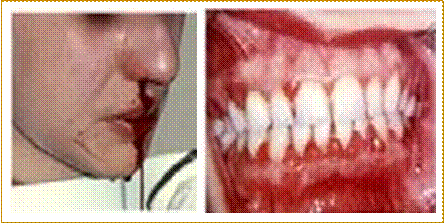
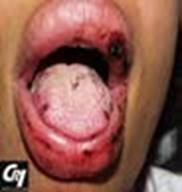
L. KAREK1, F. ALLOUN2, S. SEBTI, B. HADJ MIHOUB, L. BEDDAR1, (1) Service ORL, Hôpital Militaire Universitaire Régional, Blida, (2) Service ORL, Centre d’expertise médicale du personnel navigant, Alger.
Abstract: The incidence of oropharyngeal cancer is currently on the rise and the risk increases with certain sexual habits. Virological and epidemiological data from western countries show that a majority of tonsil and base cancer are linked to human papillomavirus (HPV) infection. The concerned patients are younger and often non-smokers and non-drinkers. These patients have a better oncological prognosis than other patients with squamous cell carcinomas of the ENT sphere. Differences in the pathophysiological mechanisms of carcinogenesis open the possibility of differentiating carcinoma treatment of oropharynx according to the presence of HPV. In addition, the role of promising preventive measures such as vaccination should be investigated.
Key-words: Upper aerodigestive tract, oropharynx, HPV, Algeria.
Résumé : L’incidence du cancer de l’oropharynx est actuellement à la hausse et le risque augmente avec certaines habitudes sexuelles. Des données virologiques et épidémiologiques provenant des pays occidentaux montrent qu’une majorité des cancers de l’amygdale et de la base de la langue sont liés à l’infection à papillomavirus humain (HPV). Les patients concernés sont plus jeunes et souvent non-fumeurs et non-buveurs. Ces patients ont un meilleur pronostic oncologique que les autres patients atteints de carcinomes épidermoïdes de la sphère ORL. Des différences dans les mécanismes physiopathologiques de carcinogenèse ouvrent la possibilité de différencier le traitement des carcinomes de l’oropharynx selon la présence d’HPV. Par ailleurs, le rôle des mesures préventives prometteuses comme la vaccination devra être investigué.
Mots-clés : Cancers des voies aérodigestives supérieures (VADS), oropharynx, HPV, Algérie.
Introduction
Il y a plusieurs différences entre les cancers HPV(+) et les cancers liés au tabac et à l’alcool au point de vue épidémiologique et histologique, mais les différences les plus importantes se trouvent probablement au niveau de la physiopathologie moléculaire. C’est probablement à cause de ces différences que le pronostic des patients HPV(+) est meilleur et qu’une désescalade thérapeutique peut être envisagée.
Caractéristiques épidémiologiques
A. Age et sexe
Les patients avec CVADS HPV positif sont généralement plus jeunes d’environ 10 ans par rapport à ceux HPV négatif, avec une prédominance du sexe masculin, ces patients sont plus susceptibles à être non-fumeurs et non-buveurs.
Gillison[1], dans une étude épidémiologique effectuée sur 253 patients atteints d’un carcinome des VADS, retrouve un âge de survenue plus précoce chez les patients HPV+ par rapport à ceux HPV– (60,5 ans versus 64 ans) avec une prédominance masculine (79 % versus 21 %) quoique, dans quelques rares études on n’a pas retrouvé une différence d’âge significative entre les deux groupes, et même un âge plus élevé chez les patients HPV positif (59,8 ans HPV +/58,2 ans HPV-, selon Hafkamp)[2].
Le jeune âge des patients CVADS HPV positif peut être expliqué par le fait que l’infection HPV survient très précocement dans la vie, dés les premiers rapports sexuels, généralement une période de 15 à 20 ans est suffisante pour l’apparition des lésions précancéreuses après une infection persistante à HPV.
