encore appelé GL-3, ou Gb3) non dégradés par cette enzyme.
Maladie de Fabry
M.A. Boubchir, Service de néphrologie, CHU Nédir-Mohamed, Tizi Ouzou
Date de soumission : 28 Novembre 2020
Abstract: Fabry’s disease is an inherited metabolic disease, of transmission linked to the X chromosome, is due to a total or partial deficiency of a lysosomal enzyme, alpha-galactosidase-A. From an early age, it leads to a progressive ubiquitous accumulation of glycosphingolipids (mainly globotriaosylceramide also called GL-3, or Gb3) not degraded by this enzyme. Cutaneous, neurological, nephrological, cardiac, gastrointestinal, ophthalmological, respiratory and cochleovestibular disorders are responsible for increased mortality and a significant deterioration in the quality of life of patients. The questioning also directs towards the diagnosis of Fabry disease if there are deaths in the family in people under 50 years of age, mainly males, cerebrovascular accidents, arrhythmias or conduction disorders heart or kidney failure. Earlier diagnosis can lead to the initiation of treatment with enzyme replacement therapy, which can prevent the occurrence of irreversible lesions. Diagnosis in male patients is based on measurement of alpha-galactosidase-A enzyme activity in leukocytes or plasma; a collapsed value confirms the diagnosis of Fabry disease. Heterozygous women show signs of the disease later and generally of less intensity. In women, only genotyping (search for the GLA gene mutation) can confirm the diagnosis. In all cases, a family investigation should be carried out after diagnosis of an index case. Regular specialist follow-up is essential for men, women and children.
Keywords: Fabry disease, Alpha-galactosidase A, Angiokeratomas, Renal failure.
Résumé : La maladie de Fabry[1] est une maladie héréditaire du métabolisme, de transmission liée au chromosome X, est due à un déficit total ou partiel d’une enzyme lysosomale, l’alpha-galactosidase-A. Elle aboutit dès le plus jeune âge à une accumulation progressive ubiquitaire de glycosphingolipides (principalement le globotriaosylcéramide encore appelé GL-3, ou Gb3) non dégradés par cette enzyme. Les atteintes cutanées, neurologiques, néphrologiques, cardiaques, gastro-intestinales, ophtalmologiques, respiratoires et cochléo-vestibulaires sont responsables d’une mortalité accrue et d’une altération significative de la qualité de vie des patients. L’interrogatoire oriente aussi vers le diagnostic d’une maladie de Fabry s’il existe dans la famille des décès chez des personnes de moins de 50 ans, principalement de sexe masculin, des accidents vasculaires cérébraux, des troubles du rythme ou de la conduction cardiaque ou des cas d’insuffisance rénale.
Un diagnostic plus précoce peut conduire à l’instauration d’un traitement par enzymothérapie substitutive à même d’éviter la survenue de lésions irréversibles. Le diagnostic repose chez le patient de sexe masculin sur la mesure de l’activité enzymatique de l’alpha-galactosidase A dans les leucocytes ou le plasma; une valeur effondrée confirme le diagnostic de maladie de Fabry. Les femmes hétérozygotes présentent des signes de la maladie plus tardivement et généralement d’intensité moindre. Chez les femmes, seul un génotypage (recherche de la mutation du gène GLA), permet d’affirmer le diagnostic. Dans tous les cas, une enquête familiale devra être réalisée après diagnostic d’un cas index. Un suivi spécialisé régulier est indispensable, chez les hommes, les femmes et les enfants.
Mots-clés : Maladie de Fabry, Alpha-galactosidase A, Angiokératomes, Insuffisance rénale.
- Introduction
La maladie de Fabry (MF) est une maladie de surcharge lysosomale progressive, liée à une anomalie du métabolisme des glycosphingolipides, de transmission liée au chromosome X, avec une atteinte plus précoce et plus sévère chez les hommes, due à un déficit en alpha-galactosidase A, enzyme nécessaire à la dégradation du globotriaosylcéramide (Gb3).
