« A propos d’une étude observationnelle monocentrique de 28 cas »
N. ZAOUI, A. BOUKABOUS, N. BACHIR, A. TERKI ; Service de Cardiologie, EHS de Cardiologie et Chirurgie cardiaque Omar YACEF Draa Ben Khedda, Université de médecine, Tizi-Ouzou Algérie.
Abstract: Introduction: Pulmonary hypertension (PH) is a rare pathology recognizing five groups of different mechanisms and etiologies. Its diagnosis is suspected by the clinical examination and the ECG and confirmed by echocardiography and cardiac catheterism; its treatment is based on general rules, first and second line treatment (diuretics, oxygenotherapy, anticoagulants and calcium-channel blockers) and specific therapies prescribed in group 1 in case of negative invasive reversibility test (ET1 antagonist, phosphodiesterase inhibitors, prostaglandin analogues). Since the COVID-19 pandemic beginning, post-mortem works report the emergence of a new form of PH related to pulmonary arterial walls thickening, confirmed by clinical observations. Many grey areas persist regarding the management of this new entity. Objective: To describe therapeutic modalities in ambulatory patients with post COVID-19 PH and to compare echocardiographic parameters evolution (PAPS and right ventricle diameter) according to the chosen therapeutic regimen. Methods: This observational, retrospective, single-center study conducted in 2021 involved 28 patients with post COVID-19 PH confirmed with echocardiography and cardiac catheterism. Patients with previous history of PH or evidence of pulmonary embolism were excluded (n = 04). According to invasive reversibility test, patients were divided in two groups, with different treatment regimen (Calcium channel blockers VS Bosentan+Iloprost) and followed for 3 months. TTE findings were compared in these two groups at first and third month of follow-up. Results: Twenty-eight patients (17 men and 11 women from 26 – 64 years) with post-COVID-19 PH (severe COVID-19 form: lung involvement > 50 % at the initial CT) and post-infection time of 28 +/- 4 days, were included. All patients had positive ESR and CRP at baseline. The average PAP in TTE was 80 +/- 15 mmHg. Mean telediastolic RV diameter in TTE was 43 +/- 7 mm/m². Cardiac catheterization revealed a positive reversibility test in nine patients (put on Diltiazem) and negative in 19 patients (put on Bosentan + Iloprost). All patients reported functional improvement, without significant improvement in echocardiographic parameters at 1 month (Δ sPAP at 7 +/- 3 mmHg and Δ RV Diameter 4 +/- 2 mm) and 3 months follow-up (Δ sPAP 10 +/-2 mmHg and Δ RV Diameter 6 +/- 2 mm). This improvement was greater in Bosentan + Iloprost group compared to Diltiazem group for sPAP and RV diameter at 3 months (Δ sPAP 13 +/- 4 mmHg VS Δ sPAP 4 +/- 2 mmHg, P at 0.07 and Δ RV 8 +/- 3 mm VS Δ RV 3 +/-2 mm, P at 0.08). Discussion: Severity of lung involvement on CT, oxygen requiring and the persistence of biological inflammation appear to be associated with a more unfavorable course. Symptomatic evolution seems favorable under treatment however a longer delay is necessary to confirm an objective TTE improvement that seem higher with specific therapeutics, suggesting rapid worsening of vascular parietal rearrangement that may explain poorer results despite a positive reversibility test. Conclusion: PH-post COVID-19 requires early identification and targeted management to improve patient prognosis. The specific treatment of PH could be, in the future, the first intension treatment of this entity, regardless of reversibility test results.
Key-words: Series, COVID 19, SARS-CoV1, PH pulmonary hypertension, precapillary, PAH, Bosentan, prostaglandin analogues.