La prédominance masculine est rapportée par la plupart des études avec un sexe ratio moyen de 3 hommes pour une femme, pour certains auteurs, cela est dû à la différence entre la survenue de l’infection HPV par voies génitale et orale[3]. L’infection génitale se produit préférentiellement entre 2 et 5 ans après le début de l’activité sexuelle et sa fréquence diminue considérablement après l’âge de 25 ans, elle laisse une immunité qui peut protéger les femmes contre une réinfection par voie orale, alors qu’elle survient plus tardivement par voie orale chez l’homme avec une fréquence stable dans le temps avec des pics entre 50 et 60 ans [4].
B. Race et distribution géographique
Le statut HPV par race est examiné par Settle[5], dans une vaste cohorte il a constaté un pourcentage significativement plus élevé de cancers HPV positif chez les patients blancs par rapport aux patients noirs (34 % contre 4 %; P = 0,0004).
Plus récemment en 2016, D’Souza[6] dans un échantillon de 1345 cas de CVADS diagnostiqués entre 1995 et 2012, a examiné le statut HPV par race et par sexe, cette analyse montre une nette prédominance de l’incidence des cancers HPV positif chez les hommes blancs.
Pour D’Souza, une augmentation des cancers oropharyngés HPV positif chez les patients blancs en raison de la différence d’accès aux soins entre les deux races, peut expliquer la disparité raciale observée dans cette cohorte. Une autre étude plus récente a montré que la fréquence chez les blancs est plus élevée malgré un apport de soins sans retard pour les deux populations[6].
La distribution géographique des cancers liés à l’HPV est aussi très variable, les cancers des VADS liés à l‘HPV sont observés surtout dans les pays développés, plus de 40 %[7] de l’ensemble de ces cancers sont observés en Europe, en Amérique du Nord, en Australie et en Nouvelle-Zélande. Les taux les plus bas sont observés en Afrique du Nord et les pays du Golf.
Pour la plupart des auteurs, cette variation d’incidence reflète le changement réel des facteurs étiologiques de ces cancers parallèlement au déclin du tabagisme.
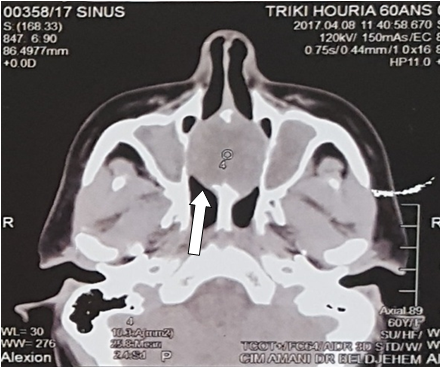
C. Localisation
La présence de Human Papilloma Virus (HPV) est avérée dans certains cancers de la cavité buccale, plus particulièrement dans les localisations oropharyngées, des taux variables sont décrits selon les études, les auteurs rapportent des taux de 45 à 100 % (D’Souza et al. 2007; Kreimer et al. 2005; Nasman et al. 2009) [8, 9,10].
Plusieurs hypothèses sont évoquées pour expliquer cette prédominance oropharyngée:
- Les cryptes linguales et amygdaliennes contiennent, comme le col de l’utérus, de profondes invaginations de la muqueuse connues pour favoriser la rétention de particules virales et la capture d’antigènes qui facilitent l’accès du virus aux cellules basales[9,11,12].
- L’épithélium pavimenteux dérive dans les deux cas (Col/VADS) du même feuillet embryonnaire endodermique.
- La présence de cytokines produites par le tissu tonsillaire peut affecter la transcription des HPV et favoriser la transformation[13].
Caractéristiques histologiques
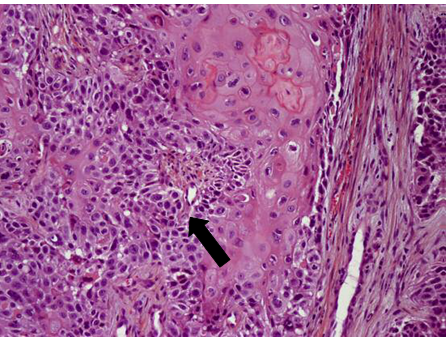
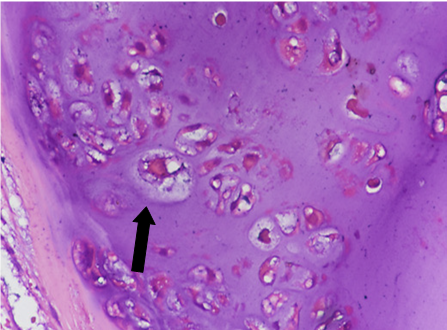
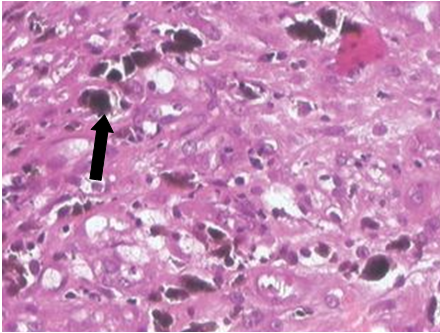
Les cancers VADS HPV positif sont le plus souvent peu différenciés et non kératinisants avec une morphologie de type basaloïde.
