Les aplasies médullaires (AM) sont des maladies rares caractérisées par une atteinte constitutionnelle ou secondaire du compartiment des cellules souches hématopoïétiques médullaires. Elles se définissent par une insuffisance médullaire quantitative
Au service d’hématologie et de thérapie cellulaire de l’EHU d’Oran
Serradj, N. Yafour, A. Krim, K. Amani, B. Entasoltan, R.A. Bouhass, A. Arabi, M.A. Bekadja,
Service d’Hématologie et de Thérapie Cellulaire. EHU 1er Novembre, Oran, Université Ahmed Benbella 1, Oran.
Date de soumission : 18 Juillet 2019
Abstract: Aplastic anaemias (AM) are rare diseases characterized by constitutional or secondary damage of the bone marrow hematopoietic stem cells compartment. There incidence in Algeria is estimated to be 2/million inhabitants. The first-line treatment of aplastic anaemia is based on immunosuppressive therapy combining, or not, Ciclosporin A (CSA) and anti-lymphocytic serum (SAL) in the majority of haematologic centres in Algeria, or on HSCT. In this work, we present a review of the diagnosis and therapeutic management of aplastic anaemia, in real life, at the haematology and cell therapy department, EHU Oran. 62 patients with AM were collected, over a period of 10 years from December 2008 to December 2018, of which 44 are evaluable. Of these, 26 patients received CSA alone, 11 received allogeneic bone marrow transplant, 1 received CSA associated with SAL, 4 received Danazol®, 2 Nilevar®, and 1 patient is in wait and see situation. Among the engrafted patients, 6 patients (55%) are in a complete response and 5 patients (45%) have died. Of the patients treated by CSA alone, 15 patients (58%) are in overall response, of which 7 in CR and 8 in PR, 8 patients are lost to sight and 3 are dead.
Key Words: Aplastic anaemia, immunosuppressant, ciclosporin, allograft, bone marrow transplant, hematopoietic stem cells.
Résumé : Les aplasies médullaires (AM) sont des maladies rares caractérisées par une atteinte constitutionnelle ou secondaire du compartiment des cellules souches hématopoïétiques médullaires. L’incidence en Algérie est de 2/million d’habitants. Le traitement de première ligne des aplasies médullaires repose soit sur le traitement immunosuppresseur associant, ou non, la ciclosporine A (CSA) et le sérum anti-lymphocytaire (SAL) dans la grande majorité des centres d’hématologie du pays, soit sur la greffe de moelle osseuse. Nous abordons dans ce travail, une revue de la prise en charge diagnostique et thérapeutique de l’aplasie médullaire dans la vraie vie, au niveau du service d’hématologie et de thérapie cellulaire de l’EHU d’Oran ; 62 patients atteints d’AM ont été colligés, sur une période de 10 années de décembre 2008 à décembre 2018, dont 47 sont évaluables. Parmi ces derniers, 26 patients ont reçu de la CSA seule, 11 ont subi une allogreffe de cellules souches hématopoïétiques, 1 patient a reçu la CSA associée au SAL, 4 patients ont reçu du Danazol®, 2 patients ont reçu du Nilevar®, et 1 patient est en abstention thérapeutique. Deux patients sont décédés avant la mise en route du traitement. Parmi les pts allogreffés, 6 pts (55%) sont en réponse complète et 5 pts (45%) sont décédés. Parmi les patients traités par CSA seule, 15 patients (58%) sont en réponse globale dont 7 en RC et 8 en RP. 8 patients sont perdus de vue et 3 patients sont décédés.
