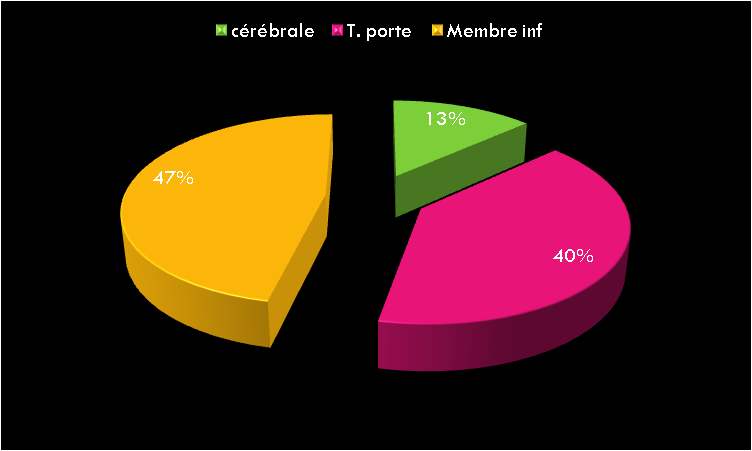Le purpura thrombopénique immunologique est la cause la plus fréquente de thrombopénie chez l’enfant. Cette entité constitue un diagnostic d’exclusion et nécessite d’éliminer les autres causes de thrombopénie.
Khemici, N. Cherif, Service de Pédiatrie B, CHU Issaad Hassani, Beni Messous, Alger.
Date de soumission : 13 Décembre 2019.
Abstract: Immune thrombocytopenic purpura (ITP) is the most common cause of thrombocytopenia in children. However, other causes of thrombocytopenia need to be ruled out to confirm the diagnosis of ITP. This disease is benign in the vast majority of cases, but its haemorrhagic potential must be assessed using Buchanan grading score. Indeed, some rare severe cases can trigger life-threatening severe bleeding symptoms. Initial management may be based on therapeutic abstention or front-line treatment with intravenous immunoglobulins or steroid therapy. There are three evolutionary modes according to the duration of evolution. Chronic ITP management is less consensual and depends on the severity of haemorrhagic symptoms and its consequences on quality of life.
Résumé : Le purpura thrombopénique immunologique est la cause la plus fréquente de thrombopénie chez l’enfant. Cette entité constitue un diagnostic d’exclusion et nécessite d’éliminer les autres causes de thrombopénie. Il s’agit d’une maladie dans la grande majorité des cas bénigne, mais son potentiel hémorragique doit être évalué de manière précise grâce notamment au score de Buchanan. La prise en charge à la phase aiguë repose principalement soit sur l’abstention thérapeutique, soit sur un traitement par immunoglobulines polyvalentes intraveineuses (IgIV) ou la corticothérapie. La prise en charge des purpuras thrombopéniques immunologiques chroniques est moins consensuelle et dépend principalement de la sévérité du syndrome hémorragique et de l’impact de la thrombopénie sur la qualité de vie de l’enfant.
Mots clés : Purpura thrombopénique immunologique, thrombopénie, hémorragies, corticoïdes, immunoglobuline.
Introoduction
Le Purpura Thrombopénique Immunologique (PTI) est une hémopathie immunologique, acquise, bénigne, caractérisée par une thrombopénie aiguë, isolée, à début brutal, survenant quelques jours ou semaines après une infection virale et/ou une vaccination.
Sur un plan évolutif, on distingue trois modes selon la durée d’évolution : aigu (< 3 mois), persistant (3-12 mois) et chronique (> 12 mois). Chez l’enfant, la majorité des PTI évoluent sur un mode aigu ou persistant (80%), contrairement à la population adulte chez laquelle le PTI évolue le plus souvent sur un mode chronique (67%) ; cependant le risque de passage à la chronicité dépend de l’âge de l’enfant : moins de 10% avant un an, un tiers entre un et dix ans et 60% après dix ans.
Diagnostic
Le diagnostic de PTI reste un diagnostic d’élimination bien qu’il constitue la cause la plus fréquente de thrombopénie chez l’enfant. Il n’existe pas d’examen complémentaire permettant d’affirmer le diagnostic. L’examen clinique attentif et l’analyse de l’hémogramme et du frottis sanguin doivent donc permettre d’exclure les diagnostics différentiels. Sur le plan clinique, le PTI est marqué par la survenue d’un syndrome hémorragique brutale, et peut concerner aussi bien la peau que les muqueuses, chez un enfant par ailleurs sans autre symptôme, sans aucune autre anomalie et l’interrogatoire peut retrouver une vaccination ou une infection virale récente. L’enfant peut présenter :
Des manifestations cutanées : les pétéchies, des ecchymoses
Des hémorragies des muqueuses : qui surviennent en l’absence de tout traumatisme sous forme d’épistaxis, gingivorragies, saignements gastro-intestinaux, ménorragies ou d’hématurie,
Des hémorragies intra articulaires
Des hémorragies intracrâniennes.
Évaluation du risque hémorragique
L’évaluation du risque hémorragique représente un temps clé de l’examen clinique car elle conditionne la prise en charge thérapeutique. Elle se base en premier lieu sur l’évaluation du score de Buchanan, qui cote le syndrome hémorragique de 0 à 5 en fonction de la gravité et de l’étendue des lésions (Tableau 1).
Tableau 1 : Le score de Buchanan
Les examens complémentaires doivent comprendre les items suivants :
La formule numération sanguine : qui objective une thrombopénie isolée (< 100.000).
Le frottis sanguin : qui objective des plaquettes avec un nombre diminué, une taille variable avec des plaquettes larges, sans anomalies morphologiques des globules rouges et blancs, sauf exceptions : en cas d’activation atypique des lymphocytes dans les PTI post-infectieux ; et parfois la présence de fragments plaquettaires ‘’microparticules’’ ou fragments mégacaryocytaires.
Le médulogramme : non nécessaire pour le diagnostic de la forme typique du PTI. Il est Indiqué si :
Formes cliniques atypiques, ou si les résultats paracliniques suggèrent une insuffisance médullaire ou une malignité : splénomégalie, douleurs osseuses, fièvre, neutropénie, lymphocytes atypiques, anémie sévère, perte pondérale,
Apparition de signes cliniques non compatibles avec PTI,
Perte de réponse au traitement qui, antérieurement, était efficace,
Réponse insuffisante ou absente aux glucocorticoïdes, immunoglobulines ou immunoglobulines anti-D,
PTI chronique (> 12 mois) n’ayant jamais reçu de traitement ou ayant une réponse transitoire au traitement,
PTI chronique (> 12 mois) avec traitement (agent thrombopoïétique) envisagé.
Splénectomie programmée (> 6 ans)
Cas de PTI chronique
Le PTI chronique représente 10 à 20% des PTI de l’enfant. Les facteurs de risque d’évolution vers la chronicité sont un âge avancé, une thrombopénie moins sévère au diagnostic, un début insidieux des symptômes, et l’absence de précession par une infection ou une vaccination. Les éléments cliniques en faveur d’un PTI chronique sont :
Une histoire jusque-là typique de PTI, dont un début brutal avec thrombopénie profonde et isolée,
Un ou des épisodes de bonne réponse au traitement de première ligne (IgIV ou corticoïdes),
L’existence de facteur de risque pour le caractère chronique,
L’absence d’antécédent familial de thrombopénie ou de syndrome hémorragique,
L’absence de signe extra-hématologique,
Des plaquettes de taille normale ou augmentée sur l’examen du frottis, sans anomalie des autres lignées.
En cas de forme atypique, le bilan doit être élargi, afin de rechercher un autre diagnostic associé, ou à l’origine de ce PTI chronique (tableau 2) :
Tableau 2 : Investigations à la recherche de pathologies associées et d’autres causes de thrombopénie
Bilan d’auto-immunité : FAN, C3, C4
Anticorps anti phospholipides
Test de Coombs
Bilan thyroïdien : TSH, T4 et Anticorps anti TPO
Dosage des immunoglobulines
Bilan hépatique
Sérologies virales : EBV, CMV, parvovirus, HIV, HCV et HBV
Recherche d’Hélicobacter pylori
PMO et/ou PBO
Prise en charge
La prise en charge thérapeutique du PTI à la phase aiguë est relativement consensuelle. En cas de score de Buchanan strictement inférieur à 3 avec un chiffre plaquettaire supérieur ou égal à 10 g/L, l’abstention thérapeutique est recommandée. Celle-ci doit être associée à une éducation des parents et une surveillance clinique des signes hémorragiques.
Si le score de Buchanan est supérieur ou égal à 3 et/ou la numération plaquettaire < 10 G/l, les thérapeutiques de première ligne sont les immunoglobulines polyvalentes et la corticothérapie courte.
Les modalités du traitement sont les suivantes :
Immunoglobulines polyvalentes : 0,8 à 1 g/kg en perfusion lente ; elles sont éventuellement répétées à J3 selon l’évolution clinique et le contrôle de la numération plaquettaire,
Corticothérapie courte (prednisone ou prednisolone). Deux schémas sont utilisés : 4 mg/kg par jour en deux prises (max 100 mg/j) pendant quatre jours, ou 2 mg/kg par jour en une ou deux prises pendant une semaine,
Les immunoglobulines anti-D : Utilisées chez les enfants à rhésus positif. Elles agissent par le détournement de l‘activité macrophagique vers les hématies sensibilisées par l’anti D, elles sont aussi efficaces chez l‘enfant et moins couteuses que les IgIV :50-75 ug/kg
En cas d’hémorragie sévère menaçant le pronostic vital ; une transfusion de plaquette associée aux immunoglobulines, corticoïdes ou Ig anti D
PTI chronique
La prise en charge thérapeutique du PTI chronique n’est pas consensuelle et dépend du syndrome hémorragique clinique, de l’asthénie et de l’impact de la thrombopénie sur la qualité de vie. Les traitements de seconde ligne doivent donc être réservés aux formes sévères.
En l’absence de critère de gravité, on privilégiera l’abstention thérapeutique associée si besoin à l’utilisation des traitements de première ligne ponctuellement en cas de récurrence de syndrome hémorragique ou de situation à risque hémorragique planifiée (intervention chirurgicale, activité sportive).
Dans les formes ne répondant pas aux traitements de première ligne (ou pour lesquels la réponse est très courte) et justifiant d’un traitement.
Traitement de première ligne : des cures périodiques des IgIV (de 04 à 10 semaines d’intervalle) sont proposée en alternance ou en association avec les corticothérapies à faibles doses.
Traitement de deuxième ligne : d’autres traitements peuvent être considérés ou préférés chez certains malades dont les symptômes ne sont pas contrôlés par les traitements de première ligne. La splénectomie est efficace dans l’augmentation du taux des plaquettes et la diminution du risque hémorragique dans 60 à 80% des cas, les immunossupresseurs comme le Rituximab à la dose de 375 mg/m² en perfusion chaque semaine pendant 04 semaines ainsi agonistes des récepteurs de la thrombopoïétine (Eltrombopag et Romiplostim) et Azathioprine.
Des thérapies adjuvantes peuvent être proposés comme un traitement hormonal pour les adolescentes avec ménorragies en utilisant les progestatifs et non les œstroprogestatifs, et les agents anti-fibrinolytiques acide aminocaproïque et acide tranexamique (Exacyl®) qui est efficace pour les gingivorragies et les épistaxis à la dose de 10 mg/kg 03 à 04 fois par jour en IV.
Évolution et pronostic
Le pronostic du PTI en général est bon avec un risque d’hémorragie sévère dans 03% des cas, d’hémorragie intracrânienne de 0,5%. 50 à 70% des enfants vont atteindre une rémission complète dans les 03 mois suivant le diagnostic du PTI et 10 à 20% des enfants vont évoluer vers le PTI chronique.
Pronostic du PTI chronique
Le pronostic du PTI chronique est lié au risque d’hémorragie sévère. Les malades avec un taux de plaquettes moins de 10.000/mm³ ont plus de risque de développer une hémorragie grave même en l’absence de saignement antérieur, mais il existe des variations de sévérité sans rapport avec le taux de plaquettes, il est aussi lié à la présence d’une maladie sous-jacente.
Conclusion
Le purpura thrombopénique immunologique (PTI) est la cause la plus fréquente des thrombopénies de l’enfant, c’est une affection souvent bénigne. Le diagnostic de sa forme aiguë repose sur un faisceau d’arguments et la rémission spontanée est obtenue dans 70 à 80% des cas. 10 à 20% évoluent vers une forme chronique, dont le traitement du PTI chronique n’est pas consensuel.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts
Références
Mc Guinn, J. B. Bussel. Disorders of platelets. in: Lanzkowsky’s manual of pediatric hematology and oncology.6th ed, 2016
David B Wilson, Immune thrombocytopenic purpura in « Nathan and Oski’s in hematology and oncology of infancy and childhood » 8th ed 2015 – Up to date 2016
Provan et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2011; 115:168-186.
La dialyse péritonéale (DP) en urgence s’impose très souvent comme la seule technique possible d’épuration extra rénale chez l’enfant dans les pays en voie de développement, essentiellement par manque de matériel d’hémodialyse adapté.
Missoum, G. Khellaf, Z. Bouderda, M. Hammouche, T. Rayane, Service de Néphrologie EHS d’Urologie-Néphrologie, Daksi, Constantine.
Date de soumission : 01 Mars 2019.
Abstract:introduction: Urgent peritoneal dialysis (PD) is very often the only possible technique for extra-renal dialysis in children in developing countries, mainly due to a lack of adequate haemodialysis equipment. Patients and methods: Over a period of 4 years (2015-2018) we identified 36 children who required emergency PD, including 14 girls and 22 boys, the average age is 42 months (range 1 month to 9 years), the average weight of 14.6 kg (3.1 kg to 25 kg). The technique used is the continuous acute DP with a Tenckhoff catheter, the volume of the intra peritoneal dialysate and the stasis times are in accordance with the European guidelines of 2014, the solutes used are the isotonic and punctually more concentrated solutions. All these parameters vary from one patient to another according to the desired objective. Results: Of our 36 young, 23 had acute kidney injury AKI (02 septic shock, 09 tubular necrosis, 10 haemolytic and uremic syndromes, 02 obstructive AKI that were difficult to derivate), 13 with end stage chronic kidney disease CKD (congenital uropathy), the mean KT/V was 3.9 and the average UF 3.6 ml/kg/h, we had only two deaths due to complications of the initial pathology (septic shock). We did not have leaks or peritonitis, the most frequent complication was paradoxically hypokalaemia (55% of cases), for AKI recovery of renal function was total in 100% of cases, for 13 cases of end stage CKD shift to chronic DP and pre-renal transplant check-up is the rule. Discussion: Our results are encouraging; the management of dialysis emergencies in children in an adult nephrology service is a real challenge. A motivated and available technical platform has adapted to their care, emergency PD in children is increasingly present in our daily lives. Conclusion: Emergency PD in children is not only a necessity imposed by the lack of paediatric haemodialysis equipment, but a method in itself and very effective.