L’aspect basaloïde se compose de cellules régulières aux noyaux ovalaires. Elles s’agencent en lobules bordés par des cellules s’organisant en palissade. Au centre des nodules, on observe souvent des plages de nécrose, réalisant des aspects de comédonécrose[14,15].
Le mécanisme exact par lequel les cancers HPV positif acquièrent une morphologie basaloïde non kératinisé, n’est pas connu. Cependant, plusieurs virus oncogènes sont connus pour interférer avec les mécanismes de régulation du cycle cellulaire. Pour Mofty[16], grâce à l’action combinée de ses oncoprotéines virales, l’HPV est capable d’entraîner une prolifération continue des cellules de l’hôte et un désaccouplement entre la prolifération et la différenciation. En effet, une activité mitotique plus élevée par rapport à la kératinisation est observée au microscope dans les tumeurs HPV positif. Ce manque de maturation des cellules tumorales se manifeste par une morphologie basaloïde non kératinisante, cet effet est similaire à l’effet de l’EBV dans les carcinomes du nasopharynx.
Caractéristiques cliniques
Cliniquement, les patients CVADS HPV positif présentent généralement des stades avancés III et IV, avec un T bas et N élevé et des métastases ganglionnaires kystiques à plusieurs niveaux. Le stade avancé des cancers liés à l’HPV est évoqué par la majorité des auteurs, Rosmarie[17], Glenn[18] et Heinrich[19], qui retrouvent une prédominance des stades III et IV avec respectivement (42%, 34%), (38%, 48%) et (29%, 46%).
Le caractère kystique est observé aussi dans différents types de cancers (cancer papillaire de la thyroïde, cancer du col de l’œsophage)[20], il a été suggéré que les kératinocytes qui envahissent les ganglions sont encore capables de reproduire un comportement de croissance et un modèle histologique similaire à leurs tumeurs d’origine. En effet, les tumeurs primitives de l’oropharynx ont tendance à s’enkyster[21,22].
Dans deux études sur les cancers oropharyngés HPV positif, menées par Regauer[23] et Thompson [24], les métastases ganglionnaires kystiques en provenance de l’amygdale étaient respectivement de l’ordre de 61% et 64%.
Ces données ont une très grande valeur dans le diagnostic étiologique des adénopathies métastatiques d’allure primitives. En effet, le caractère kystique des adénopathies cervicales oriente vers une localisation ORL, plus précisément l’oropharynx.
Caractéristiques génétiques et biologiques
Sur le plan génétique, les altérations observées dans les CVADS liés à l’HPV sont minimes par rapport à celles observées dans les CVADS liés à l’alcool et au tabagisme, ces derniers présentent de nombreuses accumulations d’altérations chromosomiques spécifiques décrites dans la littérature.
Elles concernent plus particulièrement une perte d’hétérozygotie des loci 3p, 9p, 17p, une inactivation de l’expression du gène CDKN2A (codant p16) par méthylation de son promoteur, une inactivation du gène codant pRb et des mutations ponctuelles ou des pertes alléliques en 17p13, région du gène codant p53[25].
Ces anomalies sont rarement retrouvées dans les tumeurs liées aux papillomavirus humains. Ces derniers présentent plutôt des pertes chromosomiques spécifiques en 16q24, des gains chromosomiques en 16q11, en 3q24-qter, 18q, Xp et en 11q14 [26, 27, 28]. Ces tumeurs expriment également le plus souvent une protéine p53 fonctionnelle mais dégradée par l’oncoprotéine E6[29].