Résumé : Introduction: L’hypertension pulmonaire (HTP) est une pathologie rare qui reconnaît cinq groupes de mécanismes et d’étiologies différents. Son diagnostic est suspecté par l’examen clinique et l’ECG et confirmé par l’échocardiographie et le cathétérisme cardiaque; son traitement est basé sur des règles générales, un traitement de première et de deuxième intention (diurétiques, oxygénothérapie, anticoagulants et inhibiteurs calciques) et des thérapies spécifiques prescrites dans le groupe 1 en cas de test de réversibilité invasif négatif (antagoniste ET1, inhibiteurs de la phosphodiestérase, analogues des prostaglandines). Depuis le début de la pandémie de la COVID-19, des études sur des travaux post-mortem, rapportent l’émergence d’une nouvelle forme d’HTP liée à l’épaississement des parois artérielles pulmonaires, confirmée par des observations cliniques. De nombreuses zones d’ombre persistent en ce qui concerne la gestion de cette nouvelle entité. Objectif: Décrire les modalités thérapeutiques chez les patients pris en charge en ambulatoire atteints d’HTP post COVID-19 et comparer l’évolution des paramètres échocardiographiques (PAPS et diamètre du ventricule droit) selon le schéma thérapeutique choisi. Méthodes: Cette étude observationnelle, rétrospective et monocentrique menée en 2021 a porté sur 28 patients atteints d’HTP post COVID-19 confirmée par échocardiographie et cathétérisme cardiaque. Les patients ayant des antécédents d’HTP ou des signes d’embolie pulmonaire ont été exclus (n = 04). Selon le test de réversibilité invasif, les patients ont été divisés en deux groupes, avec un schéma thérapeutique différent (inhibiteurs calciques VS Bosentan + Iloprost) et suivis pendant 3 mois. Les résultats ETT ont été comparés dans ces deux groupes aux premier et troisième mois de suivi. Résultats: Vingt-huit patients (17 hommes et 11 femmes âgés de 26 à 64 ans) atteints d’HTP post-COVID-19 (forme COVID-19 sévère: atteinte pulmonaire > 50 % à la tomodensitométrie initiale) et post-infection de 28 +/- 4 jours, ont été inclus. Tous les patients avaient une VS et une CRP positives au départ. La PAPS moyenne en ETT était de 80 +/- 15 mmHg. Le diamètre VD moyen télédiastolique en ETT était de 43 +/- 7 mm/m². Le cathétérisme cardiaque a révélé un test de réversibilité positif chez neuf patients (mis sous Diltiazem) et négatif chez 19 patients (mis sous Bosentan + Iloprost). Tous les patients ont signalé une amélioration fonctionnelle, sans amélioration significative des paramètres échocardiographiques à 1 mois (Δ PAPS à 7 +/- 3 mmHg et Δ Diamètre VD 4 +/- 2 mm) et 3 mois de suivi (Δ PAPS 10 +/-2 mmHg et Δ Diamètre VD 6 +/- 2 mm). Cette amélioration a été plus importante dans le groupe Bosentan + Iloprost par rapport au groupe Diltiazem pour la PAPS et le diamètre VD à 3 mois (Δ PAPS 13 +/- 4 mmHg VS Δ PAPS 4 +/- 2 mmHg, P à 0,07 et Δ VD 8 +/- 3 mm VS Δ VD 3 +/-2 mm, P à 0,08). Discussion: La gravité de l’atteinte pulmonaire sur la tomodensitométrie, le besoin d’oxygène et la persistance de l’inflammation biologique semblent être associés à une évolution plus défavorable. L’évolution symptomatique semble favorable sous traitement, mais un délai plus long est nécessaire pour confirmer une amélioration ETT objective qui semble plus élevée avec les thérapies spécifiques, suggérant une aggravation rapide du remodelage pariétal vasculaire qui peut expliquer des résultats non satisfaisants chez des patients avec cependant, un test de réversibilité positif. Conclusion: L’HTP post COVID-19 nécessite une identification précoce et une prise en charge ciblée pour améliorer le pronostic du patient. Le traitement spécifique de l’HTP pourrait être, à l’avenir, le traitement de première intention de cette entité, quel que soit le résultat des tests de réversibilité.