Résumé : Introduction : La dialyse péritonéale (DP) en urgence s’impose très souvent comme la seule technique possible d’épuration extra rénale chez l’enfant dans les pays en voie de développement, essentiellement par manque de matériel d’hémodialyse adapté. Patients et méthodes : Sur une durée de 4 ans (2015-2018) nous avons recensé 36 enfants ayant nécessité la DP en urgence, dont 14 filles et 22 garçons, l’âge moyen est de 42 mois (extrêmes de 1 mois à 9 ans), le poids moyen est de 14,6 kg (3,1 kg à 25 kg). La technique utilisée est la DP aiguë continue avec cathéter de Tenckhoff, le volume du dialysat intra péritonéal et les temps de stase sont conformes aux guidelines européennes de 2014, les solutés utilisés sont les isotoniques et ponctuellement les solutions plus concentrées. Tous ces paramètres varient d’un patient à un autre selon l’objectif recherché. Résultats : parmi nos 36 petits, 23 souffraient d’une insuffisance rénale aiguë (IRA), (02 chocs septiques, 09 nécroses tubulaires, 10 syndromes hémolytiques et urémiques, 02 IRA obstructives difficilement dérivables), 13 d’une insuffisance rénale chronique terminale (IRCT) de référence tardive (uropathies malformatives), le KT/V moyen était de 3,9 et l’UF moyenne de 3,6 ml/kg/h, nous n’avons eu que deux décès suite à des complications de la pathologie initiale (choc septique) ; nous n’avons pas eu de fuites, ni de péritonites, la complication la plus fréquente était paradoxalement l’hypokaliémie (55% des cas), pour les IRA, la récupération de la fonction rénale était totale dans 100% des cas, pour les 13 cas d’IRCT passage à la DP chronique et bilan pré-transplantation rénale est de règle. Discussion : Nos résultats sont encourageants, la prise en charge des urgences de dialyse chez l’enfant dans un service de néphrologie d’adulte est un vrai challenge. Tout un plateau technique motivé et disponible s’est adapté à leurs prise en charge, la DP en urgence chez l’enfant est de plus en plus présente dans notre quotidien. Conclusion : la DP en urgence chez l’enfant n’est pas seulement une nécessité imposée par le manque de matériel d’hémodialyse pédiatrique, mais une méthode à part entière et très efficace.
Mots clés : Algérie, dialyse péritonéale aiguë, enfants, urgences dialyse.
Introduction
la DP était largement acceptée pour le traitement des urémies aigues, mais sa pratique a décliné progressivement en faveur des nouvelles techniques d’épuration extra-rénale notamment l’hémodiafiltration continue [1]. Elle reste fréquemment utilisée dans les pays en développement en raison de son faible coût et de son exigence minimale en matière d’infrastructures. Ceci est particulièrement vrai pour les cas pédiatriques [2]. Un regain d’intérêt pour la DP aiguë s’est développé ces dernières années suite à plusieurs études comparant les résultats de cette technique versus hémodialyse intermittente et l’hémodiafiltration continue dans le cadre de l’IRA de l’adulte [3-5] et de l’enfant [6-12], redonnant à la dialyse péritonéale aiguë ses titres de noblesse, et c’est d’autant plus vrai dans la population pédiatrique.
Cette technique, d’accès facile surtout en urgence, est très adaptée aux enfants en défaillance multiviscérale notamment cardiovasculaire, utilisable quel que soit l’âge ou le poids, incluant les nouveau-nés et les prématurés [13,14]. Elle réunit dans le même objectif les néphrologues, les réanimateurs et les pédiatres.
Elle a longtemps été considérée comme la méthode de choix chez les enfants en IRA [15].
Elle reste dans certains centres l’unique technique salvatrice disponible d’extrême urgence [16,17].
Patients et méthodes :
C’est une étude prospective dans un centre de néphrologie adulte de l’est Algérien, s’étalant sur 4 ans (janvier 2015 à janvier 2018), et incluant ainsi 36 enfants en urgence de dialyse, jugulée exclusivement par la DP aiguë.
Le choix de la DP aiguë est basé d’un coté sur la stabilité hémodynamique, et de l’autre sur la disponibilité du matériel d’hémodialyse adapté à la tranche d’âge et au poids.
La pose chirurgicale du cathéter de Tenckhoff est effectuée sous anesthésie générale, par un chirurgien pédiatrique référant.
Nous utilisons un système en Y (l’enfant reste branché jusqu’à l’épuisement de la poche), afin d’éviter la multiplication des manipulations, et réduire au maximum le risque infectieux.
On administre 500 UI /poche d’héparine et 1 gr/poche de céphalosporine de 3ème génération dans la poche de DP de 2 litres, de façon systématique les premières 48 heures.
Nous avons utilisé la méthode manuelle (DP aiguë continue), avec 10 ml/kg/échange et une stase de 30 min les premières 24 heures ; puis augmentation progressive à un maximum de 30 ml/kg/cycle (pression intra péritonéale maximale à 10 cmH2o) pour les nourrissons ; et à 50 ml/kg/cycle (pression intra péritonéale maximale à 14 cmH2o) pour les enfants de plus de 2 ans. Ce protocole est conforme aux guidelines européennes de 2014 [18].
Le volume maximal infusé est déterminé selon la tolérance respiratoire et les symptômes cliniques, et ce pour chaque enfant.
Le temps de stase est augmenté progressivement jusqu’à 3 heures le deuxième jour.
L’usage ponctuel des poches intermédiaires ou hypertoniques est décidé au cas par cas, si les besoins de la balance hydrique de l’enfant n’étaient pas satisfaits.
Une surveillance stricte, clinique, avec bilan entrées-sorties ; et biologique avec bilan quotidien afin d’évaluer l’efficacité de la dialyse (calcul KT/V et évaluation de l’ultrafiltration UF obtenue).
Résultats :
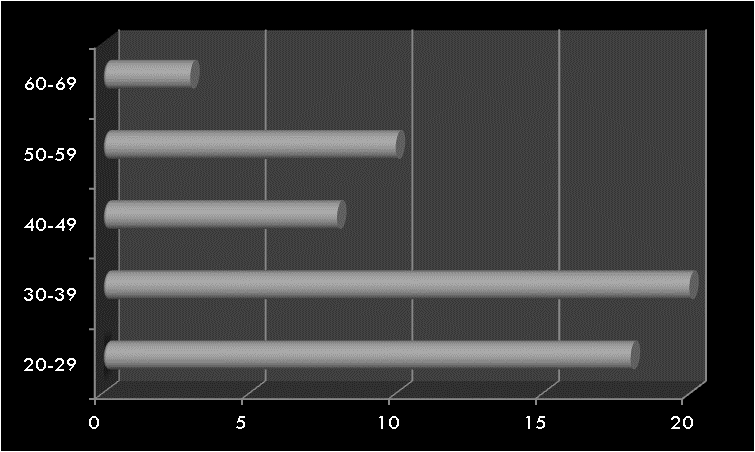
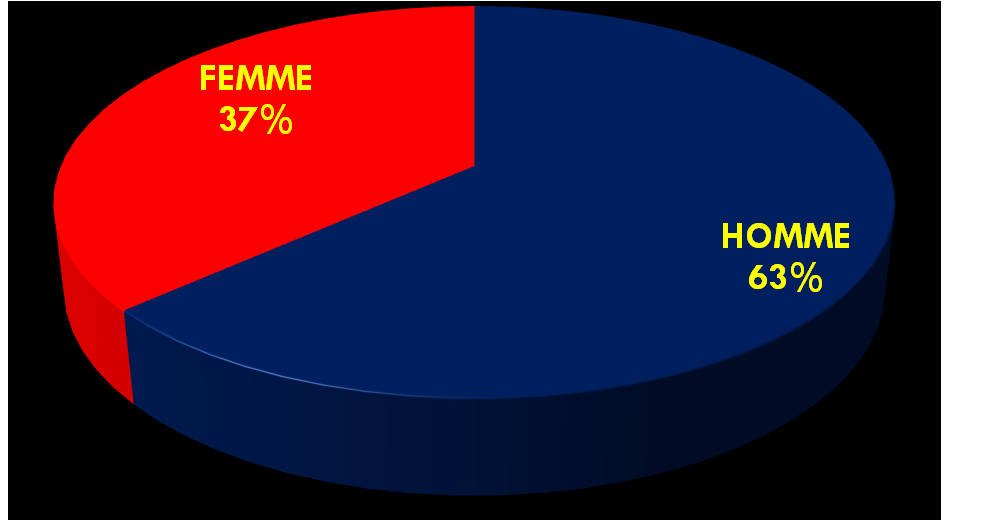
Notre cohorte inclue 36 petits, dont 14 filles et 22 garçons (sexe ratio de 1,57), l’âge moyen est de 42 mois (extrêmes de 1 mois à 9 ans), le poids moyen est de 14,6 kg (3,1 kg à 25 kg). 23 d’entre eux souffraient d’une insuffisance rénale aiguë (IRA) (02 chocs septiques, 09 nécroses tubulaires, 10 syndromes hémolytiques et urémiques et 02 IRA obstructives difficilement dérivables) ; et 13 autres d’une insuffisance rénale chronique terminale IRCT de référence tardive (uropathies malformatives).
Selon la classification de RIFLE pédiatrique, 15 patients sur les 23 IRA ont été classés au stade de défaillance (65%), et 8 patients au stade lésionnel (35%), au moment de la décision de commencer la PD aiguë.
L’amélioration de l’état clinique était à chaque fois réelle et perceptible au bout de quelques heures.
Le KT/V moyen était de 3,9, influencé par le volume intra péritonéale VIP qui semble être un caractère intra individuel (p=0.01) ; plus le VIP toléré est augmenté, meilleur est le KT/V (p=0.001) ; par l’âge, le taux semble meilleur chez les nourrissons (p=0.02) ; et par le temps de stase, le KT/V est plus élevé dans les stases longues de 3 h (p=0.01) Figure 01.
L’UF moyenne est de 3,6 ml/kg/h, influencé par le volume intra-péritonéale (VIP) ; plus le VIP toléré est augmenté, meilleur est l’UF (p=0.002) ; par l’âge, le taux semble meilleur chez les enfants de plus de 2 ans (p=0.03) ; et par le temps de stase, l’UF est plus élevée dans les stases courtes de 30 min (p=0.025) Figure 02.
Nous n’avons eu que deux décès (5%) suite aux complications de la pathologie initiale (choc septique). Nous n’avons pas eu de complications mécaniques ni infectieuses, la complication la plus fréquente était paradoxalement l’hypokaliémie (55% des cas), facilement maîtrisable par supplémentation potassique de 4 mmol/l de dialysat en intra-péritonéale.
Pour les IRA, la récupération de la fonction rénale était totale dans 100% des cas, pour les cas d’IRCT le passage à la DP chronique et bilan pré-transplantation rénale est de règle.
Figure 01 : Taux KT/V selon le VIP maximal toléré et le temps de stase (Statistiques traitées par logiciel SPSS à partir des donnés de notre cohorte)
Figure 02 : Taux UF ml/kg/h selon le VIP maximal toléré et le temps de stase (Statistiques traitées par logiciel SPSS à partir des donnés de notre cohorte)
Figure 03 : Trois enfants de notre cohorte bénéficiant de DP aigue à travers des cathéters de Tenckhoff
Discussion
En fonction des installations et des compétences disponibles, la DP, l’hémodialyse intermittente et l’hémodiafiltration sont actuellement utilisées pour les urgences de dialyse pédiatriques [19-21]. L’hémodiafiltration et l’hémodialyse nécessitent un accès vasculaire, du matériel, des compétences techniques et des ressources financières [21-23], ce qui limite leur utilisation, du fait de la non-disponibilité dans la plupart des centres des pays en développement, y compris le nôtre, d’un matériel adapté aux différentes tranches d’âge pédiatriques, en particulier pour l’enfant de moins de 15 kg. Notre série se compose de 13 IRCT et de 23 IRA résumées par 10 SHU, 09 NTA ,2 IRA urologiques et 2 IRA liées à un choc septique.Les complications mécaniques étaient inexistantes et l’infection péritonéale chaque fois évitée. L’amélioration de l’état clinique était à chaque fois réelle et perceptible au bout de quelques heures. La quantification de la dose de dialyse rendue possible en maintenant la même poche jusqu’à épuisement du dialysat.Les paramètres biologiques s’améliorent doucement sauf la déclaration fréquente d’une hypokaliémie réelle nécessitant une supplémentation intra-péritonéale adaptée.L’ultrafiltration est mesurée au fur et à mesure par un pesant calibré dédié aux seuls malades.L’efficacité du traitement est réelle dans 95% des cas, vite perceptible en attendant la reprise de la fonction rénale chez les patients aigus, surveillée par la diurèse et la mesure des solutés.Les études internationales, dont certaines sont plus étoffées (nombre de patients plus élevé), ne creusent pas l’écart avec nos résultats. Comparées aux études africaines [24-26], nos conditions de travail sont nettement meilleures dans la logistique pour des résultats similaires.La DP est une technique simple à maitriser dans une infrastructure peu exigeante, qui constitue un moyen de traitement efficace partout, y compris dans les régions les plus démunis. Par contre, elle exige un personnel médical et paramédical référant, motivé et disponible pour atteindre les mêmes résultats que l’hémodiafiltration et l’hémodialyse intermittente.
Conclusion
Nos résultats sont assez encourageants, la prise en charge des urgences de dialyse chez l’enfant dans un service d’adulte est un vrai challenge, tout un plateau technique motivé et disponible s’est adapté à leurs prise en charge, la DP en urgence chez l’enfant est de plus en plus présente dans notre quotidien.
L’originalité de ce travail se résume au fait qu’il soit le premier effectué dans notre pays. Nous pensons vivement que notre expérience peut servir d’exemple aux autres équipes de néphrologues qui pourraient sauver beaucoup d’enfants grâce à la DP utilisée de plus en plus dans le cadre aigu dans les pays en voie de développement.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
Maxwell, M.H., et al., Peritoneal dialysis. 1. Technique and applications. J Am Med Assoc, 1959. 170(8): p. 917-24.
Ademola, A.D., et al., Peritoneal dialysis in childhood acute kidney injury: experience in southwest Nigeria. Perit Dial Int, 2012. 32(3): p. 267-72.
Gabriel, D.P., et al., High volume peritoneal dialysis vs daily hemodialysis: a randomized, controlled trial in patients with acute kidney injury. Kidney Int Suppl, 2008(108): p. S87-93.
Vinsonneau, C., et al., Renal replacement therapy in adult and pediatric intensive care : Recommendations by an expert panel from the French Intensive Care Society (SRLF) with the French Society of Anesthesia Intensive Care (SFAR) French Group for Pediatric Intensive Care Emergencies (GFRUP) the French Dialysis Society (SFD). Ann Intensive Care, 2015. 5(1): p. 58.
Phu, N.H., et al., Hemofiltration and peritoneal dialysis in infection-associated acute renal failure in Vietnam. N Engl J Med, 2002. 347(12): p. 895-902.
Niang, A., A. Iyengar, and V.A. Luyckx, Hemodialysis versus peritoneal dialysis in resource-limited settings. Curr Opin Nephrol Hypertens, 2018. 27(6): p. 463-471.