Sur le plan biologique, les modifications du système immunitaire observées chez les patients atteints de cancers VADS HPV positif reste un domaine d’étude en cours.
Wansom et al. [30] ont comparé rétrospectivement le taux de cellules T dans le microenvironnement tumoral, chez des patients atteints de cancers oropharyngés classés stade III et IV, sous chimiothérapie d’induction suivie soit par radiochimiothérapie ou une chirurgie en fonction de la réponse au traitement. Les patients HPV positif avaient un taux bas du rapport CD4/CD8 avec un pourcentage significativement élevé de lymphocytes T CD8 par rapport aux patients HPV négatif avant le début du traitement. Ce taux initial bas du rapport CD4/CD8 est fortement corrélé à une meilleure réponse à l’induction par chimiothérapie et à la radiochimiothérapie concomitante avec une normalisation de ce rapport après traitement. De même, une baisse ultérieure de ce taux été corrélée à un taux élevé de récidives chez les patients HPV positif.
Le même groupe[31] a ensuite évalué le taux des lymphocytes T (CD4 et CD8) dans le sang périphérique des patients avant le début du traitement. Ils ont constaté que contrairement aux résultats tissulaires où il y avait des taux élevés des CD8, il n’y a pas de différence significative dans les taux sanguins des cellules CD4 et CD8 entre les deux populations (HPV+, HPV-). Par conséquent, il n’existe pas de corrélation entre le taux CD4/CD8 sanguin et le pronostic chez les patients HPV positif.
La modification d’expression du récepteur du facteur de croissance épidermique dans les cancers des VADS liés à l’HPV est examinée par différentes études, ce récepteur qui est une protéine monomérique transmembranaire à laquelle se fixe l’EGF ou facteur de croissance épidermique participant ainsi à la croissance et la réplication cellulaire, dans les cancers HPV positif ce facteur est détourné par les particules virales EGF like qui présentent une grande affinité pour ce récepteur aboutissant ainsi à une forte diminution de son expression. Cette baisse d’expression initiale est corrélée à un bon pronostic. En effet, le traitement par chimio-radiothérapie libère le récepteur de l’EGF des pro-oncogènes virales avec un retour d’une activité mitotique normale. Une baisse ultérieure de l’EGFR est le témoin d’une reprise tumorale chez les patients HPV positif.
La relation entre l’expression P16 et le taux de l’EGFR est examiné par Reimers et al.[32] qui ont analysé les lames de 106 patients atteints d’un cancer HPV positif. Il y avait une tendance à une relation inverse entre l’expression de la p16 et le taux de l’EGFR, les tumeurs qui expriment la p16 présentent un faible taux d’EGFR, ces patients avaient significativement un meilleur taux de survie sans récidive à 5 ans par rapport aux patients avec des taux élevés de l’EGFR et une faible expression de la p16.
Certains auteurs ont étudié le devenir des patients traités par radiothérapie anti-EGFR en fonction du statut p16[33]. Une meilleure survie a été observée chez les patients p16+ traités par radiothérapie et anti-EGFR par rapport à ceux traités par radio-chimiothérapie conventionnelle. Ce bénéfice n’est pas retrouvé dans le groupe de patients p16- (survie à deux ans 80 % versus60 %).
Les auteurs suggèrent que la radio-chimiothérapie restitue la fonction normale de l’EGFR dans les cancers HPV positif contrairement aux cancers HPV négatif ou ce récepteur est déjà muté au départ[34,35]. Actuellement plusieurs essais prospectifs testant l’effet des anti-EGFR dans le traitement des cancers liés à l’HPV sont en cours; celui du Radiation Therapy Oncology Group (RTOG) teste une irradiation avec cisplatine versuscétuximab, d’autres protocoles testeront soit une désescalade des doses soit de bas volumes d’irradiation dont les résultats peuvent êtres prometteurs.