Ce déficit enzymatique entraine une augmentation de la concentration plasmatique de Gb3 (globotriaosylcéramide) et une accumulation dans les lysosomes des organes tels que le rein, le cœur, les cellules nerveuses et les cellules endothéliales capillaires. Cette accumulation tissulaire provoque une atteinte multisystémique avec des manifestations algiques dans l’enfance, ainsi qu’une atteinte cutanée et ophtalmologique, puis des atteintes : rénale, neurologique et cardiaque qui grèvent le pronostic du patient.
- Historique
1898 : Première description par deux dermatologues Johannes Fabry et William Anderson
1947 : Découverte de dépôts lipidiques dans les vaisseaux sanguins par Pompen.
1963 : Identification du globotriaosylcéramide (Gb3), principal sphingolipide accumulé dans les tissus par Sweeley et Klionsky.
1967 : Identification de l’enzyme déficiente, l’α-galactosidase A par Brady, puis confirmation par Kint en 1970.
1986 : Séquençage du gène codant pour l’α-galactosidase A.
2001 : Le traitement de référence par Fabrazyme® (enzyme humaine recombinante) est approuvé par la commission Européenne en août 2001 et par la FDA en avril 2003.
2016 : Molécules chaperons.
- Rappels
- Génétique
Individu homozygote : Si ces deux allèles sont identiques, l’individu est dit homozygote pour le gène considéré.
Individu hétérozygote : Si les deux allèles sont différents, l’individu est dit hétérozygote pour ce gène.
Individu hémizygote : Un individu n’ayant qu’un seul allèle pour un gène donné (l’allèle du daltonisme chez un mâle par exemple).
Le gène Fabry défectueux est situé sur le chromosome X, qui est l’un des deux chromosomes qui déterminent le sexe d’un individu[2]. Chaque cellule de l’être humain contient 46 chromosomes, regroupés en 23 paires. La 23ème paire est constituée des chromosomes dits sexuels (gonosomes) : les hommes présentent un chromosome X et un chromosome Y (XY), les femmes, deux chromosomes X (XX).
Les maladies dont le gène est localisé sur le chromosome X se transmettent le plus souvent sur un mode dit “récessif lié à l’X”. Dans ce cas, la maladie se manifeste chez les sujets de sexe masculin (XY), qui possèdent une seule copie mutée du gène, alors que les femmes (XX) porteuses du gène muté sur un des deux chromosomes X sont cliniquement saines, mais conductrices de la maladie.
Seuls les garçons sont donc atteints dans la lignée maternelle. Il n’y a jamais de transmission père-fils.
|
Figure 1 : Affection liée au chromosome X |
Figure 2 : Emplacement du gène Fabry annoté en rouge – Loci Xq 22.1 sur le chromosome X |
- Lipidologie
Globotriaosylcéramide : ganglioside également appelé trihexosylcéramide, CD77, Gb3 ou GL-3 dont la structure peut être représentée par αGal–βGal–βGlc–Cer, composé :
- D’une unité céramide constituée d’un résidu de sphingosine lié à un acide gras ;
- D’un résidu de glucose lié au céramide par une liaison β-osidique ;
- D’un résidu de galactose lié au glucose par une liaison β-osidique ;
- D’un résidu de galactose terminal lié au galactose par une liaison α-osidique.
Ces composés diffèrent les uns des autres uniquement par l’acide gras combiné à la sphingosine de l’unité céramide.
Figure 3: Action de l’alpha-galactosidase
Les Gb3 sont produits dans les cellules à partir de galactose et de lactosylcéramide sous l’action de l’α-1,4-galactosyltransférase.
- Lysosome : organite cellulaire de 0,2 à 0,5 micron présent dans le cytosol de toutes les cellules eucaryotes, animales, à l’exception des érythrocytes. Il a pour fonction la digestion intracellulaire grâce à une quarantaine d’enzymes, dont des lipases, des protéases, des nucléases, des glycosidases, des phosphatases et des sulfatases.
Figure 4 : Surcharge lysosomale par déficit enzymatique [1].