Basu, B., et al., Efficacy and outcomes of continuous peritoneal dialysis versus daily intermittent hemodialysis in pediatric acute kidney injury. Pediatr Nephrol, 2016. 31(10): p. 1681-9.
Obiagwu, P.N. and A. Abdu, Peritoneal dialysis vs. haemodialysis in the management of paediatric acute kidney injury in Kano, Nigeria: a cost analysis. Trop Med Int Health, 2015. 20(1): p. 2-7.
Gaillot, T., et al., [Acute renal replacement therapy in pediatrics]. Ann Fr Anesth Reanim, 2013. 32(12): p. e231-6.
Strazdins, V., A.R. Watson, and B. Harvey, Renal replacement therapy for acute renal failure in children: European guidelines. Pediatr Nephrol, 2004. 19(2): p. 199-207.
Vasudevan, A., K. Phadke, and H.K. Yap, Peritoneal dialysis for the management of pediatric patients with acute kidney injury. Pediatr Nephrol, 2017. 32(7): p. 1145-1156.
Flynn, J.T., Choice of dialysis modality for management of pediatric acute renal failure. Pediatr Nephrol, 2002. 17(1): p. 61-9.
Valeri, A., et al., The epidemiology of peritonitis in acute peritoneal dialysis: a comparison between open- and closed-drainage systems. Am J Kidney Dis, 1993. 21(3): p. 300-9.
Zverev, D.V., A.L. Muzurov, and A.S. Doletskii, [Peritoneal dialysis in acute renal failure in children]. Anesteziol Reanimatol, 2002(1): p. 32-5.
Coulthard, M.G. and B. Vernon, Managing acute renal failure in very low birthweight infants. Arch Dis Child Fetal Neonatal Ed, 1995. 73(3): p. F187-92.
Raaijmakers, R., et al., Continuous flow peritoneal dialysis: first experience in children with acute renal failure. Clin J Am Soc Nephrol, 2011. 6(2): p. 311-8.
Coe, K. and C. Lail, Peritoneal dialysis in the neonatal intensive care unit. Management of acute renal failure after a severe subgaleal hemorrhage. Adv Neonatal Care, 2007. 7(4): p. 179-86.
Cullis, B., et al., Peritoneal dialysis for acute kidney injury. Perit Dial Int, 2014. 34(5): p. 494-517.
Askenazi, D.J., et al., Continuous renal replacement therapy for children </=10 kg: a report from the prospective pediatric continuous renal replacement therapy registry. J Pediatr, 2013. 162(3): p. 587-592.e3.
Gaillot, T., et al., [Acute renal replacement therapy in pediatrics]. Ann Fr Anesth Reanim, 2013. 32(12): p. e231-6.
Strazdins, V., A.R. Watson, and B. Harvey, Renal replacement therapy for acute renal failure in children: European guidelines. Pediatr Nephrol, 2004. 19(2): p. 199-207.
Ricci, Z. and C. Ronco, Renal replacement therapy in the critically ill: getting it right. Curr Opin Crit Care, 2012. 18(6): p. 607-12.
Bunchman, T.E., et al., Pediatric hemofiltration: Normocarb dialysate solution with citrate anticoagulation. Pediatr Nephrol, 2002. 17(3): p. 150-4.
Diarrassouba, G., et al., Acute peritoneal dialysis in African pediatric area experience of pediatric nephrology unit of Yopougon University Hospital (Abidjan, Cote d’Ivoire). Blood Purif, 2015. 39(1-3): p. 141-4.
Anochie, I.C. and F.U. Eke, Paediatric acute peritoneal dialysis in southern Nigeria. Postgrad Med J, 2006. 82(965): p. 228-30.
Esezobor, C.I., T.A. Ladapo, and F.E. Lesi, Peritoneal dialysis for children with acute kidney injury in Lagos, Nigeria: experience with adaptations. Perit Dial Int, 2014. 34(5): p. 534-8.
Les Hémorragies digestives basses constituent un symptôme relativement fréquent en consultation pédiatrique. Elles varient entre rectorragie, hématochézie, méléna et saignement occulte.
Ait Idir(1), Z. Benyahia(2),
(1) Service de pédiatrie. Centre de Consultations Spécialisées de l’Armée. Hussein Dey, Alger.
(2) Service de Pédiatrie, Hôpital Central de l’Armée, Ain Naâdja, Alger.
Abstract: Low digestive haemorrhages are a relatively common symptom in paediatric consultation. It is represented by rectorragia, haematochezia, melena and occult bleeding. The management is multidisciplinary and always begins with the assessment of the impact on the general condition before starting the etiological survey which is oriented according to the age of the child, the clinical history and physical data. The investigation must follow a rational way in order to find the source of bleeding. This symptom is often an alarming one for parents, and usually paediatricians are asked to see the patient right away.
Résumé : Les Hémorragies digestives basses constituent un symptôme relativement fréquent en consultation pédiatrique. Elles varient entre rectorragie, hématochézie, méléna et saignement occulte. La prise en charge est multidisciplinaire et commence toujours par l’appréciation du retentissement sur l’état général avant d’entamer l’enquête étiologique qui est orientée selon l’âge de l’enfant et les données anamnestico-cliniques. C’est ainsi que l’exploration suit un raisonnement logique dans le but de localiser l’origine du saignement et d’établir un plan thérapeutique ciblé. C’est une source de stress parental qui poussent les parents à poser de nombreuses interrogations.
Mots clés : Rectorragie, âge, retentissement, endoscopie, étiologie.
Introduction
Rectorragie / Hématochézie : Les hémorragies digestives basses (HDB) sont le reflet d’un saignement distal du tube digestif. Elles se manifestent généralement par des rectorragies et hématochézies. Ce dernier terme est rarement utilisé, il signifie un saignement rouge issu de l’anus, fait de sang non digéré et accompagné de selles. On la confond toujours avec le terme rectorragie qui traduit un saignement rougeâtre d’origine rectale alors qu’une hématochézie ne précise pas le site du saignement. En général et en pratique, l’appellation rectorragie est préférée et plus utilisée [1][2]. Ces deux types de saignement prennent naissance au-delà de l’angle de Treitz qui est la limite de transition entre la partie haute et la partie basse du tube digestif [2].
Méléna : élimination par l’anus de sang noir, épais et fétide, mélangé ou non aux selles. C’est du sang digéré en rapport avec hémorragie localisée au-dessus de l’angle de Treitz.
Saignement occulte fécal : présence de sang dans les selles qui n’est pas visible à l’œil nu et détectable aux bandelettes réactives.
Ces hémorragies digestives sont dites aiguës et sévères quand elles surviennent en moins de 3 jours et accompagnées de signes de retentissement hémodynamique, trouble de conscience et nécessitent une transfusion [3]. En cas d’hémorragie digestive haute massive, le sang peut arriver à l’anus non digéré et donc rouge.
Données épidémiologiques
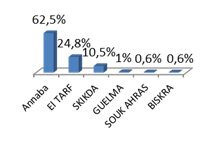
Les HDB représentent un motif non rare de consultation [4]. Habituellement, elles ne font pas l’objet d’une hospitalisation [5]. Contrairement à l’adulte où les HDB sont 5 fois moins fréquentes que les HD hautes et représentent 25% de l’ensemble des hémorragies digestives, il n’y a peu de données épidémiologiques chez l’enfant [6,7]. Aux USA entre 2006 et 2011, le Healthcare Cost and Utilization Project Nationwide Emergency Department a recensé 437.000 enfants de moins de 19 ans ayant consulté pour hémorragie digestive, 30% avaient une HDB, et seulement 11,6% ont été hospitalisés [4][5]. Une autre enquête a révélé qu’uniquement 0,3% d’enfants ayant recouru aux urgences pédiatriques avaient des rectorragies sur 40.000 consultations [4][5]. Une cohorte bangladaise récente a objectivé que seulement 1,5% soit 326 enfants avaient une rectorragie comme maitre symptôme sur 21.533 consultations [6]. Une série égyptienne a révélé que sur 91.000 consultations d’enfants âgés de moins de 18 ans entre Mars 2002 et Février 2004, 194 enfants ont consulté pour rectorragie [7].
Présentation clinique
Le diagnostic est facile quand le saignement est extériorisé mais il est indispensable de l’évoquer dans d’autres situations notamment lors d’un choc hypovolémique ou au contraire lors d’une anémie trainante inexpliquée. Les hémorragies de faible abondance doivent être recherchées notamment par des bandelettes apposées sur les selles [8][9]. Une anamnèse détaillée est de rigueur.
La couleur du sang est liée au siège de l’origine du saignement, à sa proximité de l’orifice anal, à la rapidité du transit et à la formation d’hématine qui lui confère le teint brunâtre. Il est bien établi que la durée de la présence du sang dans la lumière digestive détermine pour beaucoup, la couleur du saignement. Ainsi, après un séjour du sang < 14h, le patient présentera une rectorragie, alors qu’un séjour > 14h donnera lieu à un méléna [2].
L’abondance, de cette hémorragie, sa durée et les constantes hémodynamiques orientent le diagnostic étiologique et aident à évaluer le pronostic vital.
L’examen proctologique confirme un saignement en cours et peut aider à préciser son abondance, tout en sachant qu’un examen normal n’élimine pas le diagnostic positif. Il est réalisé chez un enfant couché soit en décubitus dorsal avec les hanches et les genoux complètement fléchis soit en décubitus latéral gauche. A noter que cet examen doit s’effectuer en la présence des parents tout en expliquant à l’enfant et le rassurant aussi.
Évaluation de la gravité : Le plus souvent – comme déjà cité – le saignement est d’importance modérée et surévalué par des parents anxieux [10,11] . Selon les données de la littérature, la résolution spontanée serait de règle comme chez l’adulte [11]. Cependant, la gravité réside dans les états de troubles hémodynamiques et d’altération de l’état général que peut provoquer un saignement en particulier aigu et abondant (rupture vasculaire) ou chronique et trainant (MICI) qui nécessiterait des mesures de réanimation adaptées [1,11].
Les examens complémentaires
Les examens complémentaires morphologiques, endoscopiques et biologiques sont orientés et ciblés [8,9]. C’est ainsi que les explorations digestives, ne sont pas toujours systématiques et sont programmées selon l’orientation mais aussi en fonction de leur accessibilité. Ces investigations ont pour but de :
Confirmer le saignement,
Situer l’origine de l’HDB ou des rectorragies,
Préciser le caractère aigu ou chronique,
Établir un bilan de retentissement.
L’endoscopie digestive est la pierre angulaire. Elle peut être à double visée diagnostique et thérapeutique. Selon les recommandations du Groupe Francophone d’Hépato-Gastro-Endoscopie et Nutrition Pédiatrique (GFHGENP), établies en 2002 et réévaluées en 2012, ainsi que de la Société Européenne d’Hépato-Gastro-Endoscopie et Nutrition Pédiatrique (ESPGHAN) 2017 ; la coloscopie est indiquée dans le cadre des HDB en l’absence de lésions ano-périnéales « banales », en l’absence de lésions gastroduodénales, et dans le cas des diarrhées chroniques glairo-sanglantes nécessitant une exploration [12,13]. Cela nous mène à dire qu’en cas de rectorragies, il est indispensable en premier lieu de faire un bon examen proctologique, puis éventuellement compléter par une rectosigmoïdoscopie pour écarter toute lésion anale ou rectale avant d’indiquer la nécessité d’une iléocoloscopie (voir annexes 2) [13,14].
L’HDB est la première indication de vidéocapsule quand l’endoscopie classique n’a pas abouti à la visualisation de la source du saignement. Ce moyen diagnostic est en pleine expansion, mais l’âge reste une contrainte, vu qu’elle ne peut s’utiliser actuellement que chez le grand enfant. D’autre part, son grand inconvénient est l’impossibilité de la pratique des biopsies [15].
L’étude anatomo-pathologique reste primordiale et prend toute son importance surtout dans l’analyse d’une masse endoluminale (polype, polypose, tumeur) [16][17] ; ou pour le diagnostic des Maladie Inflammatoire Chronique Intestinale (MICI) [18].
Pour les examens radiologiques, l’échographie abdominale reste facile d’accès, et demandée le cas échéant en première ligne. L’IRM, la TDM, l’Angio-IRM, l’écho-endoscopie ont considérablement amélioré, par leur sensibilité, le diagnostic et la prise charge, surtout dans les MICI [19], et les tumeurs colorectales.
Par ailleurs, les examens biologiques recherchent un retentissement hématologique (une anémie), un syndrome inflammatoire, ou sont spécifiques (sérologie anti-saccharomyces cerevisiae, sérologie de l’allergie aux protéines de lait de vache).
L’enquête étiologique peut même amener à pratiquer une exploration isotopique, notamment dans le cadre du diverticule de Meckel (DM).
Les étiologies
L’anamnèse, l’examen clinique et le contexte dans lequel évolue la rectorragie permettent habituellement de trouver l’étiologie, mais l’élément clé reste l’âge de l’enfant. Il convient d’éliminer une fausse rectorragie. Quelques substances (médicaments, colorant alimentaire, betteraves), ont tendance à colorer les selles.