Lassen a évoqué le caractère moins hypoxique des cancers liés à l’HPV dans un essai thérapeutique phase III testant le nimorazole, un radio-sensibilisant des cellules tumorales hypoxiques. Le groupe des patients dont la tumeur ne surexprime pas la p16 avaient un meilleur contrôle locorégional, cet effet radio-sensibilisant n’était pas retrouvé dans le groupe des patients surexprimant la p16 et donc probablement atteints de tumeur liée aux HPV à haut risque, les auteurs ont conclu que les tumeurs liées aux HPV à haut risque sont probablement moins hypoxiques que les tumeurs HPV négatif (2,4,3). Par conséquent, les traitements anti-hypoxiques tel que le nimorazole a peu d’effets sur les cancers HPV positif.
Caractéristiques thérapeutiques
Les différents articles de littérature optent pour la bonne réponse aux différents traitements des cancers HPV positif, les auteurs proposent des schémas thérapeutiques moins agressifs mais jusqu’à présent, aucun consensus thérapeutique spécifique n’est instauré et les traitements actuels ne prennent pas en compte le statut HPV.
La chimio-radiosensibilité des tumeurs HPV est évoquée par la plupart des auteurs, dans une récente étude menée aux USA par Radiation Therapy Oncology Group (RTOG)[36] sur 743 patients atteints de CVADS localement avancé stades III et IV, randomisés pour recevoir une chimio-radiothérapie concomitante, parmi eux 433 patients HPV positif confirmé par test positif de l’ADN viral. Dans cette étude, en comparaison avec les tumeurs oropharyngées HPV négatif, les patients HPV positif avaient un taux de survie à 3 ans significativement amélioré (82,4 % vs 57,1 %; P < 0,001) avec une meilleure survie sans récidives (73,7 % vs 43,4 %; P < 0,001), de même un taux de récidives loco-régionales significativement plus faible par rapport aux patients HPV négatif, mais pas de différence dans le taux de récidives à distance à 3 ans entre les deux populations. En revanche, le statut HPV était un facteur prédictif significatif des résultats, les patients HPV positif avaient une réduction de 58 % de mortalité et de 51 % de récidives et de décès combinés.
Dans une autre analyse rétrospective à partir des données SEER, la survie à long terme était significativement meilleure chez les patients HPV positif (médiane 131 mois vs 20 mois. P < 001) avec une réduction de 69 % du risque de mortalité par rapport aux patients HPV négatif.
Cette étude a également montré que le taux de survie est considérablement amélioré depuis 1984 chez les patients avec tumeur oropharyngée HPV positif, sans aucune amélioration significative en terme de survie chez les patients HPV négatif au cours de cette période. Par conséquent, l’amélioration globale des résultats observés chez les patients atteints d’un cancer oropharyngé durant cette période, semble être uniquement liée à l’amélioration des réponses des patients HPV positif.
Pour certains auteurs, l’augmentation de la réponse à la radio-chimiothérapie des cancers HPV positif peut résulter de l’inactivation induite par E6 sur la protéine p53 plutôt que des mutations dans son gène, anomalies fréquemment retrouvées chez les patients HPV négatif[37]. La chimio-radiothérapie réactive la protéine P53[38] qui reprend son pouvoir apoptotique, cela peut expliquer la meilleure réponse aux traitements chez les patients HPV positif. Toutefois, les résultats satisfaisants obtenus après chirurgie chez ces patients restent inexpliqués.
Une autre théorie évoquée par les auteurs pour expliquer la bonne réponse aux traitements chez les patients HPV positif, est celle de l’effet du champ de cancérisation[39]. Selon cette hypothèse, les tumeurs de la tête et du cou se développent dans un champ de tissu anormal en raison de l’exposition à long terme aux agents cancérigènes du tabac et de l’alcool, ce qui peut conduire à des récurrences locales ou une deuxième localisation primitive. Chez les patients atteints de cancer des VADS HPV négatif, l’étude des marges de résection autour de la tumeur a mis en évidence des tissus anormaux (dysplasies, mutations de p53). Cet effet du champ de cancérisation est absent dans les tumeurs de HPV positif où les anomalies histologiques sont confinées seulement aux tissus néoplasiques[40].