Mots-clés : Série, COVID 19, SRAS-CoV1, PH hypertension pulmonaire, précapillaire, HTAP, Bosentan, analogues de la prostaglandine.
Introduction
L’hypertension pulmonaire (HTP) est une pathologie rare et peu connue [1], définie selon le sixième Congrès mondial de l’HTP (Nice 2018), comme une PAPm ≥ 20 mmHg [2,3]. Il n’y a pas assez de données pour définir l’HTP à l’effort, l’ancienne définition de l’HTP avec une PAPm ≥ 30 mmHg a été abandonnée en 2008 en raison d’une grande variabilité de la valeur normale de la pression pulmonaire à l’effort [2, 3].
Sa prévalence est estimée à 97 cas par million d’habitants avec un ratio ♀ /♂ pour 1,8 (Royaume-Uni) [2,4]; sa mortalité est estimée entre 4,5 et 12,3 pour 10 000 habitants (USA) [2, 3, 4].
L’HTP est classée en 5 groupes selon l’étiologie et le mécanisme physiopathologique (tableau 1) [3, 5].
Le diagnostic est suspecté par la symptomatologie et l’examen clinique [1, 2, 3], spécifié par de simples explorations telles que l’ECG [2] et la radiographie thoracique et confirmé par l’échocardiographie transthoracique (TTE) et le cathétérisme cardiaque [2,6].
D’autres investigations, telles que les tests de la fonction pulmonaire, la gazométrie artérielle, la scintigraphie pulmonaire ventilation/perfusion et la tomodensitométrie thoracique, recherchent une maladie pulmonaire ou des voies respiratoires et une HTP post-embolique [4,6].
L’IRM cardiaque évalue la taille, la morphologie et la fonction de la VD si l’ETT est de mauvaise qualité [6].
La gestion des HTAP du groupe 1 est divisée en 4 volets [2, 3]:
Mesures générales: Eviction des grossesses, vaccinations anti-grippal annuelle et anti-pneumococcique tous les 5 ans, une activité physique modérée et supervisée est conseillée tandis qu’une activité intense est interdite, dans le cas où une intervention chirurgicale est prévue, la péridurale est préférable.
Traitement de première et deuxième intention: Diurétiques, oxygénothérapie de longue durée (16 à 18H/24H si la pression artérielle d’oxygène est < 60 mmHg, objective > 65 mmHg), anticoagulation orale et inhibiteurs calciques à forte dose si test de réversibilité positif au cathétérisme cardiaque.
Traitement spécifique: Effet vasodilatateur, antiprolifératif et antifibrotique avec 3 molécules :
- Prostacyclines et analogues (Iloprost inhalé et tréprostinil sous-cutané) ;
- Antagoniste de l’endothéline ET-1 (Bosentan) ;
- Inhibiteurs de la phosphodiestérase-5 (sildénafil, tadalafil).
Autres traitements: Inotropes en cas d’hypotension, transplantation cœur-poumon et atriotomie septale par ballonnet.
Dans les groupes 2 et 3, la prise en charge est basée sur le traitement de la pathologie causale; l’utilisation de thérapies spécifiques de l’HTP dans ces deux groupes n’est pas indiquée.
Dans le groupe 4, l’anticoagulation à vie et l’endartériectomie chirurgicale sont indiquées, une dilatation pulmonaire par ballonnet peut être envisagée si le risque de chirurgie est important; l’utilisation de thérapies spécifiques à l’HTP (en monothérapie ou en bithérapie) est possible.