- Épidémiologie : La maladie de Fabry, bien que rare, fait partie des maladies de surcharge lysosomale les plus fréquentes, touchant 1/40.000 naissances masculines [2]. La maladie de Fabry est pan-ethnique. L’incidence annuelle, mal connue, est estimée à 1 pour 80.000 naissances vivantes, mais ce chiffre pourrait être sous-estimé [3]. La prévalence, à la naissance, pour cette maladie s’élève à hauteur de 1/80.000 (nombre de cas dans une population donnée à un moment donné). Lorsque la maladie se développe plus tardivement, cette prévalence augmente à 1/3 000 [3].
- Tableau clinique : La surcharge tissulaire augmente avec l’âge et le tableau clinique recouvre tout un spectre de sévérités allant des formes légères (femmes hétérozygotes), aux formes graves (hommes hémizygotes) présentant toutes les manifestations caractéristiques.
Cette affection est multisystémique avec, dans sa forme classique, des manifestations algiques (débutant le plus souvent dans l’enfance ou l’adolescence).
- L’interrogatoire : Il recherchera :
- Des antécédents familiaux de MF (arbre généalogique),
- Des symptômes évocateurs, en particulier acroparesthésies, anhidrose, symptomatologie digestive, etc., des signes généraux : asthénie, altération de l’état général,
- Des palpitations, douleurs thoraciques, dyspnée,
- Des facteurs de risque cardio-vasculaire : HTA, tabagisme, dyslipidémie, diabète, obésité,
- Chez l’enfant : des manifestations douloureuses (acroparesthésies et crises douloureuses paroxystiques articulaires ou abdominales)[4].
- L’examen physique : il recherchera des signes d’atteinte spécifique
- Rénale : (quasi constantes) : l’atteinte rénale est longtemps cliniquement asymptomatique, nécessitant un dépistage systématique d’une microalbuminurie, d’un trouble de la concentration des urines, d’une hyperfiltration, d’une protéinurie, hématurie et plus rarement d’une HTA généralement modérée. Si elle n’est pas traitée précocement, la MF peut conduire à une insuffisance rénale terminale (plus tardive) [5]. Le diagnostic histologique est évident.
- MO: le cytoplasme des cellules glomérulaires (en particulier des podocytes) et tubulaires distales est intensément vacuolisé par les dépôts lipidiques.
IF: négative,
MO: n’est pas utile [6].
- Dermatologique : (très fréquentes) : apparaissent généralement entre 3 et 20 ans (angiokératomes) ; une hypohidrose, une anhidrose, résultant en une intolérance à la chaleur, sont fréquentes ;
- Acroparesthésies :
- Brûlures des extrémités,
- Régression spontanée des acroparesthésies au cours du temps,
- EMG normal,
- Possible intérêt des potentiels évoqués laser, du sudoscan,
Facteurs déclenchant : exercice, fatigue, émotions, variations de température (chaleur ou froid). Signes associés : fièvre, syndrome inflammatoire biologique.
Les angiokératomes représentent la manifestation cutanée la plus fréquente mais ne sont pas spécifiques de cette maladie, et doivent être distingués des angiokératomes isolés ou associés à des maladies de système. Autres signes cutanés: des lésions buccales, des œdèmes des membres inférieurs, etc.
- Cardiaques : L’accumulation progressive de Gb3 est à l’origine d’une vasculopathie systémique et de processus pro-fibrosants.
Les hommes hémizygotes, mais également les femmes hétérozygotes, peuvent être touchés : 48,6% des hommes et 36,4% des femmes avaient au moment du diagnostic de la MF, une HVG [7].
Dans la forme classique : hypertrophie ventriculaire, cardiomyopathie hypertrophique typiquement concentrique (mais pouvant aussi être asymétrique) survient habituellement à partir de 40 ans, souvent asymptomatique dans ses débuts, troubles de la conduction (PR court à la phase précoce puis possibilité de BAV ou dysfonction sinusale à la phase tardive) et/ou du rythme (ACFA, arythmie maligne), insuffisance cardiaque, angor, valvulopathies.