Mots clés : Aplasie médullaire, immunosuppresseurs, ciclosporine, allogreffe, greffe de moelle, cellules souches hématopoïétiques
Abréviations
AM …………………………………………………………………………………………………… Aplasie Médullaire
AMM ………………………………………………………………………………….. Aplasie Médullaire Modérée
AMS ………………………………………………………………………………………. Aplasie Médullaire Sévère
AMTS ………………………………………………………………………………. Aplasie Médullaire Très Sévère
BOM ……………………………………………………………………………………….. Biopsie Ostéo Médullaire
CAP ………………………………………………………………………………….. Culot d’Aphérèse Plaquettaire
CG ………………………………………………………………………………………………………. Culot Globulaire
CMF ………………………………………………………………………………………………… Cytométrie en Flux
CMV ………………………………………………………………………………………………….. Cytomégalo Virus
CPMC …………………………………………………………………………………… Centre Pierre & Marie Curie
CSA ……………………………………………………………………………………………………….. Cyclosporine A
CSH ………………………………………………………………………………. Cellule Souche Hématopoïétique
EBMT …………………………………………….. European Group for Blood and Marrow Transplantation
SAL ………………………………………………………………………………………… Sérum Anti Lymphocytaire
EBV ……………………………………………………………………………………………………. Epstein Barr Virus
FNS ………………………………………………………………………………….. Formule Numération Sanguine
FSP ……………………………………………………………………………………… Frottis Sanguin Périphérique
HB…………………………………………………………………………………………………………… Hémoglobine
HBC …………………………………………………………………………………………………….. Hépatite B Virus
HCV …………………………………………………………………………………………………….. Hépatite C Virus
HIV ………………………………………………………………………………… Human Immunodeficiency Virus
HSCT……………………………………………………………………………. Hematopoïetic Stem Cell Translant
HTA ………………………………………………………………………………………….. Hyper Tension Artérielle
PLQ …………………………………………………………………………………………………………….. Plaquettes
PNN……………………………………………………………………………………. Poly Nucléaires Neutrophiles
RC …………………………………………………………………………………………………. Rémission Complète
RP ……………………………………………………………………………………………………. Rémission Partielle
SMD …………………………………………………………………………………… Syndrome Myélodysplasique
TERT/TERC : Gènes codant pour la sous-unité TERT (Télomérase reverse transcriptase) / et la sous-unité ARN TERC (Télomerase RNA component) de la Télomérase humaine
Introduction
Les aplasies médullaires (AM) sont des maladies rares caractérisées par une atteinte constitutionnelle ou secondaire du compartiment des cellules souches hématopoïétiques médullaires. Elles se définissent par une insuffisance médullaire quantitative, secondaire à la disparition complète ou partielle du tissu hématopoïétique, sans prolifération cellulaire anormale ni myélofibrose. L’incidence en Algérie est de 2/million d’habitants (1).
On distingue les AM acquises, où aucun facteur étiologique n’est retrouvé, on parle alors d’aplasie médullaire idiopathique, dans lesquelles la destruction des cellules souches hématopoïétiques est probablement auto-immune associée à des mutations génétiques intrinsèques, des maladies constitutionnelles : aplasies globales, comme la maladie de Fanconi ou la dyskératose congénitale, ou bien aplasie d’une seule lignée comme l’anémie de Blackfan-Diamond (2).
Les principales complications de l’AM sont celles de la pancytopénie et le risque d’évolution clonale (3). Le traitement des aplasies médullaires est indiqué en cas d’AM sévère (critères de Camitta) ou d’augmentation des besoins transfusionnels, et repose soit sur le traitement immunosuppresseur associant ciclosporine A (CSA) et sérum anti-lymphocytaire (SAL) préférentiellement de cheval, soit la CSA seule, dans la grande majorité des centres d’hématologie en Algérie, soit sur la greffe de moelle osseuse (4).
Aujourd’hui, les agonistes du récepteur de la thrombopoïétine (Revolade®) représentent une nouvelle alternative thérapeutique dont l’efficacité est très prometteuse (5-7).
Objectifs
Nous proposons dans ce travail, une revue de la prise en charge diagnostique et thérapeutique de l’aplasie médullaire dans la vraie vie, au niveau du service d’hématologie et de thérapie cellulaire de l’EHU d’Oran, ainsi qu’une revue des perspectives thérapeutiques utilisées actuellement dans l’aplasie médullaire.
Patients et méthodes
62 patients atteints d’AM ont été colligés sur une période de 10 années, de décembre 2008 à décembre 2018. Notre étude mono centrique, rétrospective a porté sur 47 patients évaluables.
Le diagnostic a été posé sur les critères suivants : hémogramme (FNS+FSP) avec taux de réticulocytes, mettant en évidence une pancytopénie associant une anémie arégénérative sans aspect de dysmyélopoïèse ni de blastose médullaire au myélogramme et aspect d’hypoplasie ou d’aplasie médullaire totale à la biopsie ostéo médullaire.