Tableau 1: Classification de l’HTP selon le sixième Congrès mondial de l’HTP (NICE 2018) [3, 5]
| Groupes | HTP Groupe 1 | HTP Groupe 2 | HTP Groupe 3 | HTP Groupe 4 | HTP Groupe 5 |
| Mécanisme | Précapillaire | Postcapillaire | Précapillaire | Précapillaire | Mixte |
| Etiologie | HTAP (exclusion des pathologies pulmonaires et post-embolique) | Pathologies du cœur gauche | Maladies respiratoires | Occlusion de l’artère pulmonaire | Mixte |
| Détail | Héréditaire | Valvulopathies | Bronchopneumopathie Chronique Obstructive (BPCO) | Post embolique | Sarcoïdose |
| Idiopathique | Sténose des veines pulmonaires | Pneumopathies interstitielles | Obstruction non-embolique | Neuro-fibromatose | |
| Associée à: Connectivites, Hypertension portale Schistosomiase HIV Cardiopathies congénitales | Dysfonction systolique ou diastolique | Insuffisance respiratoire restrictive ou obstructive | – | Pathologie métabolique (Glycogénose, maladie de Gaucher, Dysthyroïdie) | |
| Toxique | – | Altitude | – | Hémopathies |
Contexte et justification
Depuis la fin 2019, le monde connaît une pandémie sans précédent; les données de la littérature rapportent une faible incidence d’atteinte virale chez les patients atteints d’HTP [7].
Une enquête internationale auprès des centres de référence pour l’HTP dans 28 pays, a confirmé cette faible incidence avec cependant un taux de mortalité plus élevé que dans la population générale (19 % vs 3 – 5 %) [8].
La vasoconstriction artérielle pulmonaire et l’hypoxémie pendant l’HTP, provoquent une dysfonction endothéliale et une polyglobulie, toutes deux responsables d’une libération accrue de NO endothélial et par les globules rouges.
L’excès de NO pourrait expliquer, que la dysfonction endothéliale de la phase hyper-inflammatoire de la COVID-19, avec ses conséquences thrombotiques, est moindre [9] et jouerait un rôle dans la réplication virale. En effet, Akerström et al. en 2005 ont prouvé que le NO inhibe le cycle de réplication du SARS-CoV1 du H1N1, il pourrait être identique dans la COVID-19 [8, 9].
Les travaux post-mortem ont, toutefois, rapporté que cette pandémie pourrait conduire à l’émergence d’une nouvelle forme d’HTP en mettant en évidence des parois artérielles pulmonaires épaissies chez les patients décédés de la COVID-19 [10], ces lésions histologiques n’ont pas été trouvées dans le SRAS-CoV1 de la grippe H1N1 en 2002-2004 [10, 11]. Les observations cliniques ont confirmé cette hypothèse [11, 12].
De nombreuses séries suggèrent l’utilisation du NO dans les formes les plus sévères ou dans la phase aiguë avec une proportion claire d’amélioration fonctionnelle [13].
Des zones d’ombre persistent en ce qui concerne la gestion de cette nouvelle entité. Le traitement à long terme et en l’absence de preuves, utilise actuellement le schéma thérapeutique traditionnel de l’HTAP du groupe 1 avec des mesures générales, des traitements de première intention, des inhibiteurs calciques dans les formes réversibles et des thérapies spécifiques dans les formes irréversibles avec en première intention, la bithérapie [14].
La plupart des séries décrites dans la littérature rapportent un pronostic plus sombre de cette nouvelle forme par rapport aux HTAP non COVID-19 malgré un traitement bien conduit [14].
Objectif : L’objectif de cette étude est de décrire les modalités thérapeutiques et évolutives des patients atteints d’HTP post COVID-19 et de comparer l’évolution des paramètres échocardiographiques de l’HTP (PAPS diamètre du ventricule droit) en fonction du schéma thérapeutique choisi (inhibiteurs calciques ou traitement spécifique en bithérapie: Bosentan + Iloprost).
Méthodes
Conception et contexte de l’étude : Cette étude observationnelle, rétrospective et monocentrique a été menée en 2021, pendant la pandémie de COVID-19, dans un centre de gestion de l’HTP à partir des données d’un registre prospectif recueillant les données cliniques, biologiques et l’imagerie des patients atteints d’HTP (toute forme confondue).