Des variants cardiaques, correspondant à des mutations particulières et une activité enzymatique résiduelle avec pour conséquence une symptomatologie principalement cardiaque d’apparition plus tardive, ont été décrits. Leur fréquence est mal connue [8].
|
Figure 1 : Cornée verticillée |
Figure 2 : Angiokératome |
|
Figure 3 : AVC ischémique |
Figure 4 : Cardiomyopathie hypertrophique |
- Ophtalmiques : polymorphes sans traduction clinique. Les dépôts cornéens (signe quasi-pathognomonique) sont visibles uniquement par examen à la lampe à fente.
Des tortuosités des micro-vaisseaux rétiniens et conjonctivaux sont par ailleurs classiques, thrombose de la veine centrale de la rétine.
|
o Dystrophie cornéenne (cornée verticillée) : Signe précoce pouvant être présent dès l’âge de 2 ans, n’altérant peu ou pas la vision. |
|
o Manifestation la plus fréquente : jusqu’à 90% des patients |
|
o Diagnostic différentiel : |
|
o Fucosidose (retard mental ++) o Traitements chroniques : antipaludéens de synthèse, amiodarone, phénothiazines… (liste non exhaustive). |
- Cochléo-vestibulaire, (très fréquente) : uni- ou bilatérale.
L’hypoacousie le plus souvent progressive pouvant aller jusqu’à la cophose et vertiges sont fréquemment observés,
- Pulmonaire : pseudo-asthme, dyspnée,
- Gastro-intestinales (non spécifiques) : douleurs abdominales, diarrhée, constipation, alternance diarrhée-constipation, inappétence,
- Rhumatologique : Ostéopénie/ostéoporose.
- Neurologique : Atteinte périphérique. Les signes de souffrance neurologique périphérique sont dominés par les crises douloureuses et les acroparesthésies chroniques touchant 84% des hommes et 79% des femmes. Apparaissant plus tôt chez les garçons (avant 10 ans), que chez les filles hétérozygotes [9].
Atteinte centrale (entre 20 et 40% des patients) : les accidents vasculaires cérébraux dans la majorité des cas ischémiques sont surtout vertébrobasilaires : 67% chez les hémizygotes, 60% chez les hétérozygotes [10].
La maladie est généralement reconnue après la deuxième décennie devant des complications : un ou plusieurs AIT ou AVC sans facteur de risque particulier.
|
La maladie de Fabry est responsable de : o Environ 1% des mises en dialyse inexpliquées ; o Environ 1% des cardiomyopathies hypertrophiques (CMH) inexpliquées ; o Environ 1% des accidents vasculaires cérébraux ischémiques inexpliqués [11]. |
L’examen physique, évalue l’impact sur la qualité de vie : syndrome douloureux, retentissement psychologique, scolaire ou socioprofessionnel, dépression, etc.
- Méthodes diagnostiques
La maladie de Fabry est un trouble du métabolisme des glycosphingolipides due à une activité déficiente (ou absente) d’une hydrolase, l’alpha-galactosidase-A lysosomale liée à des mutations dans le gène GLA (Xq21.3-q22).
Le diagnostic est surtout clinique et doit être évoqué devant une histoire personnelle et/ou familiale évocatrice; il est confirmé par le dosage de l’activité enzymatique au sein des leucocytes ou bien par étude moléculaire.
Le diagnostic de laboratoire définitif implique la démonstration d’un déficit enzymatique accentué chez les hommes hémizygotes. L’analyse enzymatique peut aider à détecter les hétérozygotes (souvent peu concluant dû à l’inactivation X-chromosomique aléatoire), ce qui rend les tests moléculaires (génotypage) obligatoires pour les femmes.
Actuellement, la mesure est réalisée avec un substrat synthétique, la spécificité de la réaction étant assurée par l’addition d’un inhibiteur de l’alpha-galactosidase B : la N-acétyl D-galactosamine. L’activité est non détectable ou très fortement diminuée dans les formes sévères classiques ; une activité résiduelle de 1 à 12% peut être retrouvée dans les formes plus modérées et en particulier dans le variant « cardiaque » porteur de la mutation p.Asn215Ser [4].