L’enquête étiologique a permis de rechercher la notion de toxicité médicamenteuse ou autre (phytothérapie, exposition aux insecticides/pesticides… etc.), à l’interrogatoire, l’existence d’un clone HPN, une origine infectieuse virale (HIV ; HCV ; HBC ; EBV ; CMV ; Parvovirus B19) ou bactérienne (syphilis), un bilan immunologique (anticorps anti-nucléaires, facteur rhumatoïde), ainsi que thyroïdien, un syndrome myélodysplasique (SMD), par la réalisation d’un caryotype hématologique médullaire, ainsi que la recherche d’une anémie de Fanconi par un caryotype lymphocytaire (cassures chromosomiques).
À noter qu’une exploration moléculaire a pu être faite dans certains cas évoquant une aplasie médullaire congénitale à la recherche d’une diminution de longueur des télomères (téloméropathie) tel que la dyskératose congénitale.
Un bilan pré-thérapeutique associant un groupage sanguin, la recherche d’anticorps irréguliers, un bilan rénal et hépatique, un ionogramme sanguin ainsi qu’un typage HLA de la fratrie, a été réalisé.
Au plan thérapeutique, les patients ont été traités en fonction de leur âge et du stade pronostic de la maladie selon la classification de Camitta et de l’EBMT (European Group for Blood and Marrow Transplantation).
Aplasie sévère : critères de Camitta (8, 9).
Richesse médullaire à la BOM : < 25 % ou comprise entre 25 et 50 % avec < 30 % de cellules hématopoïétiques résiduelles, et 2 ou 3 des critères suivants :
Neutrophiles < 0,5 G/L ; Plaquettes < 20 G/L ; Réticulocytes < 20 G/L.
Aplasie très sévère : critères de l’EBMT (8, 9) : idem plus neutropénie < 0,2 G/L.
Les traitements utilisés ont été d’une part les traitements de support (transfusions de culots globulaires rouges (CG), de concentrés d’aphérèse plaquettaire (CAP), antibiotiques) et d’autre part, les traitements dits spécifiques : immunosuppresseurs, allogreffe de CSH.
- La CSA est débutée à raison de 6mg/kg/j en continu avec ajustement thérapeutique en fonction de la ciclosporinémie (fourchette comprise entre 200 et 400ng/ml) et de l’apparition de signes d’intolérance cliniques : hypertrophie gingivale, HTA, acné, et biologiques : hyperbilirubinémie, élévation de la créatinine, hyperkaliémie (Cf. Tableau 1 : Toxicités de la Ciclosporine A).
- Le SAL de lapin “Thymoglobuine”, à raison de 2,5mg/kg/j durant 5 jours associé à la CSA (non disponibilité du SAL de cheval “Lymphoglobuline”).
- Les androgènes (Nilevar® à raison de 2mg/kg/j durant 9 à 12 mois et Danazol® à raison de 800mg/j durant 4 à 6 mois).
- L’allogreffe de CSH pour les AM sévères.
Tableau 1 : Toxicités de la Ciclosporine A
| Communs ne nécessitant pas de traitement | Communs intervention usuelle | Non communs pouvant requérir à une intervention | Sérieux intervention usuelle |
| · HTA · Tremblements · Hirsutisme · Hyperlipidémie | · Hypertension (non contrôlée) · Tremblements · Hyperkaliémie · Hypomagnésémie · Élévation de la Créatinine · Élévation de la BI | · Érythrose faciale (forme IV) · Érythème (forme IV) · Paresthésie · Hémolyse · Hypertrophie gingivale · Inconfort abdominal · Hyper bilirubinémie · Élévation des transaminases · Hyperglycémie · Acné · Diminution des bicarbonates | · Myosis · Troubles visuels · Encéphalopathie · Syndrome hémolytique urémique · Bullose cutanée · (Forme IV) · Gynécomastie · Convulsions |
Résultats
Caractéristiques des patients
Parmi les 47 patients évaluables, on note 22 femmes et 25 hommes, avec un sex-ratio F/H de 0,88 et un âge médian de 28 ans (16-87ans).
Manifestations cliniques au diagnostic
Le syndrome anémique était présent chez 39 patients (95%) associé chez 7 patients (15%) d’entre eux à un syndrome infectieux. Un syndrome hémorragique chez 22 patients (46%), une splénomégalie chez 3 patients (6%), et un syndrome d’hypertension portale chez 2 patients (4,25%). Un patient présentait également une fibrose pulmonaire.