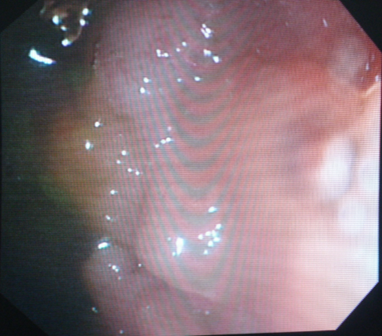
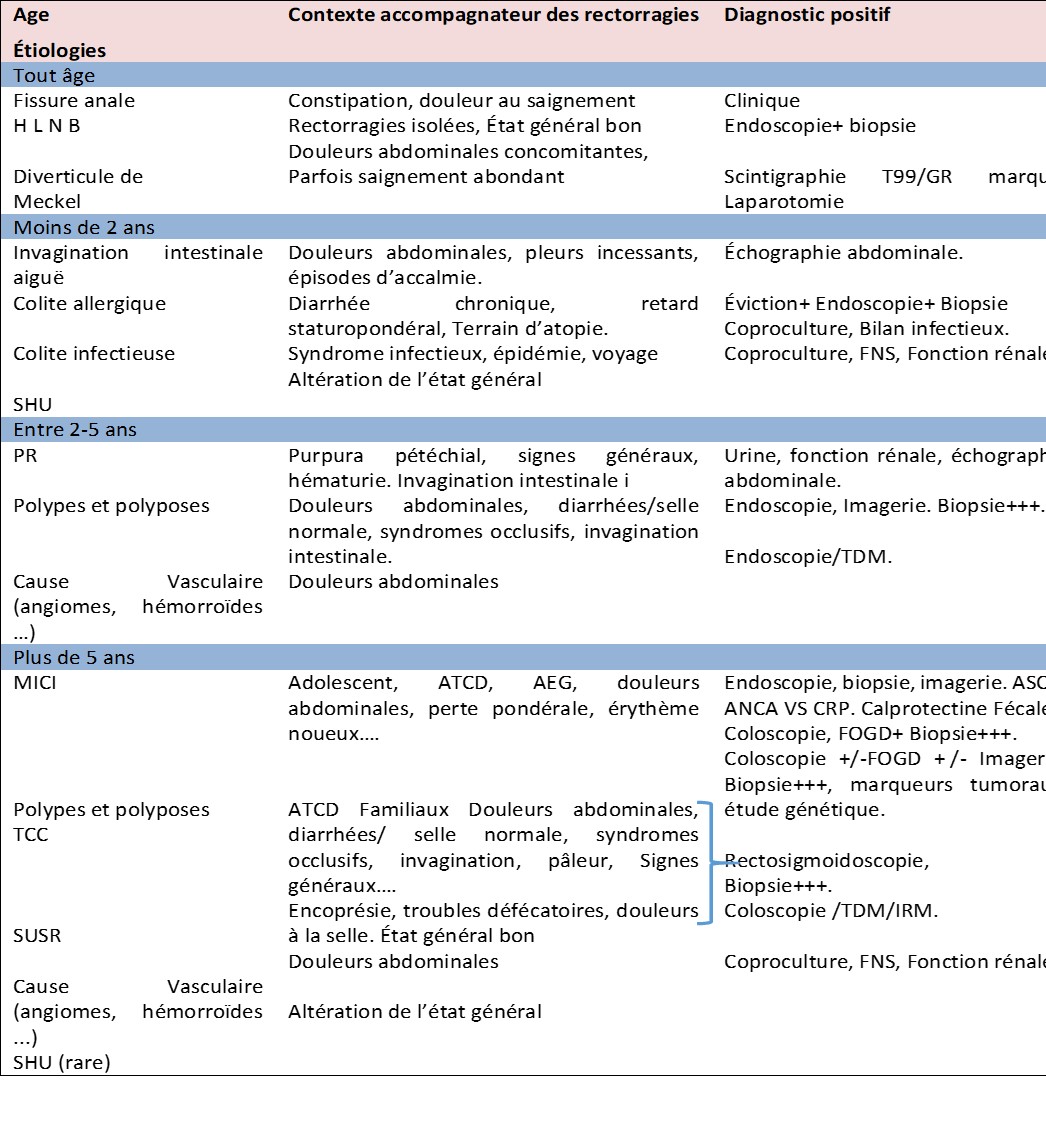
Les étiologies (tableau 1) sont nombreuses et varient selon des différents âges pédiatriques et sont relativement distinctes de celles qui touchent l’adulte [4]. Si chez ce dernier, les diverticules sont la principale cause, les polypes colorectaux (figures 1), les colites chroniques et les fissures anales constituent les causes les plus répandues en pédiatrie [6,11] .
Figures 1:Polype hyperplasique pédiculé rectal chez un enfant de 5 ans. Polype juvenile accouché chez un enfant de 6 ans.
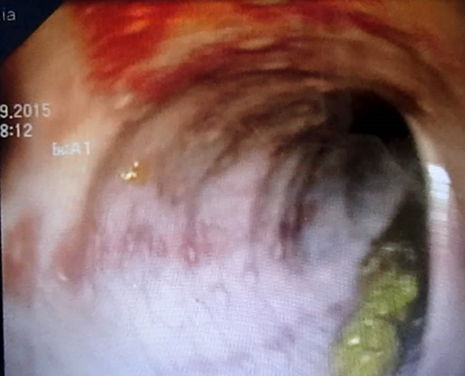
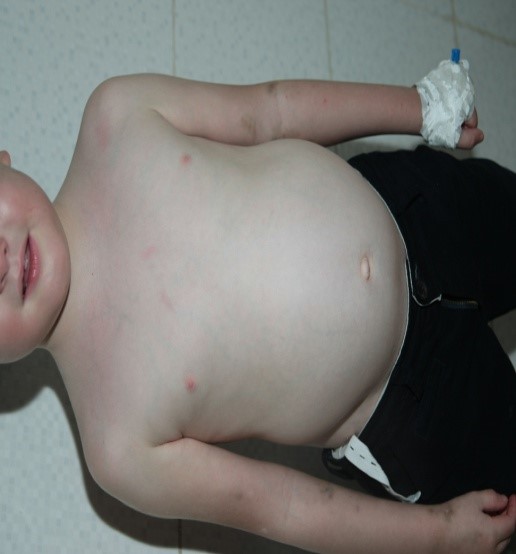
Quel que soit l’âge, les causes traumatiques, les fissures anales (figure 2), les causes infectieuses et l’hypertrophie lymphoïde nodulaire bénigne (figure 3) (HLNB) sont communément retrouvées [9].
Figure 2: Fissure anale chez un nourrison de 6 mois secondaire à la constipation.
Figure 3 : HNLB chez un garçon de 12 ans.
Pour le nourrisson (< 2ans), l’invagination intestinale aiguë, le colites allergiques (figure 4), les duplications digestives, la diarrhée infectieuse, l’HLNB et le DM, restent les causes les plus fréquentes [21].
Figure 4 : colite allergique chez un nourrisson de 6 mois suivi pour allergie aux proteins de lait de vache.
Cette ultime étiologie est considérée comme la plus fréquente anomalie congénitale du tractus gastro-intestinal [11,19].
Chez l’enfant de 2-5 ans, les fissures anales, les colites infectieuses, les polypes et l’HLNB restent des causes les plus classiques [3].
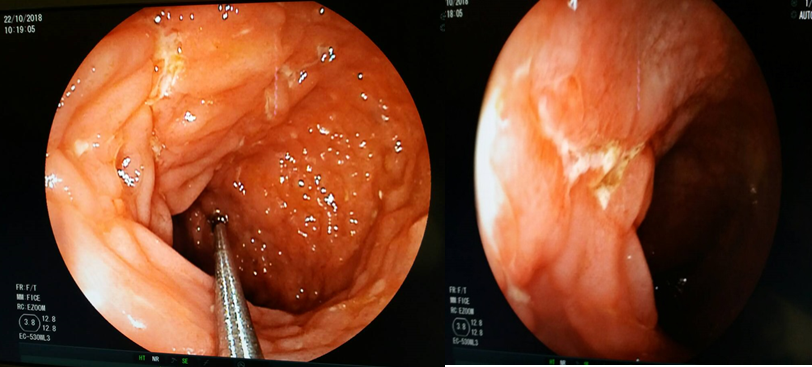
Concernant le grand enfant, dominent les polypes et polyposes, les MICI, (la maladie de Crohn, rectocolite hémorragique, colites indéterminées) (figure 5) ; les malformations vasculaires et le syndrome de l’ulcère solitaire du rectum (SUSR) [20,21].
Figure 5 : maladie de Crhon chez un adolescent de 14 ans .Remarquez l’aspect congestif de la muqueuse colique et les ulcerations longitudinales.
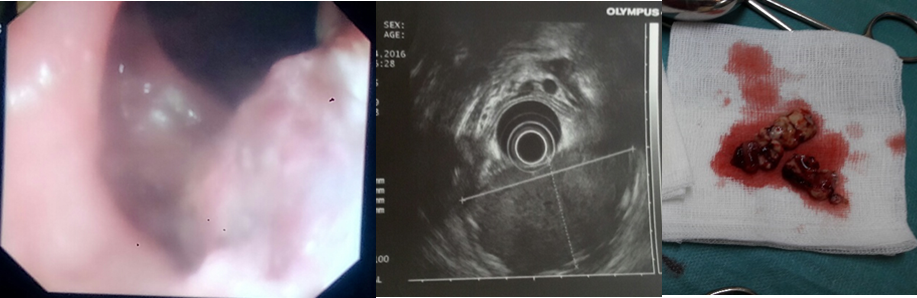
Les tumeurs colorectales (TCC) peuvent se voir, mais restent très peu fréquentes et rentrent le plus souvent dans un cadre familial, héréditaire ou syndromique (figure 6) [22,23].
Figure 6 : Aspect endoscopique et echo-endoscopique d’une tumeur neuro-endocrine rectale très friable chez un enfant de 10 ans.
Tableau 1 : Principales étiologies des rectorragies chez l’enfant et signes cliniques évocateurs (nouveau-né exclu) [2,6,9,24,25]
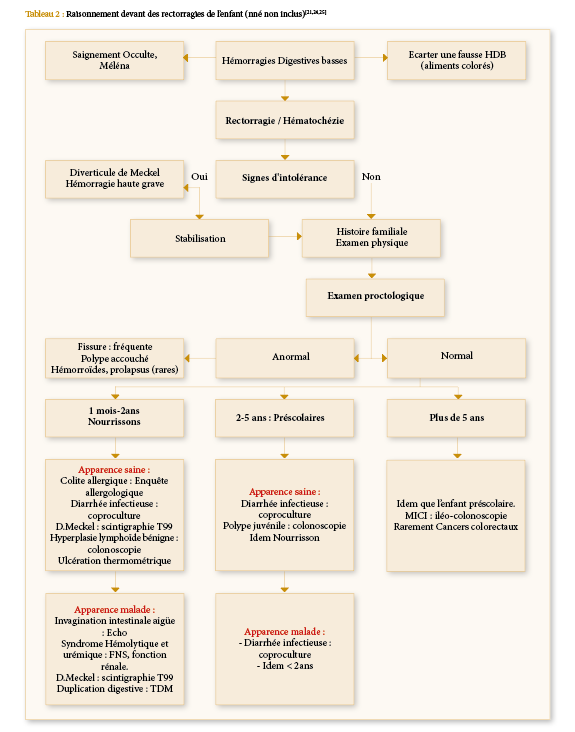
Conduite à tenir devant une rectorragie de l’enfant (nouveau-né non inclus)
Devant toute rectorragie, un raisonnement se base sur l’âge, les signes cliniques, l’examen physique, l’évolution et les examens paracliniques. L’algorithme suivant récapitule l’approche diagnostique et l’enquête étiologique.
Tableau 2: Raisonnement devant des rectorragies de l’enfant (nné non inclus)[21,24,25]
Conclusion
Les rectorragies chez l’enfant constituent un symptôme non rare et pourvoyeur de stress parental. L’interrogatoire, l’examen clinique sont indispensables pour le raisonnement du clinicien notamment l’examen proctologique. L’endoscopie est essentielle mais non systématique dans l’enquête étiologique.
En l’absence de diagnostic endoscopique, d’autres méthodes de diagnostic peuvent être utilisées, avec des degrés variables de spécificité et sensibilité.
Les causes sont généralement âge-dépendantes et différentes de celle de l’adulte. Chez les moins de 5 ans, prédominent les infections, les fissures anales, la proctocolite allergique, l’invagination intestinale, le diverticule de Meckel et les malformations vasculaires, l’HLNB.
Chez le plus de 5 ans, sont essentiellement retrouvés les polypes et les MICI. Une collaboration multidisciplinaire impliquant le gastro-pédiatre, le radiologue, l’endoscopiste, le pathologiste et le chirurgien infantile est indispensable afin de localiser la source du saignement et d’assurer une prise en charge adaptée.
Date de soumission : 04 Décembre 2019.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
E. Padilla and W. Moses, “Lower Gastrointestinal Bleeding & Intussusception,” Surg. Clin. NA, vol. 97, no. 1, pp. 173–188, 2017.
Mushtaq and S. El-hadi, “Rectal bleeding in children e causes and investigations,” Paediatr. Child Health (Oxford)., vol. 24, no. 11, pp. 491–500, 2014.
Barnert and H. Messmann, “Management of lower gastrointestinal tract bleeding,” vol. 22, no. 2, pp. 295–312, 2008.
Osman, M. Djibré, D. Da Silva, and C. Goulenok, “Management by the intensivist of gastrointestinal bleeding in adults and children,” pp. 1–17, 2012.
Pant, M. Olyaee, T. J. Sferra, R. Gilroy, O. Almadhoun, and A. Deshpande, “Brief report Emergency department visits for gastrointestinal bleeding in children: results from the Nationwide Emergency Department Sample 2006 -2011,” Curr Med Res Opin, 2014.
Alam, I. Kmd, N. Mohammad, and M. Nooruzzaman, “Per Rectal Bleeding in Children: Experiences in the Department of Paediatric Surgery in BSMMU,” pp. 20–25, 2017.
A. El-khayat, M. A. El-hodhod, F. Z. Abd, and A. M. Hamdy, “Rectal bleeding in Egyptian children,” Ann. Trop. Paediatr., vol. 26, pp. 337–344, 2006.
Benhamou and C. Dupont, “Diagnostic des hémorragies digestives du nourrisson et de l’enfant,” EMC – Tratado de Medicina. pp. 1–8, 2007.
Aroulandom, J. Lemale, and H. Chappuy, “Diagnostic des hémorragies digestives du nourrisson et de l’enfant,” vol. 13, no. 18, pp. 1–10, 2019.
R. Fleisher, “Rectal Bleeding in the Pediatric Emergency Department,” no. June, pp. 1252–1258, 1994.
Sahn and S. Bitton, “Lower Gastrointestinal Bleeding in Children,” Gastrointest. Endosc. Clin. NA, vol. 26, no. 1, pp. 75–98, 2016.
B. et al Dabadie, “Evolution des indications de la coloscopie chez l ’enfant en 2012. Mise au point,” Arch. Pédiatrie, vol. 19, pp. 1247–1251, 2012.
Mougenot, J. Cardey, and P. Vannerom, “Endoscopie digestive pédiatrique,” vol. 8, no. 13, pp. 1–10, 2019.
Hepatology et al., “Paediatric Gastrointestinal Endoscopy: European Society for Paediatric Nutrition and European Society of,” vol. 64, no. 1, pp. 133–153, 2017.
Recommendations de la SFED, “Indications et techniques de la vidéocapsule endoscopique de l’intestin grêle chez l’enfant,” Acta Endoscopica, vol. 46, pp. 63–67, 2016.
HMA, “Pathology and genetics of hereditary colorectal cancer,” Pathology, vol. 50, no. 1, pp. 49–59, 2018.
Tavano et al., “Pathologie tumorale intestinale de l’enfant,” vol. 4, no. 4, pp. 1–10, 2019.
Viala, C. Jung, N. Belarbi, D. Berrebi, and J. Hugot, “Maladies inflammatoires chroniques intestinales de l’enfant : maladie de Crohn , rectocolite hémorragique,” EMC – Pediatría, vol. 11. Elsevier, pp. 1–10, 2019.
Gallinet and F. Sauvat, “Diverticule de Meckel et pathologie du canal omphalomésentérique,” vol. 12, no. 16, pp. 1–6, 2019.
Sun, T. Hull, and G. Ozuner, “Facteurs de risque et caractéristiques cliniques du prolapsus rectal chez le sujet,” J. Chir. viscérale, vol. 151, no. 6, pp. 1–6, 2014.
Ait Idir, A. Tibouk, S. Kordjani, and N. Zidane, “Le syndrome de l’ulcère solitaire du rectum chez l’enfant : une entité pas si fréquente. Cas clinique et mise au point,” Batna J Med Sci, vol. 6, no. 1, pp. 65–67, 2018.