Le déficit étant ubiquitaire, tous les milieux biologiques permettent le diagnostic : plasma, taches de sang séchées sur papier buvard, leucocytes, lymphoblastes, fibroblastes, biopsies tissulaires. Tout déficit dans le plasma doit être vérifié dans les leucocytes en raison d’exceptionnels faux positifs et faux négatifs. L’analyse du gène codant pour l’alpha-galactosidase A (gène GLA), qui n’est pas indispensable au diagnostic chez l’hémizygote, est nécessaire à la réalisation de l’enquête familiale [4].
Chez les femmes, le diagnostic est plus délicat : S’il existe un cas masculin dans la famille, le diagnostic d’hétérozygotie repose sur la mise en évidence de la mutation précédemment identifiée chez le malade. Dans le cas où aucun cas familial n’est connu ou étudié, le diagnostic de certitude est plus difficile à établir :
o L’activité enzymatique de l’alpha-galactosidase-A peut être normale chez une femme hétérozygote du fait d’une inactivation préférentielle de l’X muté.
o L’excrétion du Gb3 urinaire (en dehors du cas de la mutation p.Asn215Ser et des malades ayant eu une greffe rénale), est généralement augmentée modérément mais peut être normale chez les hétérozygotes.
o Le séquençage complet du gène GLA est donc souvent nécessaire pour confirmer le diagnostic [4].
Annonce du diagnostic : Il s’agit d’une double annonce : celle de la maladie chronique grave et celle de l’origine génétique. Les circonstances de l’annonce vont avoir une incidence directe sur le processus d’adaptation personnelle et familiale à la maladie à long terme. Il convient donc de prendre le temps de l’annonce et de revoir le patient dans un deuxième temps pour répondre de nouveau à ses questions. Pour un enfant, il est souhaitable que les deux parents soient présents.
L’explication de l’origine génétique doit être fournie avec tact et prudence. Il faut faire comprendre le mode de transmission par les mères lorsqu’elles sont reconnues comme conductrices (hétérozygotes) : probabilité qu’un garçon soit atteint : 50%, qu’une fille soit transmettrice : 50%, en évitant toute notion de culpabilité maternelle, et souligner le fait que les garçons d’un père atteint seront toujours sains, ses filles toujours conductrices, mais non forcément malades.
Diagnostic(s) différentiel(s) : Chez l’enfant, éliminer les autres causes de douleur, telle que la polyarthrite rhumatoïde et des douleurs de croissance doivent être exclues. Chez l’adulte, la sclérose en plaque est à éliminer.
Il est rappelé que le diagnostic doit être confirmé avant toute initiation d’une enzymothérapie substitutive :
- Chez l’homme, par la mise en évidence d’un déficit en alpha-galactosidase-A (activité enzymatique absente ou fortement diminuée) dans les leucocytes. L’activité enzymatique dans le plasma a néanmoins une bonne valeur indicative (faux positifs);
- Chez la femme : par un génotypage (recherche de la mutation du gène GLA), après consentement éclairé écrit pour étude des caractéristiques génétiques, permettant seul d’affirmer si elle est ou non hétérozygote. En effet, une activité enzymatique normale ne permet pas d’exclure un statut d’hétérozygote. Le dosage du Gb3 urinaire peut également aider au diagnostic.
Diagnostic anténatal. Le diagnostic prénatal est basé sur la détermination de l’activité enzymatique ou par des tests ADN des villosités choriales ou des cellules amniotiques cultivées uniquement pour les fœtus mâles. Un diagnostic préimplantatoire est possible. Deux approches possibles:
Première approche: grossesse spontanée. Prise de sang maternel : Diagnostic du sexe fœtal à 10 SA
Garçon : Biopsie de placenta à 12 SA : Dosage enzymatique +/- recherche de la mutation. Demande de diagnostic prénatal quel que soit le sexe fœtal => biopsie de placenta directement sans diagnostic de sexe fœtal préalable.
Deuxième approche : Diagnostic préimplantatoire.
Conseil génétique : La maladie de Fabry est transmise par le chromosome X. L’existence de variantes atypiques et les nouvelles options thérapeutiques compliquent le conseil génétique.