Bilan diagnostic
Tous les patients ont présenté des anomalies à l’hémogramme, allant de la thrombopénie isolée à la pancytopénie sévère, avec un taux médian de GB de 2,27 G/L dont 0,5 G/L de polynucléaires neutrophiles, un taux médian de plaquettes de 16 G/L et un taux médian de réticulocytes de 26 G/L.
Le frottis de sang périphérique était désertique chez la quasi-totalité des patients, la biopsie ostéo-médullaire a objectivé une aplasie médullaire totale chez 45 patients (95,75%) et une hypoplasie médullaire chez 2 patients (4,25%) dont un a présenté une amégacaryocytose associée.
Selon le score de Camitta, 21 patients (44%) ont présenté une AM modérée, 18 patients (38%) une AM sévère et 7 patients (15%) une AM très sévère. Une patiente a présenté une hypoplasie médullaire.
Le choix du traitement entrepris a été fait selon l’âge, le score pronostic, la présence d’un donneur HLA compatible dans la fratrie et les besoins transfusionnels.
Bilan étiologique
Le bilan étiologique était positif chez 9 patients (19%) dont 3 patients (33%) avaient une AM liée à une cause toxique (exposition aux insecticides domestiques), 1 patient (11%) une tuberculose pulmonaire, 3 patients (33%) présentaient une hémoglobinurie paroxystique nocturne dans sa forme aplasiante (10) et 2 patientes (22%) étaient enceintes au moment du diagnostic.
L’étude cytogénétique n’a pu être réalisée que chez 18 patients (38%) :
- Un caryotype hématologique médullaire chez 4 patients (8,5%) : normal chez 3 d’entre eux et la mise en évidence chez un patient de deux délétions, del 6p et del 12p, ce qui a permis de redresser le diagnostic en myélodysplasie.
- Un caryotype lymphocytaire a été réalisé chez 14 patients, parmi lesquels une anémie de Fanconia été retrouvée chez l’un d’entre eux.
- La biologie moléculaire a été réalisée chez 2 patients qui présentaient une dyskératose congénitale ayant mis en évidence une mutation de type TERT/TERC.
Traitement
Au moment de l’évaluation, un patient était en abstention thérapeutique et 2 patients sont décédés avant la mise en route du traitement. Au total 44 patients étaient évaluables sur le plan thérapeutique et se répartissaient comme suit :
- Aplasies modérées (n=20 patients ) : 4 patients ont reçu du Danazol®, 1 patient du Nilevar®, et 15 patients ont été traités par de la CSA en monothérapie. Du point de vue évolutif, 6 patients sont en RC, 6 patients en RP, 2 patients sont perdus de vue et 1 patient est décédé (diagnostic de myélodysplasie par caryotype).
- Aplasies sévères (n=18 patients) : ont été traitées par CSA (7 patients), CSA + SAL (1 patient), Nilevar® 1 patient, ou par allogreffe (9 patients). Les résultats montrent, 2 décès (patients CSA+SAL et Nilevar), 1 patient RC, 2 patients RP, et 4 patients sont perdus de vue. Parmi les 9 patients ayant subi une allogreffe de cellules souches périphériques, 5 patients sont décédés, 3 patients en RC et 1 patient en rechute.