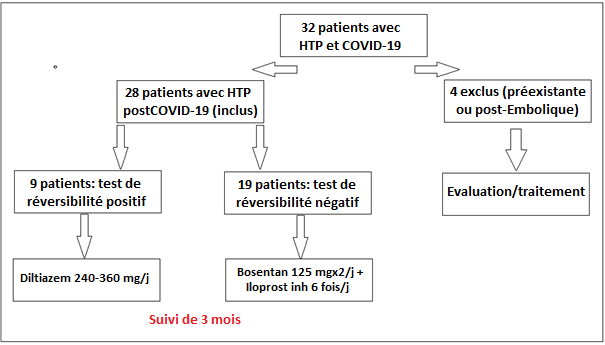
Participants: Ont été inclus dans l’étude, tous les patients référés à notre centre pour HTP post COVID-19 et confirmés à l’ETT et au cathétérisme cardiaque (total de 32 patients).
Ont été exclus de l’étude, les patients ayant des antécédents d’HTP ou des signes d’embolie pulmonaire (n = 04).
Selon le test de réversibilité invasif, les patients ont été divisés en deux groupes, avec un schéma thérapeutique différent (Diltiazem forte dose VS Bosentan + Iloprost) délivré par l’hôpital selon le protocole habituel de prise en charge de l’HTP dans notre centre et suivi pendant 3 mois.
L’état fonctionnel et les résultats échocardiographiques ETT ont été comparés dans ces deux groupes aux premier et troisième mois de suivi.
Tous les participants ont donné rétrospectivement leur consentement pour le partage des données de ce travail.
Variables et évaluation: Le statut fonctionnel et le périmètre de marche ont été évalués par interrogatoire.
Les paramètres échocardiographiques ont été mesurés sur un échocardiographe GE:
Le diamètre du VD (VDd) a été mesuré en télédiastole en fenêtre apicale à 4 cavités et indexé automatiquement à la surface corporelle [2,3].
La PAP systolique (PAPS) a été estimée par le flux de régurgitation tricuspide avec la formule: PAPS = 4 × vitesse de la fuite tricuspide² + pression de l’oreillette droite (POD) [2,3].
Le cathétérisme cardiaque a été effectué en salle GE Optima, le test de réversibilité a été effectué chez tous les patients, soit par NO (20 ppm), soit par Iloprost inhalé (2,5 μg), selon les préférences de l’opérateur et a été considéré comme positif pour tout test montrant une diminution de 10 mmHg de PAP (valeur absolue < 40 mmHg) sans diminution du débit cardiaque [2].
Gestion des biais d’étude :
Biais de sélection: Afin de réduire ces biais et de rendre la population étudiée aussi représentative que possible de la pratique quotidienne, nous n’avons pas limité les points d’origine des patients dont le recrutement a été successif. Pour rappel, notre centre est un centre convergence pour les patients atteints d’HTP.
Biais de vérification: Tous les patients inclus dans l’étude ont bénéficié du test de référence obligatoire (mesure de la PAPS au cathétérisme cardiaque).
Biais d’interprétation: Face au risque de contamination et à la charge de travail imposée par la pandémie, nous n’avons pas effectué d’évaluation en double aveugle pour les deux tests de notre étude (ETT et cathétérisme cardiaque), mais chaque paramètre a été confirmé par le même opérateur sur deux mesures différentes.
Analyse statistique: Toutes les données ont été recueillies sur le logiciel EPI-INFO 7. Les résultats ont été exprimés en pourcentage pour les variables qualitatives et en moyenne ± écart-type (ET) pour les variables quantitatives.
Les analyses bivariées de l’évolution des paramètres ETT selon le schéma thérapeutique ont été réalisées selon le χ² et le test de Fisher pour les variables qualitatives et le test de Student pour les variables quantitatives.
La valeur P < 0,10 a été considérée comme statistiquement significative.
Résultats
Participants et données descriptives: Vingt-huit patients (17 hommes et 11 femmes), âgés de 26 à 64 ans (âge moyen 47 +/- 3,4 ans), atteints d’HTP post COVID-19 confirmée par ETT et cathétérisme cardiaque droit, ont été inclus (figure 1). Tous les patients ont présenté une forme sévère de COVID-19 (atteinte pulmonaire > 50 % à la tomodensitométrie initiale) et ont nécessité une hospitalisation et une oxygénation à haut débit (> 10 l/min).