Tougeron, “Carcinogenèse colorectale, données fondamentales,” Colloids Surfaces A Physicochem. Eng. Asp., vol. 9, no. 3, pp. 1–15, 2019.
Bonnet and R. Guimbaud, “Polyposes et cancers colorectaux familiaux,” Colloids Surfaces A Physicochem. Eng. Asp., vol. 10, no. 4, pp. 1–7, 2019.
A. Lane and I. D. Sugarman, “Investigation of rectal bleeding in children,” Paediatr. Child Health (Oxford)., vol. 20, no. 10, pp. 465–472, 2010.
Shaoul, “Practical Algorithms in Pediatric Gastroenterology,” 2014.
Le tableau clinique de la ß-thalassémie majeure apparaît généralement entre 6 et 24 mois, avec classiquement la triade d’hémolyse associant : une anémie microcytaire sévère, un ictère et une hépatosplénomégalie.
Aggoune, Service de Pédiatrie, EPH Hassen Badi, El Harrach, Alger.
Date de soumission : 13 Juillet 2019.
Abstract: The clinical presentation of ß-thalassaemia major usually occurs between 6 and 24 months of age with severe microcytic anaemia, mild jaundice, and hepatosplenomegaly. ß-thalassaemia intermedia should be suspected in individuals who, at a later age, present similar but milder clinical findings. Growth failure is one of the most common co-morbidities witnessed in children and adolescents with TM and an important cause of poor body image in these children. The prevalence of growth failure and short stature in children with thalassemia varies from 30- 50% in most studies. The causes are multifactorial. Despite iron chelation, these children develop endocrine complications such as hypothyroidism, hypoparathyroidism, diabetes mellitus, short stature, delayed puberty due to hypopituitarism or hypogonadism as well as bone disease, cardiomyopathies, liver related complications due to iron overload. Additionally, they are at a risk of transfusion transmitted infections too. They need to be monitored for these complications and treated appropriately by specialists.
Key words: Thalassemia, child and adolescent, complications, small size, chelation.
Résumé : Le tableau clinique de la ß-thalassémie majeure apparaît généralement entre 6 et 24 mois, avec classiquement la triade d’hémolyse associant : une anémie microcytaire sévère, un ictère et une hépatosplénomégalie. La ß-thalassémie intermédiaire doit être suspectée chez les personnes qui, à un âge plus avancé, présentent des résultats cliniques similaires mais plus modérés. Le retard de la croissance est l’une des comorbidités les plus courantes observées chez les enfants et les adolescents atteints de ß-thalassémie, et présente une cause importante de mauvaise image corporelle chez ces enfants. La prévalence de l’échec de croissance et de la petite taille chez les enfants atteints de thalassémie varie de 30 à 50% dans la plupart des études. Les causes sont multifactorielles. Malgré le traitement chélateur, ces enfants développent des complications endocriniennes telles que l’hypothyroïdie, l’hypoparathyroïdie, le diabète sucré, une puberté retardée due à un hypopituitarisme ou à un hypogonadisme, ainsi que des maladies osseuses, des cardiomyopathies et des complications hépatiques dues à une surcharge en fer. En outre, ils courent également un risque d’infections transmises par la transfusion. Un monitoring doit être proposé de manière appropriée par des spécialistes afin de guetter ce genre de complications.
Mots clés : Thalassémie, enfant et adolescent, complications, petite taille, chélation.
Introduction
Selon l’OMS, le risque de naissance d’enfants malades b-thalassémiques est de 1 pour 10.000 naissances, et celui d’une union entre b-thalassémiques hétérozygotes est de 2 pour mille mariages. Il naît entre 300 et 400.000 enfants porteurs d’une hémoglobinopathie sévère parmi lesquels 100.000 thalassémiques majeurs (2).
En Algérie, nous ne disposons pas encore de chiffre précis concernant la prévalence ou l’incidence des thalassémies. Une communication fait état d’une prévalence de 750 cas en 2006 (3). Si l’on se réfère aux chiffres référence de l’OMS, la prévalence pour notre pays serait de 2%, dans notre service plus de 85 malades thalassémiques sont suivis. Ces maladies constituent un réel problème de santé publique, lié à une prise en charge difficile, et à un taux de mortalité élevé.
Parmi les complications qui guettent l’enfant thalassémique, l’hémochromatose cardiaque post-transfusionnelle est la plus grave, et la principale cause de mortalité des malades atteints d’une b-thalassémie. À elle seule, elle est à l’origine de la moitié des décès (5).
Toxicité du fer
La quantité de fer apportée par les transfusions et l’hyper absorption intestinale du fer suite à l’érythropoïèse inefficace, sont les deux facteurs responsables de la surcharge martiale.Chaque concentré érythrocytaire apporte environ 200 mg de fer. L’organisme ne disposant d’aucun mécanisme régulateur permettant d’éliminer le fer en excès, le fer apporté par chaque transfusion s’accumule progressivement dans l’organisme des patients. Une surcharge en fer apparaît après 10 à 20 transfusions de concentrés érythrocytaires.
En l’absence de chélation, la surcharge en fer entraîne des atteintes tissulaires et, à terme, des défaillances viscérales et des décès. Chez les patients thalassémiques, le traitement est habituellement initié précocement dès 10 à 20 concentrés érythrocytaires, et cela dès que le patient a une ferritine supérieure à 1.000 ng/ml (18).
Complications de la thalassémie
La surcharge martiale dans les thalassémies est secondaire au fer, apporté par les transfusions érythrocytaires et par l’hyper-absorption digestive, au cours de ces maladies. Elle expose aux complications viscérales, majorées en cas de comorbidités associées. Les principales complications de l’hémochromatose post-transfusionnelle sont :
Les atteintes cardiaques
Comme pour les hépatocytes, le fer déposé dans les cardiomyocytes induit une augmentation de la peroxydation des lipides membranaires du sarcolemme et des mitochondries par formation de radicaux libres. Ceci aboutit à une altération de l’activité de la pompe Na+/K+-ATPase, une augmentation de la fragilité des lysosomes et une entrave à l’activité de la chaîne respiratoire mitochondriale. La surcharge en fer a pour conséquence ultérieure une dilatation cavitaire et un épaississement des parois ventriculaires. L’altération de la relaxation ventriculaire gauche apparaît en premier lieu, en rapport avec la surcharge myocardique en fer (31).
Aujourd’hui, les complications cardiaques rendent compte d’au moins 70% des décès dans la thalassémie majeure, en raison de la gravité de cette complication. Les patients soumis à des transfusions répétées, doivent faire l’objet d’une surveillance cardiaque régulière, avec les méthodes les plus précises telles que, la mesure de la fraction d’éjection systolique, l’échocardiographie.
L’utilisation de l’IRM cardiaque, permet également de suivre l’évolution de la surcharge en fer intra myocardique. L’évaluation du temps de relaxation de T2* par IRM cardiaque, est un excellent moyen non invasif d’évaluation du dépôt de fer myocardique et une technique utile après une réponse au traitement de chélation du fer.
Les atteintes hépatiques
Après l’atteinte cardiaque, l’atteinte hépatique est la seconde cause de mortalité chez les patients atteints de surcharge en fer, post-transfusionnelle.
Plus de 70 % du fer en excès est stocké dans le foie. L’appréciation du contenu hépatique en fer (CHF) est par conséquent l’examen de référence dans l’appréciation de la surcharge martiale.
La toxicité hépatique du fer s’exprime sous la forme d’une nécrose hépatocytaire, suivie d’une fibrose hépatique pouvant évoluer vers une cirrhose et un cancer du foie. Le degré de surcharge et le délai d’exposition sont 2 facteurs qui participent à cette évolution.
Les infections virales, notamment par le virus de l’hépatite C, constituent un facteur aggravant.
L’imagerie en résonance magnétique (IRM) est maintenant la méthode non invasive la plus utilisée dans l’évaluation quantitative de la surcharge en fer des organes. Elle utilise le caractère paramagnétique du fer stocké (ferritine et hémosidérine) responsable d’un raccourcissement du temps de relaxation, surtout prononcé en pondération T2, entraînant une diminution du signal en T2 c’est- à- dire un aspect plus noir de l’organe riche en fer.
L’IRM peut s’avérer plus précise que la biopsie chez les patients présentant des dépôts de fer hépatiques hétérogènes (comme en cas de cirrhose), car elle mesure le fer dans la totalité de l’organe. C’est une technique sensible et spécifique, mais dont la méthode n’est pas standardisée.
Les complications endocriniennes
Sont observées dès l’âge de 12-15 ans, et contribuent à la morbidité de la thalassémie majeure à partir de la fin de la deuxième décennie. Les plus fréquentes sont le déficit en gonadotrophines et l’hypothyroïdie. Un retard statural est fréquent, et peut être associé à un retard pubertaire, ce dernier étant d’habitude plus sévère chez le garçon. La puberté peut demeurer incomplète (39). Le déficit statural n’est pas secondaire à un déficit en hormone de croissance, mais il s’accompagne de taux circulants bas du facteur IgF1 en raison de l’atteinte hépatique. Le retard pubertaire est en grande partie responsable du déficit statural après l’âge de 10 ans.
Chez la plupart des malades, on observe une hypothyroïdie clinique ou biologique et une hypoparathyroïdie. Chez les enfants plus âgés, un diabète insulino-dépendant par insuffisance pancréatique endocrine peut apparaitre.
Les atteintes osseuses
Les signes osseux peuvent apparaître dans les premières années de la vie dans les formes majeures :
Au niveau du crâne, le diploé est épaissi, la table externe très amincie, la déformation siège d’abord en frontal puis s’étend à l’ensemble de la voûte. Les travées osseuses perpendiculaires à la table externe vont par leur disposition radiaire réaliser un aspect en « poils de brosse ».
Les os de la face sont concernés par l’hyperplasie médullaire qui donne une expansion osseuse avec des déformations caractéristiques en faciès de rongeur. On observe un hypertélorisme, une absence ou un retard de pneumatisation des sinus maxillaires, un prognathisme supérieur. Au niveau des os longs la transparence osseuse est excessive ; les diaphyses sont déformées, élargies ; les métaphyses sont larges, les corticales sont amincies de par leur versant interne, les travées osseuses restantes paraissent épaissies et la spongieuse a un aspect granité ou réticulé.
Les vertèbres ont une transparence excessive, elles peuvent parfois être tassées ; les arcs costaux antérieurs sont parfois dédoublés et les arcs postérieurs peuvent présenter un renflement en bulbe à leur partie interne, témoignant de l’hyperplasie médullaire. Au niveau des membres, malgré l’ostéoporose, les fractures pathologiques sont relativement rares chez ces patients (43).
L’ostéopénie et l’ostéoporose sont deux autres problèmes osseux dont souffre l’enfant thalassémique, et les mécanismes intriqués sont divers. Les auteurs retiennent habituellement comme ostéoporotiques, les sujets ayant un Z-score inférieur à -2, parfois -2,5 ; et comme ostéopéniques les sujets ayant un Z-score entre -1 et -2,5.
Les complications buccales et maxillo-faciales
Chez les patients thalassémiques, du fait des apports transfusionnels fréquents, des dépôts pigmentaires ferriques sont observés au niveau des téguments et notamment des muqueuses buccales. L’examen dentaire peut révéler chez le jeune enfant des retards d’éruption dentaire, ainsi que des hypominéralisations de l’émail et la dentine La pathologie carieuse est plutôt imputée à la mauvaise pratique des instructions d’hygiène aggravée par la respiration buccale (53).
Les complications pulmonaires
Il existe un syndrome restrictif pulmonaire, avec diminution de la compliance thoracique, ainsi que la capacité vitale et les volumes de réserve expiratoire chez le splénectomisé. Ceci est expliqué par l’existence d’un état d’hypercoagulabilité ainsi qu’une activation des plaquettes. Ce phénomène contribue à l’installation d’une hypertension artérielle pulmonaire, plus importante chez le thalassémique (57). Le dépistage régulier de la fonction pulmonaire devrait être adopté dans le suivi clinique des patients.
Les complications transfusionnelles
Allo immunisations anti érythrocytaires
Apparition d’allo-anticorps anti-érythrocytaires : l’apparition d’allo-immunisation anti-érythrocytaire par transfusion est l’effet indésirable receveur le plus déclaré. Les auto-anticorps sont le plus souvent associés à l’existence d’allo-anticorps anti-érythrocytaires.
Incompatibilités immunologiques érythrocytaires non ABO : Aujourd’hui les effets indésirables immédiats, graves causés par des anticorps irréguliers anti érythrocytaires sont plus nombreux que les accidents ABO (59). Ces effets indésirables sont peu connus. Ils sont causés par des faux-négatifs lors de la recherche d’agglutinines irrégulières, par erreur d’étiquetage du tube, ou bien d’anticorps apparus entre le moment du prélèvement et celui de la transfusion.
Incompatibilités ABO
L’incompatibilité ABO en cas de transfusion de plasma résulte très souvent d’une mauvaise interprétation de l’universalité du groupe sanguin O.
Il est important que cette notion soit rayée du vocabulaire transfusionnel.
Le tableau clinique classique de l’hémolyse intravasculaire est en fait variable selon la précocité du diagnostic, la quantité d’hématies incompatibles transfusées, la puissance de l’anticorps, la vitesse de la transfusion et l’état clinique du patient (61).
Incompatibilités par allo-immunisation anti-HLA ou HPA
Ces anticorps ne sont pas recherchés généralement en pré-transfusionnel, leur présence ou apparition sont imprévisibles en l’état actuel (63). Ils sont acquis par grossesse ou par transfusion.
Réaction fébrile non hémolytique (RFNH)
Elle est en règle sans gravité et doit constituer un diagnostic d’exclusion. Bien que son incidence ait été réduite de moitié par la déleucocytation, la RFNH reste la réaction per ou post-transfusionnelle la plus fréquente, et représente en moyenne annuelle plus de 40 % de l’ensemble des effets indésirables transfusionnels immédiats déclarés.
Risque allergique
Il s’agit dans 90 % des cas de signes cutanés variés, souvent sans autre manifestation clinique. Dans 10 % des cas, il n’y a pas de signes cutanés et l’allergie se manifeste par une gêne. Plus rarement, des complications allergiques graves de type bronchospasme sévère ou choc anaphylactique apparaissent, pouvant engager le pronostic vital du patient.
Œdème aigu pulmonaire (OAP)
L’OAP de surcharge ou OAP cardiogénique est la complication la plus fréquente de la transfusion, bien que son incidence soit vraisemblablement sous-estimée, mais heureusement plus rare dans la population pédiatrique. Il est classique de considérer comme prédisposantes certaines particularités du receveur : cardiopathie, petit poids, insuffisance rénale oligoanurique.
Incompatibilité par les protéines du plasma
Elle est essentiellement représentée avec l’accident anaphylactique causé par les anticorps anti-IgA du receveur, présents chez certains sujets déficitaires en IgA, ou par les anticorps dirigés contre les IgA, apportées dans le plasma résiduel du CGR, chez certains polytransfusés.