Le dépistage familial s’impose chez tous les apparentés à risque en tenant compte du mode de transmission lié à l’X : par exemple il n’existe pas de transmission père-fils et toutes les filles des hommes atteints de la maladie de Fabry sont porteuses obligatoires.
- Plus de 800 mutations pathogènes ont été décrites dans le gène GLA (Xq22.1, 7 exons).
- Un conseil génétique et un test génétique doivent être proposés à tout patient à risque, par un clinicien possédant une expertise spécifique, au sein d’une équipe pluridisciplinaire comportant au moins un généticien et un psychologue.
- Le dépistage génétique pré-symptomatique des apparentés dans la maladie de Fabry peut être proposé, en l’absence de symptômes, à partir de l’âge de 16 ou 18 ans chez les garçons (en lien avec les recommandations européennes ou françaises pour l’initiation du TES).
- Certaines mutations de GLA, appelées “variants cardiaques” peuvent donner des formes cliniques avec atteinte cardiaque isolée.
- Il peut être intéressant de rechercher une maladie de Fabry devant les cas d’HVG non expliqués par des conditions de charge ou une autre étiologie (HTA, sténose aortique, cœur d’athlète, obésité, CMH sarcomérique, autres maladies infiltratives et métaboliques), a minima par dosage enzymatique chez les hommes de 30 ans et plus.
- Suivi des patients atteints de la maladie de Fabry
La maladie de Fabry nécessite un suivi régulier clinique, biologique et morphologique, par un médecin expert dans les maladies métaboliques, qui coordonne le suivi par les différents spécialistes d’organe (rein, cœur, SNC, yeux). Le suivi cardiologique régulier doit être conseillé et répété tous les ans en cas d’atteinte cardiaque ou de symptômes cardiovasculaires.
- Surveillance cardiologique :
- Évaluation clinique, ECG et ETT : à réaliser tous les ans, ou en cas d’apparition/aggravation de symptômes,
- Holter ECG de 24h et épreuve d’effort : à considérer tous les 2 à 3 ans chez les patients stables, ou en cas d’apparition/aggravation des symptômes,
- IRM cardiaque pour suivi de la fibrose, en fonction de l’atteinte myocardique
- Scintigraphie d’effort, coronarographie, en fonction de l’orientation clinique et paraclinique
- Suivi biologique par dosage urinaire annuel des Gb3 ou lysoGb3 pour évaluer l’observance et l’efficacité de la TES
- Surveillance de la fonction rénale par mesure isotopique du DFG, ionogramme sanguin, créatininémie, protéinurie, créatininurie,
- Surveillance neurologique clinique +/- IRM cérébrale si apparition de nouveaux signes neurologiques ou suspicion d’AIT,
- Évaluation de la douleur et de la qualité de vie,
- Surveillance ORL avec audiogramme +/- PEA,
- Suivi ophtalmologique.
- Pronostic
Avec l’âge, la détérioration progressive peut mener à la défaillance organique. L’insuffisance rénale terminale et des complications cardio- et cérébro-vasculaires, mettant en danger le pronostic vital, réduisent l’espérance de vie des hommes et des femmes de 20 et 15 ans respectivement par rapport à la population générale [2].
Les femmes sont pour la plupart symptomatiques à des degrés divers, mais les manifestations cliniques sont généralement plus atténuées que chez l’homme, et de survenue plus tardive. L’atteinte rénale est moins fréquente et moins sévère. Ceci s’explique probablement par l’inactivation au hasard d’un chromosome X dans chaque cellule. Il n’existe pas d’étude contrôlée, randomisée versus placebo, apportant un niveau de preuve quant à l’intérêt des enzymes de substitution administrées chez la femme hétérozygote asymptomatique ou pauci-symptomatique.
En cas d’hémodialyse ou de dialyse péritonéale :
- La survie des patients dialysés atteints de maladie de Fabry est moins bonne que celle des autres dialysés, diabétiques exclus, possiblement du fait des complications cardiaques ou cérébro-vasculaires inhérentes à la maladie ;
- La mise sous traitement par enzymothérapie substitutive est recommandée, aucune adaptation posologique n’est nécessaire chez les patients présentant une insuffisance rénale.