Les patients ont été référés à notre service avec un délai post-infection COVID-19 moyen de 28 +/- 4 jours (15 – 45 jours).
La moitié des patients avaient un taux de dimères positif (moyenne à 2300 μg /l) lors de l’infection initiale, ces données étaient manquantes chez huit patients tandis que les six autres avaient un taux normal (< 500 μg /l).
Tous les patients avaient une VS et une CRP positives au départ, témoins d’une inflammation persistante.
La PAPS moyenne à l’échocardiographie était de 80 +/- 15 mmHg.
Le VDd moyen était de 43 +/- 7 mm/m² avec dilatation chez 21 patients (75 %).
Le cathétérisme cardiaque a révélé une HTP chez tous nos patients avec une PAP moyenne de 58 +/- 8 mmHg.
Le test de réversibilité est revenu positif chez 9 patients, mis sous Diltiazem 240 à 360 mg répartis en 3 fois par jour et négatif chez 19 patients ayant nécessité une combinaison: Bosentan 125 mg x2 /jour avec Iloprost inhalé 6 fois/jour.
Le traitement a été initié à l’hôpital, puis les patients ont été autorisés à quitter l’hôpital en fonction de leur évolution clinique.
Analyse : Tous les patients (100 %) ont été examinés 1 et 3 mois après le début du traitement et ont signalé une amélioration fonctionnelle et du périmètre de marche sans amélioration significative des paramètres échocardiographiques à 1 mois (ΔPAPS à 7 +/- 3 mmHg et ΔVDd 4 +/- 2 mm) et à 3 mois (Δ PAPs 10 +/-2 mmHg et ΔVDd 6 +/- 2 mm) (tableau 2 et tableau 3).
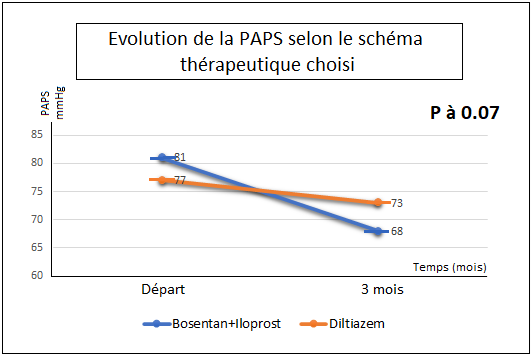
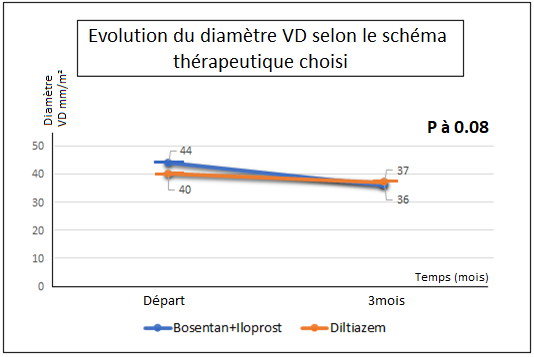
Cependant, cette amélioration était meilleure dans le groupe Bosentan + Iloprost par rapport au groupe Diltiazem pour la PAPS échocardiographique et la VDd à 3 mois (ΔPAPS 13 +/- 4 mmHg VS ΔPAPS 4 +/- 2mmHg, P 0.07 et ΔVDd 8 +/- 3mm VS ΔVDd 3 +/-2mm, P 0.08) (tableau 4 – figures 2-3).