Il faut prévoir pour les polytransfusés :
Une détermination du phénotype étendu avant la toute première transfusion
La mention systématique de la pathologie sur l’ordonnance de produits sanguins labiles
La prescription de la qualification phénotypée, tant que l’espérance de vie est raisonnable
L’organisation du prélèvement pour recherche d’agglutinines irrégulières, voire identification des anticorps irréguliers, le plus près possible de la transfusion, mais, en faisant en sorte que les résultats soient disponibles au moment prévu pour la délivrance des produits sanguins labiles
Une bonne coordination avec le CTS, en cas d’allo-immunisation connue.
Nouvelles menaces
Les virus majeurs de la transfusion semblent être bien maîtrisés, et le XXème siècle voit apparaître d’autres agents infectieux, nouvellement identifiés : Virus du Nil occidental ou West Nile virus (WN-V), Parvovirus B19, Cytomégalovirus (CMV) (HHV-5), Hépatites virales.
Splénectomie
Au cours des thalassémies, la rate augmente le plus souvent de volume pendant la première décennie, et peut être à l’origine de diverses complications qui grèvent la morbidité et la mortalité des thalassémies majeures. Le risque infectieux existe également en cas de thalassémie par une défaillance de l’immunité.
L’intérêt de la splénectomie dans la prévention et le traitement de ces différentes complications, au cours de la drépanocytose ou de la thalassémie, a été démontré. Il a été aussi documenté, que les patients porteurs de β-thalassémie présentent un état d’hypercoagulabilité attribué d’une part à un dysfonctionnement du système d’anti-coagulation endothélial naturel, et d’autre part, l’implication de la thrombomodulation et des protéines C et S (72). Le taux de complications thromboemboliques chez ces patients, est compris entre 9 et 29 % avec une forte prévalence dans les formes bêta thalassémiques intermédiaires.
Conclusion La prise en charge d’un enfant atteint de thalassémie, nécessite une évaluation régulière. La prévention est la meilleure approche car l’efficacité de la chélation intensive pour inverser les complications établies est inconnue. Ainsi, prévenir l’anémie grâce à un programme de transfusion régulier, une chélation optimale, un maintien d’un état nutritionnel adéquat, une reconnaissance et un traitement rapide des comorbidités sont les pierres angulaires de la prévention des complications liées à la thalassémie.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
J. Weatherall et J. B. Clegg. Hémoglobinopathies congénitales : un problème de santé publique d’importance croissante. Bulletin de l’ Organisation mondiale de la santé : la revue internationale de santé publique : recueil d’ articles 2002 ; 6 : 153 ;
Belhani. Epidémiologie de la β Thalassémie en Algérie. Revue Algérienne d’hématologie.2009; NO1.
Zurlo MG, De Sstefano P, Borgna Pignatti C et al. Survival and causes of death in Thalassaemia major. Lancet 1989; 2: 27-30.
Ángel Remacha, Cristina Sanz, Enric Contreras, Cristina Díaz de Heredia et al. Guidelines on haemovigilance of post-transfusional iron overload. Blood Transfus. Jan 2013; 11(1): 128–139.
Vassilios Ladis, Giorgos Chouliaras, Vasilios Berdoukas, Christos Kattamis et al. Relation of chelation regimes to cardiac mortality and morbidity in patients with thalassaemia major: an observational study from a large Greek Unit. Eur J Haematol. Oct 2010; 85(4): 335–344.
Vincenzo De Sanctis, Ashraf T. Soliman, Heba Elsedfy, et al. Growth and endocrine disorders in thalassemia: The international network on endocrine complications in thalassemia (I-CET) position statement and guidelines. Indian J Endocrinol Metab. 2013 Jan-Feb; 17(1): 8–18.
Naselli A, Vignolo M, Di Battista E, Garzia P, Formi GL, Traverso T, et al. Long-term follow-up of skeletal dysplasia in thalassaemia major. J Pediatr Endocrinol Metab. 1998;11:817–25.
Jaideep Singh, Reader, Nitin Singh, Senior Lecturer, Amit Kumar, Senior Lecturer, et al. Dental and Periodontal Health Status of Beta Thalassemia Major and Sickle Cell Anemic Patients: A Comparative Study. J Int Oral Health. Oct 2013; 5(5): 53–58.
Prapaporn Pornsuriyasak, Kulanee Vongvivat, Khanchit Likittanasombat, et al. Pulmonary function abnormalities in non-splenectomized and splenectomized adult hemoglobin E/ ß-thalassemia patients and their correlation with pulmonary hypertension. Thalassemia Reports. 2013; volume 3:E5.
Le syndrome de Chediak-Higashi (SCH) est une maladie génétique autosomique récessive rare, caractérisée par un albinisme oculo-cutané, une immunodéficience de l’activité cytotoxique des lymphocytes T et des cellules NK, responsables d’infections récurrentes, une prédisposition aux saignements et une détérioration neurologique tardive.
Khelafi, MS. Ladj, S. Benali Khodja, R. Belbouab, M. Noumi, S. Sokhal, R. Boukari, Service Pédiatrie CHU Mustapha, Alger
Date de soumission : 03 Avril 2019.
Abstract: Chediak-Higashi syndrome (SCH) is a rare autosomal recessive genetic disorder characterized by oculo-cutaneous albinism, immunodeficiency by cytotoxic activity of T lymphocytes and natural killer cells responsible for recurrent infections, predisposition to bleeding, and late neurological deterioration. According to the International Union of Immunological Societies, the SCH is a primary immunodeficiency by immune dysregulation belonging to familial lymphohistiocytic haematophagocytosis syndromes (HFH) with hypopigmentation [1]. The LYST-CHS1 (Lysosomal Trafficking Regulator Gene) gene was identified on the long arm of chromosome 1 in 1q42-q43 [2,3]. This gene encodes the CHS protein whose exact function remains imprecise. About 500 cases have been reported [4,5]. The diagnosis is oriented by the clinical signs and facilitated by the study of the microscopic aspect of the hair which highlights the presence of pigment aggregates. But the pathognomonic sign of the disease is the presence of giant intracytoplasmic granulations in most cells of the organism [6], especially in peripheral blood or bone marrow. Approximately 85% of patients develop an acceleration phase characterized by a syndrome of lymphohocytic hemophagocytosis (HLH), which occurs during the first decade, rarely present at the onset of the disease [7]. It is fatal in the absence of treatment [8]. Currently, the only effective therapeutic option is bone marrow transplantation, which improves haematological and immune abnormalities, but does not prevent subsequent neurological deterioration. The prognosis remains poor in the absence of a bone marrow transplant, the death often occurring before the age of ten years.
Résumé : Le syndrome de Chediak-Higashi (SCH) est une maladie génétique autosomique récessive rare, caractérisée par un albinisme oculo-cutané, une immunodéficience de l’activité cytotoxique des lymphocytes T et des cellules NK, responsables d’infections récurrentes, une prédisposition aux saignements et une détérioration neurologique tardive. Selon International Union of Immunological Societies, le SCH est une immunodéficience primaire par dysrégulation immunitaire appartenant aux syndromes d’hématophagocytose lymphohistiocytaires familiale (HLF) avec hypopigmentation [1]. Le gène LYST-CHS1 (Lysosomal Trafficking Regulator Gene) a été identifié sur le bras long du chromosome 1 en 1q42-q43 [2,3]. Ce gène code pour la protéine CHS dont la fonction exacte reste imprécise. Environ 500 cas ont été signalés [4,5]. Le diagnostic est orienté par les signes cliniques et facilité par l’étude de l’aspect microscopique du cheveu qui met en évidence la présence d’agrégats pigmentaires. Mais le signe pathognomonique de la maladie est la présence de granulations intracytoplasmiques géantes dans la plupart des cellules de l’organisme [6], notamment dans le sang périphérique ou la moelle osseuse. Environ 85% des patients développent une phase d’accélération caractérisée par un syndrome d’hémophagocytose lymphohistiocytaire (HLH), qui survient au cours de la première décennie, rarement présent au début de la maladie [7]. Elle est fatale en l’absence de traitement [8]. Actuellement, la seule option thérapeutique efficace est la greffe de moelle osseuse, qui améliore les anomalies hématologiques et immunitaires, mais n’empêche pas la détérioration neurologique ultérieure. Le pronostic reste mauvais en l’absence de greffe de moelle osseuse, le décès survenant souvent avant l’âge de dix ans.
Mots clés : syndrome de Chediak-Higashi, dysrégulation immunitaire, LYST-CHS1, albinisme oculo-cutané, immunodéficience, détérioration neurologique, HLH, greffe de moelle osseuse.
Introduction
Le syndrome de Chediak-Higashi (SCH) est une maladie génétique autosomique récessive rare caractérisée par un albinisme oculo-cutané, une immunodéficience par défaut d’activité cytotoxique des lymphocytes T et des cellules natural killer, responsable d’infections récurrentes, une prédisposition au saignement, et une détérioration neurologique tardive. Selon l’International Union of Immunological Societies, le SCH est un déficit immunitaire primaire par dysrégulation immunes faisant partie des syndromes d’hematophagocytoses lymphohistiocytaires familiaux (FHL) avec hypopigmentation (1).
Le gène LYST-CHS1 (Lysosomal Trafficking regulator gene) de la maladie a été identifié 1996 sur le bras long du chromosome 1 en 1q42-q43 (2 ,3). Ce gène code pour la protéine CHS dont la fonction exacte reste imprécise.
Environ 500 cas ont été rapportés (4,5). Le diagnostic est orienté par les signes cliniques et facilité par l’étude de l’aspect microscopique des cheveux qui met en évidence la présence d’agrégats de pigment ; mais le signe pathognomonique de la maladie est la présence de granulations intracytoplasmiques géantes dans la plupart des cellules de l’organisme (6), surtout au niveau du sang périphérique ou de la moelle osseuse. Environ 85% des patients développent une phase d’accélération caractérisée par un syndrome d’hémophagocytose lymphohistiocytaire (HLH), qui survient pendant la première décennie, rarement présente au début de la maladie (7) ; elle est mortelle en l’absence de traitement (8).
Actuellement, la seule option thérapeutique efficace est une greffe de moelle osseuse, qui améliore les anomalies hématologiques et immunitaires, mais n’empêche pas la détérioration neurologique ultérieure. Le pronostic reste mauvais en l’absence de greffe de moelle, le décès survenant souvent avant l’âge de dix ans.
Patients et méthodes
Il s’agit de quatre enfants suivis dans le service de pédiatrie du CHU Mustapha d’Alger pour syndrome de Chediak-Higashi entre 2014 et 2017. Le diagnostic a été porté sur les manifestations cliniques, la présence de granulations géantes intra-leucocytaires. Une étude génétique faite chez trois enfants a confirmé le diagnostic.
Résultats
Il s’agit de trois garçons et une fille dont l’âge au diagnostic est en moyenne de 3,2 ans (7 mois – 6 ans). Une consanguinité est retrouvée dans tous les cas ainsi qu’une forme familiale (2 frères). Trois patients ont des antécédents d’infections répétées, de fièvre inexpliquée avec un décès par septicémie dans la forme familiale. Les enfants qui ont été vaccinés n’ont pas présenté d’incidents particuliers. Au plan clinique, l’albinisme oculo-cutané est présent chez 3 enfants et une mélanodermie avec un iris très pigmenté dans 1 cas (Tableau 1).
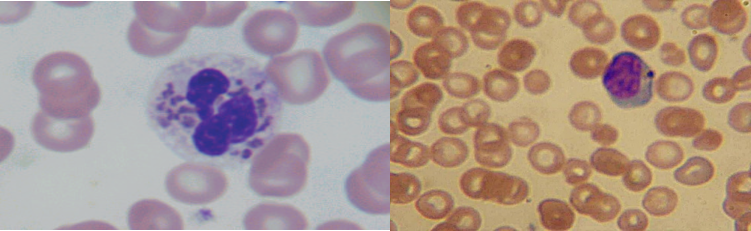
Le frottis sanguin périphérique a permis de faire le diagnostic en montrant les granulations géantes intra-cytoplasmiques chez tous les patients. L’étude microscopique des cheveux a retrouvé des dépôts de mélanine en mottes irrégulières au niveau de la tige pilaire en faveur du syndrome de Chediak-Higashi. Un syndrome d’hémophagocytose lymphohistiocytaire (HLH) (Tableau 2) est présent dans 3 cas puis dans un cas 8 mois après le diagnostic.
Deux enfants ont présenté des signes neurologiques à type de convulsions et de nystagmus horizontal associé à une dyslexie dans un cas. Le diagnostic d’activation lymphohistiocytaire (HLH) est porté selon les critères de la Hystiocyte Society 2004, confirmant la phase accélérée de la maladie. L’étude de l’activité des lymphocytes NK est abaissée dans un cas. La confirmation génétique a été faite chez 3 patients (haplotype homozygote dans 2 cas et partiellement homozygote dans un cas). Le LCR est normal chez tous les patients et un patient a eu une sérologie EBV positive a IgM (VCA). Tous les patients avec des signes d’activation lymphohistiocytaire ont été mis protocole HLH 2004. L’évolution est marquée par une bonne tolérance du traitement. Un enfant a bénéficié d’une greffe de moelle allogénique, mais est décédé quelques mois après, les trois autres patients sont toujours vivants et stables sur le plan clinique.
Tableau 1 : Données cliniques, cytologique et génétiques
Cas 1
Cas 2
Cas 3
Cas 4
Âge
7 mois
6 ans
2 ans
3,5 ans
Sexe
♂
♂
♀
♂
Consanguinité
+
+
+
+
Infections répétées
+
+
+
+
Cas familial
+
+
–
–
Albinisme cutané
Présent
Présent
Présent
Mélanodermie
Cheveux gris argentés
+
+
+
+
Albinisme oculaire
Absent
Hétérochromie
Absent
Iris très pigmenté
Photophobie
–
+
–
–
Nystagmus horizontal
–
–
–
Présent
Signes neurologiques
–
–
–
+
Granulations géantes (Frottis sanguin)
+
+
+
+
Génétique
Haplotype homozygote
Haplotype homozygote
Haplotype partiellement homozygote
–
Cas 1(a 15 mois)
Cas 2
Cas 3
Cas 4
Fièvre
+
+
+
+
Splénomégalie
+
+
+
+
Signes neurologiques
–
–
–
+
Neutrophiles (/mm3)
570
700
120
450
Hémoglobine (g/dl)
5.8
9.7
9.5
8.7
Plaquettes (/mm3)
56 000
104 000
28 000
66 000
Hémophagocytose (PMO)
+
+
+
+
Fibrinogène (g/l)
1.9
2.76
1.34
?
Triglycérides (g/l)
9.5
2.28
5
2.9
Ferritine (ng/ml)
1388
512
1150
977
Activité NK
?
normale
Abaissée (1%)
?