- Aplasies très sévères (n=6 patients) : ont été traitées, 4 par CSA seule (4 patients) et par allogreffe (2 patients). Un seul patient est décédé dans le groupe CSA, les autres patients sont vivants en RP ou en RC. (Cf. Tableau 2)
Tableau 2 : Critères de réponse complète ou partielle
| Réponse complète | Réponse partielle |
| HB> 10 g/dl | HB> 8 g/dl |
| PNN> 1500/mm3 | PNN> 500/mm3 |
| PLQ > 100000/mm3 | PLQ > 30 000/mm3 |
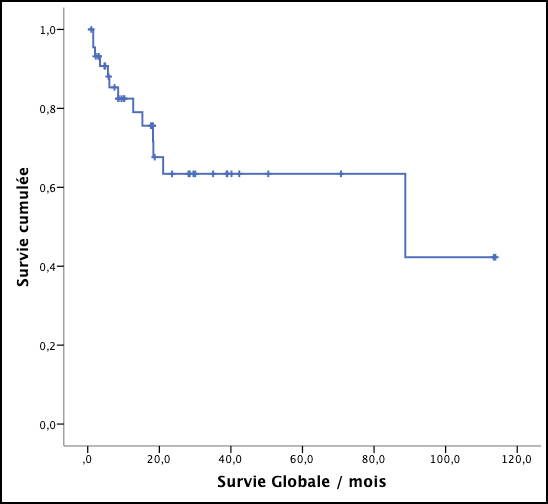
La survie globale des patients, tous traitements confondus, est estimée à 42% à 114 mois (cf. figure 1) avec une médiane de suivi de 12 mois (extrêmes 1 à 114 mois). Tous les patients ayant une aplasie médullaire modérée (AMM) et traités par CSA sont vivants, avec une survie globale de 100%, et une médiane de suivi 19 mois (5 à 113 mois), à l’exception du patient dont le diagnostic a été rectifié en myélodysplasie. La survie globale des aplasies médullaires sévères (AMS) est de 50% à 114 mois avec une médiane de suivi de 15 mois (1 à 114 mois) (cf. figure 2). Dans ce groupe, la CSA semble faire mieux que l’allogreffe avec une survie globale de 86 % vs 32%, cependant les chiffres sont à prendre avec précaution du fait de la petite taille de la série (17 patients). Concernant les AM très sévères (AMTS), la survie globale semble meilleure pour le groupe allogreffé par rapport à celui traité par la CSA : 100% vs 75% à 8 mois avec un plateau jusqu’à 3,5 ans (cf. figure 3).
Figure 1 : Survie globale des AM tous traitements confondus : estimée à 42% à 114 mois
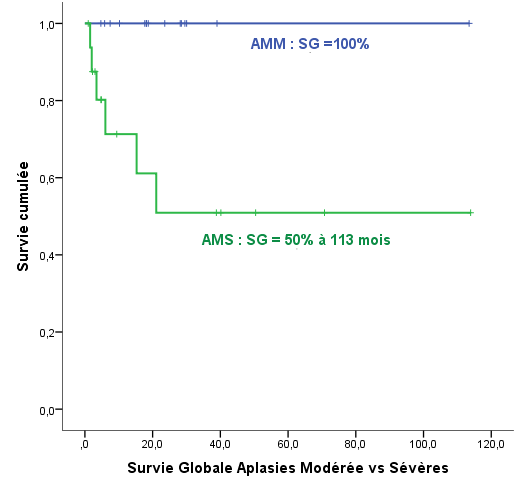
Figure 2 : Survie globale des AMM vs AMS. (Myélodysplasie exclue dans le calcul de la SG des AMM)
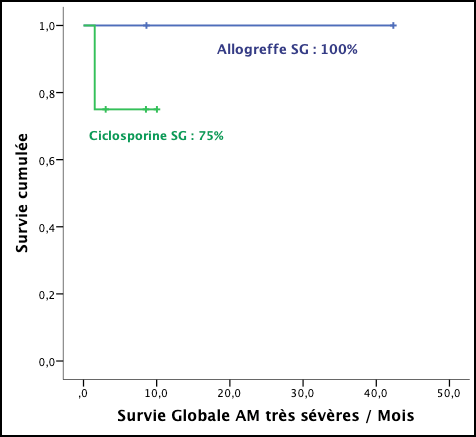
Figure 3 : Survie globale des six AM Très Sévères. 100% des patients traités par allogreffe à 42 mois vs 75% à 8 mois pour les patients traités par ciclosporine
Discussion
Nos résultats sont le reflet de notre contexte de travail et sont concordants avec ceux présentés par les différentes équipes nationales, au cours du XIV Congrès National de la Société Algérienne d’Hématologie et de Transfusion Sanguine (SAHTS) organisé en octobre 2017 à Constantine ; ceci en particulier pour les AMM avec 70% de SG à 10 ans (11). Concernant les AMS, l’équipe du CPMC rapporte une SG de 87% pour 32 aplasies (30 AMS et 2 AMTS) traitées pas CSA seule (12), nous enregistrons à notre niveau 86% de SG à 114 mois pour 8 AMS également traitées pas CSA dont un décès : patient traité par CSA+SAL. Quant aux AMTS notre série est trop restreinte pour être statistiquement exploitable.