Conclusion
L’avènement des stratégies thérapeutiques actuelles qui ciblent les CVADS, telles que l’immunothérapie anti-EGFR, le transfert adoptif des lymphocytes humains, la radiothérapie par modulation d’intensité et la chirurgie robotique trans-orale, découlent en grande partie des différentes caractéristiques cliniques et biologiques observées dans les cancers HPV positif.
La découverte des cancers HPV positif a permis aussi d’élaborer des critères biologiques efficaces pour le suivi post-thérapeutique des CVADS, parmi ces critères, l’immunohistochimie P16 reste le moyen le plus simple et le moins coûteux pour le suivi et la sélection des patients aptes à la vaccination.
Références
- MAURA L. GILLISON, WAYNE M. KOCH, RANDOLPH B. CAPONE, MICHAEL SPAFFORD, WILLIAM H. WESTRA, LI WU, MARIANNA L. ZAHURAK, RICHARD W. DANIEL, MICHAELVIGLIONE, DAVID E. SYMER, KEERTI V. SHAH, DAVID SIDRANSKY Evidence for a Causal Association Between Human Papillomavirus and a Subset of Head and Neck Cancers Journal of the National Cancer Institute, Vol. 92, No. 9, May 3, 2000
- Hafkamp HC, Manni JJ, Haesevoets A, et al. Marked differences in survival rate between smokers and nonsmokers with HPV 16-associated tonsillar carcinomas. Int J Cancer 2008; 122: 2656–64.
- GIULIANO AR, NYITRAY AG, KREIMER AR, PIERCE CAMPBELL CM, GOODMAN MT, SUDENGA SL, ET AL. EUROGIN 2014 roadmap: differences in human papillomavirus infection natural history, transmission and human papillomavirus-related cancer incidence by gender and anatomic site of infection. Int J Cancer 2015; 136: 2752e60
- GILLISON ML, BROUTIAN T, PICKARD RK, TONG ZY, XIAO W, KAHLE L, ET AL. Prevalence of oral HPV infection in the United States, 2009-2010. JAMA 2012; 307:693e703
- KATHLEEN SETTLE, MARSHALLR. POSNER, LISAM. SCHUMAKER, MING TAN, MOHAN SUNTHARALINGAM, OLGA GOLOUBEVA, SCOTTE. STROME, ROBERTI. HADDAD, SHITALS. PATEL, EARLV. CAMBELL III, NICHOLAS SARLIS, JOCHEN LORCH AND KEVIN J. Cullen Racial Survival Disparity in Head and Neck Cancer Results from Low Prevalence of Human Papillomavirus Infection in Black Oropharyngeal Cancer Patients DOI: 10.1158/1940-6207.CAPR-09-0149
- GYPSYAMBER D’SOUZA, PHD; WILLIAM H. WESTRA, AN AL Differences in the Prevalence of Human Papillomavirus (HPV) in Head and Neck Squamous Cell Cancers by Sex, Race, Anatomic Tumor Site, and HPV Detection Method JAMA Oncol.doi:10.1001/jamaoncol.2016.3067
- CATHERINE DE MARTEL, MARTYN PLUMMER, JEROME VIGNAT AND SILVIA FRANCESCHI Worldwide burden of cancer attributable to HPV by site, country and HPV type International Agency for Research on Cancer, Lyon, France, Int. J. Cancer: 141, 664–670 (2017)
- D’SOUZA G, FAKHRY C, SUGAR EA, ET AL. Six-month natural history of oral versus cervical human papillomavirus infection. Int J Cancer 2007; 121: 143–50.
- AIMEE R. KREIMER, GARY M. CLIFFORD, PETER BOYLE AND SILVIA FRANCESCHI Human Papillomavirus Types in Head and Neck Squamous Cell Carcinomas Worldwide: A Systematic Review DOI: 10.1158/1055-9965.EPI-04-0551 Published February 2005
- NASMAN, A., ATTNER, P., HAMMARSTEDT, L., ET AL., 2009. Incidence of human papillomavirus (HPV) positive tonsillar carcinoma in Stockholm, Sweden: an epidemic of viral induced carcinoma? Int. J. Cancer 125, 362–366.
- PSYRRI A, DIMAIO D. Human papillomavirus in cervical and headand-neck cancer. Oncology 2008; 5: 24-31.