Fig 1 . Un de nos patient avec une hypopigmentation cutanée
Fig. 2. Deux de nos patients avec un aspect « gris argenté » *des cheveux
Fig 3. Tige pilaire de l’un de nos patients avec dépôt de mélanine
Fig.4 Frottis sanguin : granulations géantes au niveau des leucocytes et lymphocytes (In Indian J Hematol Blood Transfus (Sept 2014) 30(Suppl 1):S223–S226)
Discussion
Maladie génétique autosomique récessive très rare, le syndrome de Chediak-Higashi atteint toutes les races et tous les groupes d’âge. Moins de 500 cas ont été rapportés (4,5). Le plus souvent il s’agit de cas rapportés ou de petites séries publiées. La prévalence est difficile à déterminer en raison des cas rapportés plus d’une fois, et d’autres non déclarés. La plus grande série publiée (15 cas) a été rapportée au Japon sur une période de dix ans (9). Une consanguinité, présente chez tous nos patients, est rapportée dans 50 à 85% des cas (5).
Le gène LYST/CHS1 de la maladie code pour la protéine CHS cytosolique dont la fonction reste imprécise. Elle aurait un rôle dans l’exocytose de protéines à partir des endosomes multivésiculaires tardifs. Plus d’une soixantaine de mutations ont été rapportées dans la littérature (faux-sens, non-sens, délétion, insertions) (10). Les mutations du gène LYST/CHS1 entrainent une fonction anormale de la protéine CHS avec une altération du transport intra cytoplasmique, une séquestration de protéines dans les structures intra cytoplasmiques géantes et le blocage de la fonction sécrétoire notamment celle des leucocytes et des mélanocytes. Des corrélations phénotypes/génotypes ont étés rapportées, ainsi une mutation de type délétion est corrélée avec la survenue précoce et fulminante de la phase d’accélération, alors qu’une mutation de type faux-sens est corrélée avec un meilleur pronostic avec absence de phase d’accélération et de détérioration neurologique (11).
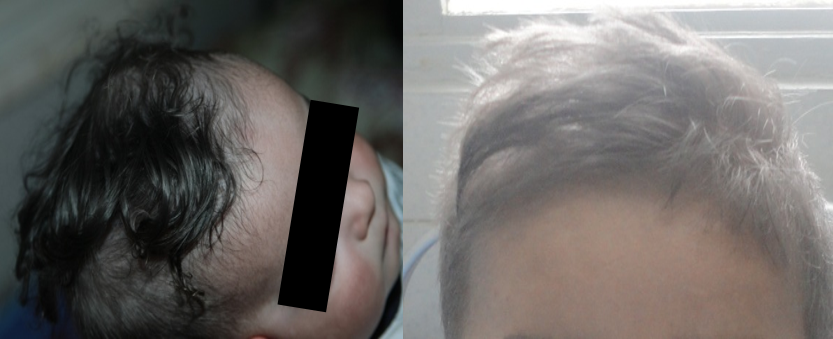
Les signes cliniques débutent après la naissance, ou avant l’âge de 5 ans. L’albinisme oculo-cutané (AOC) est un signe important d’orientation diagnostique, présent chez trois de nos patients (Fig.1), il se caractérise par une hypopigmentation qui affecte généralement la peau, les cheveux et les yeux. Elle est en rapport avec l’agrégation pathologique et une répartition inégale des mélanosomes.
L’AOC peut être présent dès la naissance, et concerner les trois organes ou certains d’entre-deux, total ou partiel ou même être absent (12). Parfois une hyperpigmentation comme chez l’un de nos patients peut exceptionnellement être vue, faisant retarder le diagnostic (13,14). La plupart des patients présentent une photosensibilité. Certains patients ont un phénotype atypique avec une forme atténuée où l’AOC est subtile, voire absent, et qui sont probablement méconnues (11,15). Rarement d’autres lésions cutanées sont observées comme une hyperhydrose, un érythème polymorphe.
Au niveau des cheveux l’hypopigmentation leur donne une couleur blonde, grise (Fig. 2) ou blanche, souvent avec un éclat argenté ou métallique. Les yeux sont de couleur bleue et l’hypopigmentation de l’iris peut être associée à une diminution de la pigmentation de la rétine, et des manifestations oculaires comme la photophobie, diminution de l’acuité visuelle, nystagmus, et le strabisme.
Les infections fréquentes et récurrentes sont habituelles pendant l’enfance. Souvent graves, elles sont en rapport avec un défaut de cytotoxicité des lymphocytes T, de la fonction NK, et une diminution de l’activité chimiotactique et bactéricide des granulocytes (16,17). Les infections pyogènes sont les plus fréquentes, en particuliers au niveau de la peau, des voies aériennes supérieures et des muqueuses. Les germes les plus souvent isolés sont le staphylocoque aureus, streptocoques ß-hémolytiques et le pneumocoque.
Les atteintes de la cavité buccale, à type de gingivite, hémorragie gingivale, chute précoce des dents, aphtes, et ulcérations buccales ont été décrites. La parodontite a été identifiée comme une manifestation de la dysfonction immunitaire.
La tendance au saignement chez ces patients est en rapport avec un déficit du pool de stockage des granules denses et un défaut d’agrégation des plaquettes. Les manifestations hémorragiques sont en général bénignes et ne nécessitent habituellement pas de traitement.
La phase accélérée de la maladie représente la complication la plus importante et dangereuse du SCH. Elle est responsable d’un taux élevé de moralité en quelques mois (18). Elle peut survenir à tout âge mais surtout durant la première décennie (85%) (19).
Nos patients ont développé cette phase accélérée avant l’âge de 6 ans. Rarement, elle en est la première manifestation (7,20). Sa survenue précoce est associée à l’existence d’une mutation génétique de type délétion (11) ; et a une activité effondrée ou absente des LT cytotoxiques (21).
Elle se manifeste par un syndrome d’hématophagocytose lymphohystiocytaire (HLH) dont les facteurs déclenchant cette accélération ne sont pas clairs. Le rôle de l’infection à EBV retrouvé chez l’un de nos patients a été soulevé sans que cette relation ne soit établie (22). Le diagnostic de la maladie de la phase accélérée repose sur les critères de la Histiocytic Society 2004 (23).
Les manifestations neurologiques surviennent dans environ 50% des cas et peuvent apparaître à tout moment dans l’enfance ou à l’âge adulte, ils sont variables : neuropathie périphérique, coma, convulsions, ataxie, troubles cognitifs, troubles de l’équilibre, mouvements anormaux et altérations mentales. La greffe de moelle osseuse n’empêche pas leur apparition ultérieure (24).
Le diagnostic de SCH doit être précoce, souvent fait aux environs de 6 ans mais dans environ 25% des cas le diagnostic est tardif après l’âge de 10 ans (12), pour nos patients. La moyenne d’âge au diagnostic était de 3,2 ans. Il est suspecté sur les éléments cliniques, facilité par l’étude microscopique des cheveux (Fig.3) qui montre des agrégats de pigments de mélanine retrouvés chez tous nos patients, cet aspect permet le diagnostic différentiel avec d’autres types d’hypopigmentation cutanée. Mais le signe pathognomonique de la maladie est la présence de granulations géantes intracytoplasmiques (Fig.4) dans la plupart des cellules de l’organisme (6) mais souvent elles sont identifiées au niveau du sang périphérique comme ce fut le cas de nos patients ; ou au niveau de la moelle osseuse. Le diagnostic est confirmé par un test génétique de recherche de la mutation LYST. Le diagnostic anténatal de la maladie est possible au niveau des cellules des villosités choriales, liquide amniotique, leucocytes du cordon fœtal (25).
Comme pour la maladie de Chediak-Higashi, d’autres déficits immunitaires génétiques s’accompagnent d’un albinisme oculo-cutané partiel, telles que la maladie de Griscelli et le syndrome Hermansky-Pudlak. La distinction peut se faire sans ambiguïté par l’aspect différent des amas de pigment dans la gaine du cheveu, beaucoup plus fins dans le cas du CHS, et surtout par la présence des granulations géantes intracytoplasmiques observées uniquement dans le CHS. Toutefois, dans certains cas de leucémies myéloïdes, on peut voir des granulations géantes dites anomalie pseudo-Chediak-Higashi (26).
Le traitement de la maladie de Chediak-Higashi est multidisciplinaire et repose sur la prise en charge des complications de la maladie, le traitement de la « phase accélérée » ou HLH et surtout la greffe de cellules souches hématopoïétiques. Le traitement symptomatique de la maladie de Chediak-Higashi repose sur une antibiothérapie efficace contre les infections et des transfusions de dérivés du sang pour lutter contre l’anémie et les complications hémorragiques. Les troubles oculaires doivent être corrigés. Les yeux et la peau doivent être protégés des rayons UV. Les vaccinations sont généralement bien tolérées comme ce fut le cas pour nos patients, et doivent être faites. L’hygiène et les soins buccodentaires sont primordiaux. La survenue de symptômes neurologiques et leur progression doivent être pris en charge assez tôt par un spécialiste en réadaptation. En cas de phase d’accélération (HLH, un traitement combinant corticoïdes, VP16, cyclosporine et des injections intrathécales de MTX (protocole HLH 2004) (23) est instauré pour obtenir une rémission. Celle-ci survient dans 75% des cas dans les huit semaines (27), mais les rechutes sont fréquentes et la réponse au traitement diminue au fil du temps. Une fois la rémission obtenue la greffe est recommandé. Chez les patients SCH avec HLH par EBV l’adjonction du Rituximab pourrait améliorer le traitement (28). En cas de HLH réfractaire, une autre option thérapeutique incluant un anticorps monoclonal l’anti CD52 (Alemtuzumab) (29) est possible comme traitement de seconde ligne avant la greffe de moelle osseuse.
La greffe allogénique de moelle osseuse (GMO) est le seul traitement efficace actuel qui guérit les anomalies hématologiques et immunologiques, mais sans résultats sur l’albinisme oculo-cutané ni sur la détérioration neurologique ultérieure (9,24,30) . Le régime de conditionnement pré-greffe comprend une combinaison d’étoposide, le busulfan, le cyclophosphamide (31). La réduction d’intensité du conditionnement pré-greffe avec fludarabine, melphalan, et alemtuzumab a entrainé une augmentation de la survie dans le HLH primaire ou forme familiale avec une plus faible toxicité (32,33). La greffe de moelle osseuse est d’autant plus efficace qu’elle est effectuée avant la survenue de la phase accélérée (31). Les patients ayant une diminution profonde de la fonction cytotoxique des lymphocytes T (CTL) ont un risque élevé de développer un syndrome d’activation lymphohistiocytaire (HLH), aussi leur dépistage peut constituer une indication à une greffe de moelle précoce (21). Le taux de survie globale après une greffe de moelle est 60-70% (30-32).
Le pronostic reste mauvais en l’absence de greffe de moelle, le décès survient fréquemment durant la première décennie par infections ou développement d’un HLH de la phase accélérée (34).
Environ 10% des patients survivent à la petite enfance, et développeront des troubles neurologiques graves à l’adolescence et au début de l’âge adulte.
Conclusion
Le syndrome de Chediak-Higashi est une maladie rare, dont le diagnostic est suspecté chez un enfant présentant un albinisme oculo-cutané avec des infections récurrentes. La majorité des formes cliniques sont précoce “infantiles” mortelles en l’absence de traitement. Une minorité de patients présentent une forme “atténuée” de la maladie survivent après l’enfance mais développent une maladie neuro-dégénérative associée. Dans tous les cas un diagnostic précoce doit être posé par un examen simple, le frottis de sang périphérique qui montre la présence de granulations géantes intracytoplasmiques pathognomonique de cette affection. Le seul traitement actuel efficace des anomalies hématologiques et immunologiques reste la greffe de moelle osseuse allogénique, mais sans impact sur les manifestations cutanées ou la détérioration neurologique ultérieure. Elle est d’autant plus efficace qu’elle est réalisée avant la survenue d’un syndrome HLH. En cas de d’apparition de la phase accélérée (HLH), un traitement selon le protocole HLH 2004 est instauré afin d’obtenir une rémission avant la greffe de moelle osseuse.
Le pronostic de la forme infantile est mauvais, le décès survenant fréquemment dans la première décennie de la vie par infections ou développement de HLH. La recherche de facteurs prédictifs de développement de HLH pourrait aider à poser l’indication d’une greffe de moelle osseuse précoce.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Bibliographie
Picard C,Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME et al – Primary Immunodeficiency Diseases: an Update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol 2015 Nov;35(8):696-726).
Barbosa MDFS et al – Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature 1996, 382:262–265
Nagle DL et al – Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet 1996, 14:307–311
Kaplan J, De Domenico I, Ward DM – Chediak-Higashi syndrome. Curr Opin Hematol 2008, 15:22–29
Reddy RR, Babu BM, Venkateshwaramma B, Hymavathi Ch – Silvery hair syndrome in two cousins: Chediak-Higashi syndrome vs Griscelli syndrome, with rare associations. Int J Trichology 2011; 3(2):107-11
C. Stinchcombe, L. J. Page, et G. M. Griffiths – Secretory lysosome biogenesis in cytotoxic T lymphocytes from normal and Chediak Higashi syndrome patients Traffic 2000; 1(5):435-444)
Pooja Jaiswal, Yogesh Kumar Yadav, Nilam Bhasker, Rashmi Kushwaha – Accelerated Phase of Chediak-Higashi Syndrome at Initial Presentation: A Case Report of an Uncommon Occurrence in a Rare Disorder Journal of Clinical and Diagnostic Research. 2015 Dec, Vol-9(12): ED13-ED14)
Introne WJ, Westbroek W, Golas GA, Adams D: Chediak-Higashi syndrome. In Gene Reviews™. Edited by Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K. Seattle (WA): University of Washington, Seattle; 2009 [updated 2012] [http://www.ncbi.nlm.nih.gov/books/NBK5188/]
Nagai K, Ochi F, Terui K, Maeda M, Ohga S, Kanegane H, et al – Clinical characteristics and outcomes of Chediak–Higashi syndrome: a nationwide survey of Japan. Pediatr Blood Cancer 2013; 60: 1582–6)
Maria L Lozano*, Jose Rivera, Isabel Sánchez-Guiu and Vicente Vicente Orphanet Journal of Rare Diseases 2014, 9:132
Westbroek W, Adams D, Huizing M, Koshoffer A, Dorward H, Tinloy B, et al – The severity of cellular defects in Chediak–Higashi syndrome correlate with the molecular genotype and clinical phenotype. J Invest Dermatol 2007; 127: 2674–7
Barak Y, Nir E. Chédiak-Higashi – Syndrome. Am J Pediatr Hematol Oncol 1987; 9(1):42-55
Ho M-C, Hsieh Y-T – Mixed hyperpigmentation and hypopigmentation of iris and choroid in Chediak-Higashi syndrome. J AAPOS 2013, 17:558–560
Pujani M, Agarwal K, Bansal S, Ahmad I, Puri V, Verma D, Pujani M – Chediak Higashi syndrome – a report of two cases with unusual hyperpigmentation of the face. Turkish J Pathol 2011, 27(3):246–248
Karim MA, Suzuki K, Fukai K, Oh J, Nagle DL, Moore KJ et al – Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak Higashi syndrome.Am J Med Genet.2002; 108: 16–22.