Conclusion
L’amélioration de nos résultats passe nécessairement, outre un diagnostic plus précoce des syndromes d’aplasie médullaire, par la disponibilité régulière de moyens permettant d’affiner l’approche diagnostique, en particulier au niveau cytogénétique et moléculaire, ainsi que la disponibilité de moyens thérapeutiques tels que la Lymphoglobuline (SAL du cheval) moins hématotoxique que la Thymoglobuline (SAL du lapin) et les agonistes des récepteurs de la thrombopoiétine (Eltrombopag).
En effet, l’apport du Eltrombopag pourrait engendrer un gain significatif en matière de réponse globale pour les patients réfractaires et/ou en rechute avec en perspective de l’associer en 1ère intention au traitement immunosuppresseur chez les patients de plus de 40 ans ou n’ayant pas de donneur HLA compatible (13-16) (cf. Figure 4)
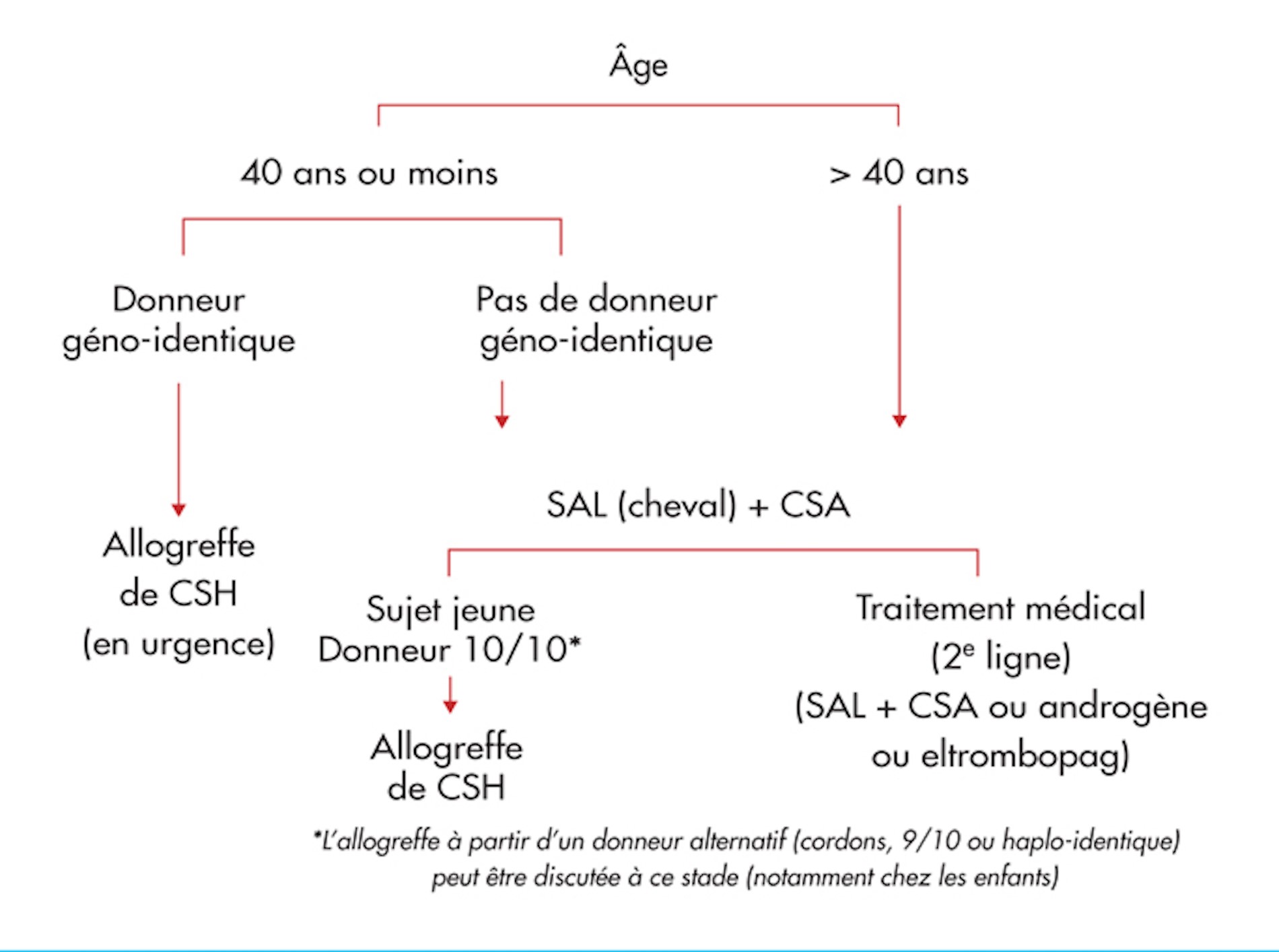
Figure 4 : Algorithme de prise en charge de l’aplasie médullaire idiopathique
SAL : Sérum Anti-Lymphocytaire ; CSA : ciclosporine ; CSH : cellules souches hématopoïétiques.Selon : R. Peffault de Latour & A. Tichelli, Traitement de l’aplasie médullaire idiopathique en 2015, Revue d’oncologie hématologie pédiatrique, volume 3, Septembre 2015.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
- Mehdid. Approche epidémiologique des Aplasies Médullaires en Algérie sur 10 ans (2007-2016). In: d’Hématologie XCN, editor. Constantine.2017.
- Tony Marchand ML. Aplasies Médullaires. Hematologie 2014;20:329-41.
- Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108(8):2509-19.
- Bacigalupo A. Treatment strategies for patients with severe aplastic anemia. Bone marrow transplantation. 2008;42 Suppl 1:S42-S4.
- Townsley DM, Scheinberg P, Winkler T, Desmond R, Dumitriu B, Rios O, et al. Eltrombopag Added to Standard Immunosuppression for Aplastic Anemia. New England Journal of Medicine. 2017;376(16):1540-50.