- RINGSM E, PETERS E, HASEGAWA M, POSNER M, LIU M, KELTÖSEY KT. Human papillomavirus type 16 and squamous cell carcinoma of the head and neck. Clin Cancer Res 2002; 8 : 3187-92.
- International Agency for Research on cancer. Mechanistic consideration in the molecular epidemiology of head and neck cancer. IARC monographs on the evaluation of the carcinogenic risk of chemicals to humans, Volume 40. Lyon: IARC; 2004. pp. 393–414
- WEINEKE J, THOMPSON LDR, WENIG BM. Basaloid squamous cell carcinoma of the nasal cavity and paranasal sinuses. Cancer1999;85:841-54.
- BAHAR G, FEINMESSER R, POPOVTZER A, ULANOVSKY D, NAGERIS B, MARSHAK G, ET AL. Basaloid squamous carcinoma of the larynx. Am J Otoryngol 2003; 24:204-8.
- EL-MOFTY. SK, PATIL S. Human papillomavirus (HPV)-related oropharyngeal nonkeratinizing squamous cell carcinoma: characterization of a distinct phenotype. Oral Surg Oral Med Oral Pathol Oral RadiolEndod 2006; 101:339–345.
- ROSEMARIEMICK, MS, EVERETT E. VOKES, MD, RALPH R. WEICHSELBAUM, MD, WILLIAM R. PANJE, MD Chicago. Illinois Prognostic Factors in Advanced Head and Neck Cancer Patients Undergoing Multimodality Therapy https://doi.org/10.1177/019459989110500109
- GLENN W. JONES, FRCPC, GEORGE BROWMAN, FRCPC, MICHAEL GOODYEAR, FRCPC, D. MARCELLUS, FRCPC, AND D. IAN HODSON, frcpc comparison of the addition of t and n integer scores with tnm stage groups in head and neck cancer 1993; 15:497-503
- HEINRICH IRO, FRANK WALDFAHRER, Evaluation of the Newly Updated TNM Classification of Head and Neck Carcinoma with Data From 3247 patients
- GRANSTROM G, EDSTROM S. The relationship between cervical cysts and tonsillar carcinoma in adults. J Oral MaxillofacSurg 1989; 47:16–20.
- THOMPSON LD, HEFFNER DK. The clinical importance of cystic squamous cell carcinomas in the neck: a study of 136 cases. Cancer 1998; 82:944–956
- VERMA K, MANDAL S, KAPILA K. Cystic change in lymph nodes with metastatic squamous cell carcinoma. ActaCytol 1995; 39:478–480.
- REGAUER S, MA NNWEILER S, ANDERHUBER W, ET AL. Cystic lymph node metastases of squamous cell carcinoma of Waldeyer’s ring origin. Br J Cancer 1999; 79:1437–1442.
- THOMPSON LD, HEFFNER DK. The clinical importance of cystic squamous cell carcinomas in the neck: à study of 136 cases. Cancer 1998; 82:944–956.
- PEREZ-ORDONEZ B, BEAUCHEMIN M, JORDAN RC. Molecular biology of squamous cell carcinoma of the head and neck. J ClinPathol 2006; 59:445–53.
- SMEETS SJ, BRAAKHUIS BJ, ABBAS S, SNIJDERS PJ, YLSTRA B, VAN DE WIEL MA, ET AL. Genome-wide DNA copy number alterations in head and neck squamous cell carcinomas with or without oncogene-expressing human papillomavirus. Oncogene 2006; 25:2558–64.
- JUNG AC, BRIOLAT J, MILLON R, DE REYNIES A, RICKMAN D, THOMAS E, ET AL. Biological and clinical relevance of transcriptionally active human papillomavirus (HPV) infection in oropharynx squamous cell carcinoma. Int J Cancer 2009; 126:1882–94.
- DAHLGREN L, MELLIN H, WANGSA D, HESELMEYER-HADDAD K, BJORNESTAL L, LINDHOLM J, ET AL. Comparative genomic hybridization analysis of tonsillar cancer reveals a different pattern of genomic imbalances in human papillomavirus-positive and -negative tumors. Int J Cancer 2003; 107:244–9.