Tableau 2: Evolution des paramètres échocardiographiques à 1 mois dans la population générale de l’étude
| Départ | 1 mois | Variation: Δ | P | |
| PAPS | 80 +/- 15 mmHg | 73 +/- 12 mmHg | 7 +/- 3 mmHg | 0.62 |
| Diamètre VD | 43 +/- 7 mm/m² | 39 +/- 5 mm/m² | 4 +/- 2 mm/m² | 0.43 |
Tableau 3: Evolution des paramètres échocardiographiques à 3 mois dans la population générale de l’étude
| Départ | 3 mois | Variation: Δ | P | |
| PAPS | 80 +/- 15 mmHg | 70 +/- 13 mmHg | 10 +/- 2 mmHg | 0.41 |
| Diamètre VD | 43 +/- 7 mm/m² | 37 +/- 5 mm/m² | 6 +/- 2 mm/m² | 0.29 |
Tableau 4: Comparaison de l’évolution échocardiographique à 1 et 3 mois entre les 2 groupes de l’étude
| Bosentan + Iloprost | Variation | Diltiazem | Variation | P | |
| PAPS départ | 81 +/- 14 mmHg | – | 77 +/- 12 mmHg | – | – |
| PAPS 1 mois | 73 +/- 11 mmHg | 8 +/- 3 | 76 +/- 10 mmHg | 1 +/- 2 | 0.18 |
| PAPS 3 mois | 68 +/- 10 mmHg | 13 +/- 4 | 73 +/- 10 mmHg | 4 +/- 2 | 0.07 |
| VD départ | 44 +/- 6 mm/m² | – | 40 +/- 4 mm/m² | – | – |
| VD 1 mois | 40 +/- 4 | 4 +/- 2 | 38 +/- 3 mm/m² | 2 +/- 1 | 0.23 |
| VD 3 mois | 36 +/- 3 mm/m² | 8 +/- 3 | 37 +/- 2 mm/m² | 3 +/- 2 | 0.08 |
Discussion
Limites : Le petit nombre de patients de notre série ne permet pas d’identifier les facteurs prédictifs d’installation d’HTP après une infection à la COVID-19; cependant, il semble que la gravité de l’atteinte pulmonaire sur la tomodensitométrie ainsi que le recours à l’oxygénothérapie et la persistance de l’inflammation biologique après une infection COVID-19, sont associés à une évolution plus défavorable.
Le niveau élevé de dimères semble également être associé à l’installation de l’HTP, mais les données manquantes dans notre série empêchent toute conclusion dans ce sens.
Principaux résultats et interprétation: L’évolution symptomatique semble favorable sous traitement; cependant, un temps de suivi plus long semble nécessaire pour confirmer une amélioration objective de la PAPS et du VDd qui est plus favorable avec les thérapies spécifiques par rapport aux inhibiteurs calciques suggérant une évolution persistante et rapide du phénomène de remodelage pariétal vasculaire qui peut expliquer des résultats échocardiographiques moins satisfaisants malgré un test de réversibilité positif.
Nous sollicitons d’autres centres à coordonner efficacement pour la transmission de leurs résultats et ce, pour disposer d’un système important de collecte de données.
Conclusion
L’HTP-post COVID-19 nécessite une identification précoce et une prise en charge ciblée pour améliorer le pronostic du patient. Le traitement spécifique de l’HTP pourrait être, à l’avenir, le traitement de première intention de cette entité, quels que soient le résultat des tests de réversibilité.
Ce que nous en savons
- L’HTP post COVID-19 est une entité de plus en plus décrite dans la littérature; elle fait partie du groupe 1 et est due à un épaississement des parois des artères pulmonaires ;
- Le traitement de cette forme utilise les médicaments dédiés à l’HTP du premier groupe ;
- Le pronostic semble être plus sombre que dans la population générale.
Ce que cette étude ajoute
La gravité de l’atteinte pulmonaire et la persistance de l’inflammation après la phase infectieuse semblent prédisposer à l’apparition de l’HTP.
- Les patients sous traitement spécifique ont une meilleure évolution que les patients sous inhibiteurs calciques et ce, malgré un test de réversibilité positif, ce qui justifie l’épaississement pariétal persistant et rapidement évolutif.