- Danielle M. Townsley BD, Phillip Scheinberg, Ronan Desmond, Xingmin Feng,, Olga Rios BW, Janet Valdez, Thomas Winkler, Marie Desierto, Harshraj Leuva,, Colin Wu KRC, Andre Larochelle, Cynthia E. Dunbar and Neal S. Young. Eltrombopag Added to Standard Immunosuppression for Aplastic Anemia Accelerates Count Recovery and Increases Response Rates. Blood. 2015; 126:LBA-2;.
- Desmond R, Townsley DM, Dumitriu B, Olnes MJ, Scheinberg P, Bevans M, et al. Eltrombopag restores trilineage hematopoiesis in refractory severe aplastic anemia that can be sustained on discontinuation of drug. Blood. 2014;123(12):1818-25.
- Aplasies Médullaires Protocole National de Diagnostic et de Soins pour une Maladie Rare, (2009).
- Yoon HH, Huh SJ, Lee JH, Lee S, Kim SH, Kwon HC, et al. Should we still use Camitta’s criteria for severe aplastic anemia? The Korean journal of hematology. 2012;47(2):126-30.
- Viviane Gournay CB. Aplasies médullaires, hémoglobinurie paroxystique nocturne, insuffisances médullaires constitutionnelles. Hématologie. 2018;24(3).
- Allouda HL, S. Gherras, N.Boulaziz, H. Aftisse, N. Dali, F. Ait Ahmed, Dj. Si-Tayeb, L. Lamri, H. Ait Ali. Prise en Charge des Formes Modérées de l’Aplasie Médullaire Acquise. In: d’Hématologie XCN, editor. Communication Orale ed. Constantine.: SAHTS; 2017.
- Hamladji, R. Ahmed Nacer, N. Benya, F. Tensaout, N. Ait Amer. Résultats du Traitement de l’Aplasie Médullaire Sévère Acquise (AMS) par la Ciclosporine. SAHTS. XIVème Congrès National d’Hématologie. Constantine.2017.
- Peslak SA, Olson T, Babushok DV. Diagnosis and Treatment of Aplastic Anemia. Curr Treat Options Oncol. 2017;18(12):70.
- Bacigalupo A. How I treat acquired aplastic anemia. BLOOD. 2017;129(11).
- Young NS. Aplastic Anemia. N Engl J Med. 2018;379(17):1643-56.