- LICITRA L, PERRONE F, BOSSI P, SUARDI S, MARIANI L, ARTUSI R, ET AL. High-risk human papillomavirus affects prognosis in patients with surgically treated oropharyngeal squamous cell carcinoma. J ClinOncol 2006; 24:5630–6.
- DERRICK WANSOM, BA; EMILY LIGHT, MS; FRANK WORDEN, MD; ET AL Correlation of Cellular Immunity With Human Papillomavirus 16 Status and Outcome in Patients With Advanced Oropharyngeal Cancer Arch Otolaryngol Head Neck Surg. 2010; 136 (12): 1267-1273
- DERRICK WANSOM BA,EMILY LIGHT MS,DAFYDD THOMAS MD,FRANCIS WORDEN MD,MARK PRINCE MD,SUSAN URBA MD,DOUGLAS CHEPEHA MD,BHAVNA KUMAR MS,KITRINA CORDELL DDSAVRAHAM EISBRUCH MD,JEREMY TAYLOR PHD,JEFFREY MOYER MD,CAROL BRADFORD MD,NISHA D’SILVA BDS, MSD, PHD,THOMAS CAREY PHD,JONATHAN MCHUGH MD,GREGORY WOLF MD,UM HEAD NECK SPORE Program Infiltrating lymphocytes and human papillomavirus-16–associated oropharyngeal cancer DOI: 10.1002/lary.22133
- REIMERS N, KASPER HU, WEISSENBORN SJ, STUTZER H, PREUSS SF, HOFFMANN TK, SPEEL EJ, DIENES HP, PFISTER HJ, GUNTINAS-LICHIUS O, ET AL.: Combined analysis of HPV-DNA, p16 and EGFR expression to predict prognosis in oropharyngeal cancer. International journal of cancer 2007, 120(8):1731-1738.
- PAJARES B. Human papillomavirus (HPV)-related head and neck squamous cell carcinoma (HNSCC) and outcome after treatment with epidermal growth factor receptor inhibitors (EGFR inhib) plus radiotherapy (RT) versus conventional chemotherapy CT) plus RT. J ClinOncol2011 ; 29: 5528
- KUMAR B, CORDELL KG, LEE JS, PRINCE ME, TRAN HH, WOLF GT, ET AL. Response to therapy and outcomes in oropharyngeal cancer are associated with biomarkers including human papillomavirus, epidermal growth factor receptor, gender, and smoking. Int J RadiatOncolBiol Phys 2007; 69:S109–11.
- GUPTA AK, LEE JH, WILKE WW, QUON H, SMITH G, MAITY A, ET AL. Radiation response in two HPV-infected head-and-neck cancer cell lines in comparison to a non- HPV-infected cell line and relationship to signaling through AKT. Int J RadiatOncolBiol Phys 2009; 74:928–33.
- GILLISON HJ ML, WESTRA W, CHUNG C, JORDAN R, ROSENTHAL D, NGUYEN-TAN P, SPANOS WJ, REDMOND KP, ANG K: Survival outcomes by tumor human papillomavirus (HPV) status in stage III-IV oropharyngeal cancer (OPC) in RTOG 0129. J Clin Oncol2009, 27:15s.
- [PEREZ-ORDONEZ B, BEAUCHEMIN M, JORDAN RC. Molecular biology of squamous cell carcinoma of the head and neck. J ClinPathol 2006; 59:445–53.
- BUTZ K, GEISEN C, ULLMANN A, SPITKOVSKY D, HOPPE-SEYLER F. Cellular responses of HPV-positive cancer cells to genotoxic anti-cancer agents: repression of E6/E7- oncogene expression and induction of apoptosis. Int J Cancer 1996; 68:506–13
- BRAAKHUIS BJM, LEEMANS RC, BRAKENHOFF RH. Expanding fields of genetically altered cells in head and neck squamous carcinogenesis. Semin in Cancer Biol 2005; 15:113-120.
- GILLISON HJ ML, WESTRA W, CHUNG C, JORDAN R, ROSENTHAL D, NGUYEN-TAN P, SPANOS WJ, REDMOND KP, ANG K: Survival outcomes by tumor human papillomavirus (HPV) status in stage III-IV oropharyngeal cancer (OPC) in RTOG 0129. J ClinOncol2009, 27:15s