Consentement éclairé :
Tous les participants ont donné leur consentement éclairé pour participer rétrospectivement à cette étude et partager les résultats.
Liste des abréviations
ETT : Echocardiographie transthoracique
HTAP : Hypertension artérielle pulmonaire
HTP : Hypertension pulmonaire
NO : Nitricoxyd
PAPM : Pression artérielle pulmonaire moyenne
PAPS : Pression artérielle pulmonaire systolique
PH : Pulmonary hypertension
POD: Pression de l’oreillette droite
RV: Right ventricle
sPAP: Systolic pulmonary artery pressure
TEE : Transesophageal echocardiography
VD : Ventricule droit
Références
- Ryan JJ, Thenappan T, Luo N, Ha T, Patel AR, Rich S et al. Archer SL. The WHO classification of pulmonary hypertension: A case-based imaging compendium. Pulm Circ. 2012 Jan-Mar; 2(1):107-21.
- Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A et al. ESC Scientific Document Group. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016 Jan 1; 37(1):67-119.
- Barberà JA, Román A, Gómez-Sánchez MÁ, Blanco I, Otero R, López-Reyes R et al. Guidelines on the Diagnosis and Treatment of Pulmonary Hypertension: Summary of Recommendations. Arch Bronconeumol (Engl Ed). 2018 Apr; 54(4):205-215.
- Hoeper MM, Humbert M, Souza R, Idrees M, Kawut SM, Sliwa-HahnleK et al. A global view of pulmonary hypertension. Lancet Respir Med. 2016 Apr; 4(4):306-22.
- Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. EurRespir J. 2019 Jan 24; 53(1): 1801913.
- Mandras SA, Mehta HS, Vaidya A. Pulmonary Hypertension: A Brief Guide for Clinicians. Mayo Clin Proc. 2020 Sep; 95(9):1978-1988.
- Castiglione L, Droppa M. Pulmonary Hypertension and COVID-19. Hamostaseologie. 2021 Dec 21. doi: 10.1055/a-1661-0240.
- Sulica R, Cefali F, Motschwiller C, Fenton R, Barroso A, Sterman D. COVID-19 in Pulmonary Artery Hypertension (PAH) Patients: Observations from a Large PAH Center in New York City. Diagnostics (Basel). 2021 Jan 15; 11(1):128.
- Horn E, Chakinala MM, Oudiz R, Joseloff E, Rosenzweig EB. Author rebuttal to response regarding “Letter to the Editor regarding ‘Could pulmonary arterial hypertension patients be at lower risk from severe COVID-19?'”. Pulm Circ. 2020 Jun 23; 10(3)
- Suzuki YJ, Nikolaienko SI, Shults NV, Gychka SG. COVID-19 patients may become predisposed to pulmonary arterial hypertension. Med Hypotheses. 2021 Feb; 147: 110483.
- Tudoran C, Tudoran M, Lazureanu VE, Marinescu AR, Pop GN, Pescariu AS et al. Evidence of Pulmonary Hypertension after SARS-CoV-2 Infection in Subjects without Previous Significant Cardiovascular Pathology. J Clin Med. 2021 Jan 7; 10(2):199.
- Khan A W, Ullah, I, Khan K S, Tahir M J, Masyeni S, Harapan, H. (2021). Pulmonary arterial hypertension post COVID-19: A sequala of SARS-CoV-2 infection? Respiratory Medicine Case Reports, 33: 101429.
- Zamanian RT, Pollack CV Jr, Gentile MA, Rashid M, Fox JC, Mahaffey KW et al. Outpatient Inhaled Nitric Oxide in a Patient with Vasoreactive Idiopathic Pulmonary Arterial Hypertension and COVID-19 Infection. Am J RespirCrit Care Med. 2020 Jul 1; 202(1): 130-132.
- Pagnesi M, Baldetti L, Beneduce A, Calvo F, Gramegna M, Pazzanese V et al. Pulmonary hypertension and right ventricular involvement in hospitalised patients with COVID-19. Heart. 2020 Sep; 106(17): 1324-1331.