- Fontbrune FSd. Aplasies Medullaires Traitements. Bordeaux Filiaire Maladies Rares Immuno-Hématologiques MaRIH; 2016.
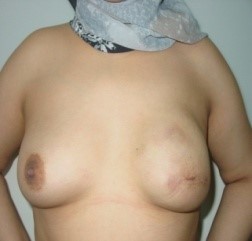


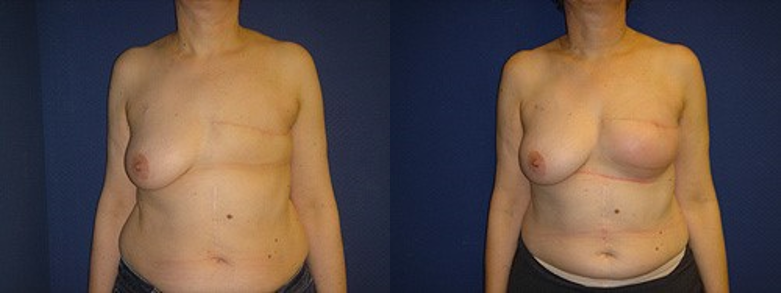
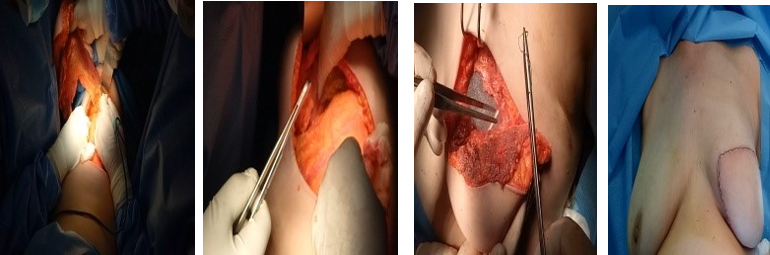
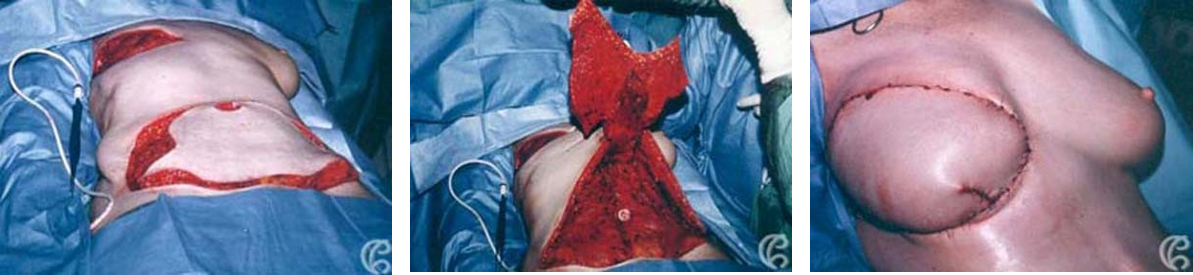
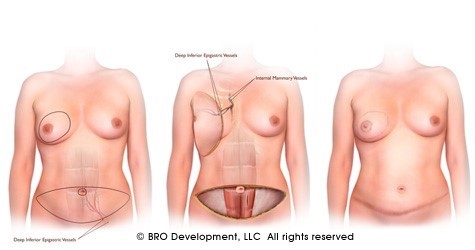
 (Figure 5) : RM par LGD+ Prothèse Selimed 410 cc chez une patienteRM par LGD+ Prothèse Selimed 430 cc chez une patiente 36 ans après 38 mois de sa mastectomie.Lambeau extérorisé 29 ans après 15 mois de sa mastectomie.Lambeauextérorisé par la cicatrice de la mastectomie (2 cicatrices)par le sillon sous mammaire (3 cicatrices : trident); Photos du service de sénologie chirurgie B CPMC
(Figure 5) : RM par LGD+ Prothèse Selimed 410 cc chez une patienteRM par LGD+ Prothèse Selimed 430 cc chez une patiente 36 ans après 38 mois de sa mastectomie.Lambeau extérorisé 29 ans après 15 mois de sa mastectomie.Lambeauextérorisé par la cicatrice de la mastectomie (2 cicatrices)par le sillon sous mammaire (3 cicatrices : trident); Photos du service de sénologie chirurgie B CPMC