En cas de greffe rénale :
- La survie des transplantés rénaux est identique dans la maladie de Fabry et dans les autres maladies rénales hors diabète ;
- Le greffon n’est pas recolonisé par le processus pathologique, il possède une activité enzymatique normale,
- Le greffon ne suffit pas pour substituer l’ensemble de la fonction enzymatique et les morbidités cérébro- et cardio-vasculaires persistent ;
- La mise sous traitement peut se justifier.
- Traitement
La prise en charge est multidisciplinaire; elle implique un traitement symptomatique et un traitement spécifique, responsables d’une amélioration à la fois de la survie et de la qualité de vie des sujets atteints. L’enzymothérapie substitutive (ETS) par l’alpha-galactosidase-A recombinante constitue la pierre angulaire du traitement spécifique et peut être associée à d’autres types de traitements comme le galactose et les molécules chaperonnes. La thérapie génique représente elle aussi une approche en plein essor.
Fabrazyme, stabilise la fonction rénale chez 90% des patients sur une période médiane de suivi de 52,2 mois [12].
La dose de 1mg/kg permet une meilleure clairance du GL-3 au niveau des podocytes comparée aux doses plus faibles [13].
Chez la femme hétérozygote, il est recommandé d’initier un traitement par enzymothérapie substitutive en cas de survenue de symptômes cliniques, en particulier en présence de cardiomyopathie, de maladie rénale débutante évolutive, de maladie rénale avérée (protéinurie > 1 g/24 h, existence de lésions vasculaires étendues à la ponction-biopsie rénale, insuffisance rénale modérée à sévère), d’AIT, d’AVC, ou d’atteinte cochléaire sévère.
Chez l’enfant de moins de 18 ans : Il n’existe pas d’étude contrôlée, randomisée versus placebo publiée apportant un niveau de preuve quant à l’intérêt des enzymes de substitution administrées chez l’enfant de moins de 18 ans. Il n’y a pas de preuve scientifique permettant de conclure que le traitement enzymatique substitutif ralentisse l’évolution de la maladie chez l’enfant. En l’absence de données, il n’y a pas lieu de traiter l’enfant de moins de 6 ans. Aucun schéma posologique ne peut être recommandé actuellement dans cette sous-population. Chez l’enfant, en l’état actuel des connaissances [14] :
- Il n’est pas justifié de traiter les enfants asymptomatiques ;
- Il est recommandé d’instaurer un suivi médical annuel en particulier rénal, cardiaque et auditif ;
- Il est justifié d’envisager un traitement par enzymothérapie substitutive dans certaines situations cliniques en rapport avec la maladie :
- Grandes crises douloureuses réfractaires à un traitement antalgique bien conduit (carbamazépine, diphénylhydantoïne, gabapentine, amitryptiline),
- Atteinte rénale organique débutante,
- Atteinte cardiaque,
- Atteinte cochléo-vestibulaire (hypoacousie objectivée par l’audiogramme, crises vertigineuses d’origine vestibulaire),
- Accident vasculaire cérébral ischémique.
Posologies :
- Fabrazyme® : 1 mg/kg de poids corporel, administré une fois toutes les 2 semaines par perfusion intraveineuse. La vitesse de perfusion initiale ne doit pas dépasser 0,25 mg/mn (15 mg/h) afin de minimiser l’éventualité de la survenue de réactions liées à la perfusion ;
- Replagal® : 0,2 mg/kg de poids corporel, administré une fois toutes les 2 semaines par perfusion intraveineuse de 40 minutes [4].
Traitement symptomatique: il améliore la qualité de vie des patients.
- Le traitement de la douleur par l’éviction des facteurs déclenchant et les antalgiques,
- La néphroprotecteur par inhibiteurs de l’enzyme de conversion et antagonistes des récepteurs de l’angiotensine puis prise en charge de l’insuffisance rénale terminale,
- La neuroprotection par aspirine et la cardioprotection par ß-bloquants +/- anti-arythmiques, les pacemakers ou un défibrillateur implantable, et contrôle des facteurs de risque cardiovasculaires.
Molécules chaperonnes : Traitement oral par des molécules pharmacologiques chaperonnes (Migalastat, Amicus Therapeutics). Ces molécules de bas poids moléculaire se lient spécifiquement à certaines formes mutantes de l’α-galactosidase-A pour les stabiliser et ainsi restaurer leur activité lysosomale.
Conclusion
Affection génétique rare et grave de manifestation multisystémique causée par un gène défectueux (le gène GLA). La maladie de Fabry est, en effet, encore souvent diagnostiquée avec retard. La maladie de Fabry est une maladie progressive, destructrice et potentiellement mortelle.
Le défaut du gène provoque une quantité insuffisante de l’hydrolase alpha-galactosidase-A qui est nécessaire à la dégradation quotidienne du globotriaosylcéramide (Gb-3). Lorsque le métabolisme correct de ce lipide ne se produit pas, le Gb-3 s’accumule dans la majorité des cellules de l’organisme.
L’accumulation progressive de lipides qui en résulte entraîne des dommages cellulaires qui provoquent un large éventail de symptômes légers à graves, y compris des conséquences potentiellement mortelles telles que l’insuffisance rénale, l’arrêt cardiaque et les accidents vasculaires cérébraux, souvent à un âge relativement précoce.
La maladie de Fabry doit être envisagée dès l’enfance, chez les garçons, devant un ou plusieurs des signes cliniques.
Un diagnostic et un traitement précoces peuvent éviter la survenue de nombreuses complications.
.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts en rapport avec cet article.
Références
- Lidove O, Sené T. Diagnostic illustré de la maladie de Fabry. Service de Médecine interne – Rhumatologie Centre de Référence Maladies Lysosomales Site Diaconesses – Croix Saint Simon, Paris. ÉDITÉ PAR PHASE 5 – NOVEMBRE 2017
- Mac Dermot KD, et al. Anderson Fabry disease: clinical and impact of disease in a cohort of 98 hemizygous males. Journal of Medical Genetics 2001; 38: 750 – 760
- Germain DP. Fabry disease. Orphanet J Rare Dis 2010;5:30.
- Protocole national de diagnostic et de soins PNDS Maladie de Fabry. Novembre 2010 undefined application/pdf/2010-12/ald_17_pnds_fabry_vd.pdf
- Warnock et al. Clin J Am Soc Nephrol 5: 371- 378, 2010-9
- Dussol B. Résultats de l’enzymothérapie substitutive sur l’atteinte rénale dans la maladie de Fabry. La presse médicale, Mars 2007.
- Kampmann et al. Int J Cardiol 2008; 130: 367-73
- Seydelmann N, Wanner C, Störk S, Ertl G, Weidemann F. Fabry disease and the heart. Best Pract Res Clin Endocrinol Metab 2015;29:195-204.
- Mehta A et al. Fabry disease defined: baseline, clinical manifestations of 366 patients in the Fabry outcome Survey. Eur J Clin invest 2004; 34: 236-42
- Mitsias P, Levine SR. Cerebrovascular complications of Fabry’s disease. Ann Neurol 1996; 40: 8-17;
- Rolfs A et al. Prévalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet 2005; 366: 794-6
- Germain DP et al. J Am Soc Nephrol. 2007; 18 (5): 1547-1557
- Tondel et al. J Am Soc Nephrol 2013; 24 (1): 137-48
- Robert J. Hopkin, John L. Jefferies, Dawn A. Laney, Victoria H. Lawson, Michael Mauer, Matthew R. Taylor, William R. Wilcox, on behalf of the Fabry Pediatric Expert Panel. The management and treatment of children with Fabry disease: A United States-based perspective. Mol Genet Metab 2015.
[1] Synonyme(s) : ALD 17, FD, Maladie d’Anderson-Fabry, Angiokératome (angiokératose) diffus de Fabry, Angiokératose diffuse universelle, Angiokeratoma corporis diffusum universale, Déficit en alpha-galactosidase A, Sphingolipidose héréditaire de Fabry, Thésaurismose lipoïdique héréditaire.
[2] La maladie de Fabry peut également apparaître dans une famille comme une mutation génétique spontanée initiale (de novo).