Yenan T. Bryceson, Daniela Pende, Andrea Maul-Pavicic, Kimberly C. Gilmour , Heike Ufheil, Thomas Vraetz et al – A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes Blood 2012, 119:2754-2763;
MC Dinauer -Disorders of Neutrophil Function: An Overview Methods Mol Biol.2014, 1124, 501-515
Pullarkat ST – Accelerated phase of Chediak-Higashi syndrome. Blood 2012; 119(1): 5.
Lozano et al -Towards the targeted management of Chediak-Higashi syndrome Orphanet Journal of Rare Diseases 2014, 9:132.
Gajendra S, Das RR, Chopra A, Singh A, Seth R – Accelerated Phase at Initial Presentation in Chédiak-Higashi Syndrome: Is It Really Uncommon? Pediatr Hematol Oncol 2014, 31:382–385.
Birthe Jessen, Andrea Maul-Pavicic, Heike Ufheil, Thomas Vraetz, Anselm Enders, Kai Lehmberg, et al – Subtle differences in CTL cytotoxicity determine susceptibility to hemophagocytic lymphohistiocytosis in mice and humans with Chediak-Higashi syndrome Blood 2011, 118:4620–4629
Wendy J Introne, Wendy Westbroek, Gretchen A Golas, et David Adams – Chediak HigashiSyndrome Gene Reviews® Last Update: January 15, 2015
Henter JI, Home A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004 – Diagnostic and therapeutic guidelines for haemophagocytic lymphohistiocytosis. Paediatr Blood Cancer. 2007;48:124-31
Tardieu M, Lacroix C, Neven B, Bordigoni P, de Saint Basile G, Blanche S, Fischer A – Progressive neurologic dysfunctions 20 years after allogeneic bone marrow transplantation for Chediak-Higashi syndrome Blood 2005;106:40–2.
Chang H, Yi QL – Acute Myeloid Leukemia With Pseudo–Chediak-Higashi Anomaly Exhibits a Specific Immunophenotype With CD2 Expression. Am J Clin Pathol 2006; 125: 791-4.
Filipovich AH. Hemophagocytic lymphohistiocytosis and related disorders.Curr Opin Allergy Clin Immunol.2006;6:410–5.
Ogimi C, Tanaka R, Arai T, Kikuchi A, Hanada R, Oh-ishi T: Rituximab and cyclosporine therapy for accelerated phase Chediak-Higashi syndrome. Pediatr Blood Cancer 2011, 57:677–680
Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, Lee ND, Khan SP, Lawrence J, Mo JQ, Bleesing JJ, Filipovich AH, Jordan MB: Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer 2013, 60:101–109
Eapen M, DeLaat CA, Baker KS, Cairo MS, Cowan MJ, Kurtzberg J, Steward CG, Veys PA, Filipovich AH: Hematopoietic cell transplantation for Chediak-Higashi syndrome. Bone Marrow Transplant 2007, 39:411–415
Haddad E, Le Deist F, Blanche S, Benkerrou M, Rohrlich P, Vilmer E, Griscelli C, Fischer A. Treatment ofChediak-Higashi syndrome by allogenic bone marrow transplantation: report of 10 cases Blood 1995, 85: 3328–33.
Marsh RA, Vaughn G, Kim M-O, Li D, Jodele S, Joshi S, et al – Reduced-intensity conditioning (RIC) significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood 2010, 116:5824–5831.
Marsh RA, Jordan MB, Filipovich AH: Reduced-intensity conditioning haematopoietic cell transplantation for haemophagocytic lymphohistiocytosis: an important step forward. Br J Haematol 2011, 154:556–563.
Cooper N, Rao K, Gilmour K, et al. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic Blood 2006: 107: 1233–1236
Laura Dotta, Silvia Parolini, Alberto Prandini, Giovanna Tabellini, Maddalena Antolini, Stephen F Kingsmore and Raffaele Badolato – Clinical, laboratory and molecular signs of immunodeficiency in patients with partial oculo-cutaneous albinism Orphanet Journal of Rare Diseases 2013, 8:168
Les besoins en médicaments de la population néonatale sont importants et, pour certaines classes médicamenteuses comme les antibiotiques, relativement plus élevés que ceux des enfants plus âgés
1 Service de Néonatologie, EHS Mère-Enfant ; et Faculté de Médecine, Tlemcen.
2 Département de pharmacie, Faculté de Médecine, Université Abou Bakr Belkaid, Tlemcen.
* Laboratoire de biologie moléculaire appliquée et d’immunologie, Université Abou Bakr Belkaid, Tlemcen.
Date de soumission : 25 Mars 2019.
Abstract: Background and aims. The aim of our study was to assess the consumption and to analyse antibiotics prescription in the neonatal department of the Mother and Child Hospital at Tlemcen. Methodology. Descriptive cohort study conducted between November 1st, 2013 and April 30th, 2014 (6 months) at the neonatal department of the Mother and Child Hospital in Tlemcen. It consisted of a daily regular monitoring of antibiotic prescription until the exit of the new-born. Each prescription was studied separately in order to better assess the adequacy of dosing. Data were collected using an appropriate questionnaire, and were analysed using SPSS 17 software. Results. 238 of 701 patients hospitalized neonates (for at least 1 day) received an antibiotic prescription, which corresponds to about 1 to 1.5 admissions per day on which the new-born had received antibiotics; the number of antibiotic prescriptions was 636, corresponding to 3.5 prescriptions/day. The highest prescription rate was recorded among infants less than 7 days (76% of cases). Early onset sepsis was the most common cause of prescription (66.8% of cases). Gentamicin was the most prescribed antibiotic. But formulated in terms of total daily defined dose ampicillin (336.3) had the largest volume of consumption, followed by cefotaxime (133.5). The doses and duration of antibiotic therapy were pretty much in line with the recommendations. The total direct cost of antibiotics used during this period was 85 058.93 dinars (≈803 Euros). Conclusion. Writing therapeutic protocols and decision trees as well as creating a hospital committee for antibiotic use should help to better rationalize prescriptions.
Keywords. Antibiotics, infection, new-born.
Résumé : Introduction. Les besoins en médicaments de la population néonatale sont importants et, pour certaines classes médicamenteuses comme les antibiotiques, relativement plus élevés que ceux des enfants plus âgés. Pour ces derniers, l’utilisation irrationnelle, en plus du risque d’erreurs de prescription, peut négativement impacter la flore individuelle du nouveau-né receveur (par exemple apparition d’entérobactéries résistantes aux céphalosporines) et/ou la flore des autres nouveau-nés du service. Objectifs. Évaluer la consommation et analyser les prescriptions des antibiotiques dans le service de néonatalogie de l’EHS Mère et Enfant de Tlemcen. Patients et méthodes. Il s’agissait d’une étude de cohorte descriptive réalisée au niveau du service de néonatologie de l’EHS Mère et Enfant de Tlemcen. Elle a consisté en un suivi régulier quotidien de la prescription des antibiotiques jusqu’à la sortie du nouveau-né, et a eu lieu entre le 01 Novembre 2013 et le 30 Avril 2014 (6 mois). Étaient inclus, tous les nouveau-nés âgés de 0 à 28 jours admis pendant la période d’étude, quel que soit le motif d’hospitalisation, et ayant reçu un traitement antibiotique. Chaque prescription a été étudiée à part, pour mieux apprécier l’adéquation des posologies avec les règles de prescription des antibiotiques en période néonatale. Les données ont été collectées à l’aide d’un questionnaire approprié, et analysées à l’aide du logiciel SPSS 17. Résultats. Durant la période d’étude 238 malades sur 701 nouveau-nés hospitalisés (pour au moins un jour) ont bénéficié d’une prescription d’antibiotique, ce qui correspond à environ 1 à 1,5 entrées par jour où le nouveau-né avait reçu une antibiothérapie ; le nombre des prescriptions d’antibiotique était de 636, correspondant à 3,5 prescriptions/jour. Le taux le plus élevé de prescription a été enregistré chez les nouveau-nés de moins de 7 jours (76,05% des cas). L’infection materno-fœtale a été le motif de prescription le plus fréquent (66,8% des cas). La gentamycine avec 34,6% était l’antibiotique le plus prescrit. Mais formulé en termes de dose définie journalière totale, le volume de consommation le plus important était celui de l’ampicilline (336,3), suivi du Cefotaxime (133,5). Les doses et les durées d’antibiothérapies étaient dans la majorité des cas conformes avec celles préconisées dans la littérature. Le coût direct total des antibiotiques utilisés pendant cette période était de 85.058,93 dinars (≈803 Euros). Conclusion. La rédaction des protocoles thérapeutiques et d’arbres décisionnels ainsi que la création d’un comité pour l’antibiothérapie à usage hospitalier devraient permettre de mieux rationaliser les prescriptions des antibiotiques.
Mots clés. Antibiotiques, infection, nouveau-né.
Introduction
Les besoins en médicaments de la population néonatale sont importants et, pour certaines classes médicamenteuses comme les antibiotiques, significativement plus élevés que ceux des enfants plus âgés [1]. Parmi les raisons expliquant la prescription élevée d’antibiotiques on peut citer : 1. Les difficultés diagnostiques que pose l’infection materno-foetale (IMF), puisque durant les premiers jours de vie, les symptômes de l’infection sont peu spécifiques [2]. Aussi, les recommandations pour le diagnostic et la prise en charge, notamment celles publiées en 2002 par l’ANAES (Agence nationale d’accréditation et d’évaluation en santé), tiennent compte de cette particularité en précisant que “tout nouveau-né qui va mal, sans raison apparente, est a priori suspect d’infection” [3] ; et 2. La susceptibilité des nouveau-nés et plus particulièrement les prématurés aux infections nosocomiales du fait qu’ils sont en partie immuno-incompétents [4].
Ainsi, dans la crainte du sepsis, les néonatologistes commencent très souvent à administrer des antibiotiques de manière prophylactique et empirique. Or, l’usage irrationnel des antibiotiques, en plus du risque d’erreurs de prescription, peut négativement impacter la flore individuelle du nouveau-né receveur (par exemple apparition d’entérobactéries résistantes aux céphalosporines) et/ou la flore des autres nouveau-nés hospitalisés.
L’utilisation appropriée des antibiotiques revêt donc une grande importance en pratique clinique [5]. Cela peut aider les soignants à être plus efficaces, rationnels et rentables. En Algérie, nous ne disposons que de très peu d’informations sur l’utilisation des antimicrobiens en période néonatale. L’objectif de notre étude était d’évaluer la consommation et d’analyser les prescriptions des antibiotiques dans le service de néonatalogie de l’EHS Mère et Enfant de Tlemcen.
Patients et méthodes
Il s’agissait d’une étude de cohorte descriptive réalisée au niveau du service de néonatologie de l’EHS Mère et Enfant de Tlemcen. Elle a consisté en un suivi régulier quotidien de la prescription des antibiotiques jusqu’à la sortie du nouveau-né, et a eu lieu entre le 01 Novembre 2013 et le 30 Avril 2014 (6 mois). Étaient inclus, tous les nouveau-nés âgés de 0 à 28 jours admis pendant la période d’étude, quel que soit le motif d’hospitalisation, et ayant reçu un traitement antibiotique.
Chaque prescription a été étudiée à part, pour mieux apprécier l’adéquation des posologies avec les règles de prescription des en période néonatale en s’appuyant sur les deux sources de référence internationales couramment utilisées et bien établies que sont le Néofax [6] et le Nelson’s Pocket Book of Paediatric Antimicrobial Therapy (Nelson’s) dans sa 22ème édition [7].
Pendant cette période d’étude le protocole du service concernant le diagnostic et la prise en charge des infections bactériennes materno-foetales était en grande partie adapté des recommandations de l’HAS de 2002 [3]. La Dose Définie Journalière (DDJ) a été utilisée comme indicateur de la consommation ATB. Elle est définie par l’OMS comme la dose moyenne journalière d’un médicament dans son indication principale pour un adulte de 70 kg[1] [8]. Les données ont été collectées à l’aide d’un questionnaire standardisé, et analysées à l’aide du logiciel SPSS 17.
Résultats
Durant la période d’étude 238 (33,9%) malades sur 701 nouveau-nés hospitalisés (pour au moins un jour) ont bénéficié d’une prescription d’antibiotique, ce qui correspond à environ 1 à 1,5 entrées par jour où le nouveau-né avait reçu une antibiothérapie ; le nombre des prescriptions d’antibiotique était de 636, correspondant à 3,5 prescriptions/jour. Les principales caractéristiques de la population d’étude sont présentées au tableau I. L’âge moyen des patients à l’admission était de 4,66 ± 7,6 ; 76% avaient un âge ≤ 7 jours, et 26,1% sont nés par voie haute. L’âge gestationnel moyen était de 37,1 ± 4,2 semaines avec une médiane de 39 semaines (38,24% étaient prématurés), et un sex-ratio de 1,98. La majorité des nouveau-nés (65,5%) étaient des « in-born ».
La répartition des cas selon le type (IMF ou nosocomiale) et le site de l’infection néonatale sont présentées au tableau II. L’IMF a constitué le motif principal de prescription des antibiotiques (septicémie dans 44,5% des cas, suivie des infections pulmonaires précoces dans 15,1% ; et des infections pulmonaires tardives primitives dans 12,2%). L’infection urinaire d’origine materno-fœtale et tardive, l’omphalite et l’infection pulmonaire d’origine nosocomiale, ont représenté des infections minoritaires avec un taux de 0,4% chacune. Dans 3,8% des cas, l’antibiotique a été prescrit à titre « prophylactique », essentiellement en préopératoire pour des urgences chirurgicales.
Les différents antibiotiques prescrits sont présentés au tableau III. La gentamycine était l’antibiotique le plus prescrit (34,6%), devançant l’ampicilline (29,2%) et le Céfotaxime (28,5%). Mais formulé en termes de dose définie journalière totale (tableau IV), le volume de consommation le plus important était celui de l’ampicilline (336,3), suivi du Céfotaxime (133,5).
Les antibiotiques ont été prescrits en association dans 93,70% des cas (double dans 31,93%, triple dans 57,56% et quadruple dans 3,78%). La Josamycine (53,3%), l’Oxacilline (20%) et l’Imipenème (13,3%) étaient les antibiotiques les plus prescrits en monothérapie. La voie veineuse directe était la voie la plus utilisée (70,13%), suivie par la voie veineuse lente (23,3%), la voie intramusculaire (4,25%), et la voie orale (2,35%).
Les doses et les durées d’antibiothérapies étaient, dans la majorité des cas, conformes avec celles préconisées dans la littérature. L’adéquation des posologies avec les règles de prescription en période néonatale au cours des IMF précoces est présentée dans le tableau V.
Le coût direct total des antibiotiques utilisés pendant cette période était de 85.058,93 dinars (tableau VI). L’imipenème, même s’il n’a été prescrit que dans 1,1% des cas, a représenté 40,6% du coût direct total.
Discussion
La consommation mondiale d’antibiotiques a augmenté de 65% entre 2000 et 2015, particulièrement dans les pays à revenu intermédiaire ou faible. L’Algérie a été citée parmi le top 5 des pays dans le monde, à coté de la Tunisie, la Turquie, et la Romanie où le taux de consommation d’antibiotiques a été le plus élevé en 2015, mais on ne dispose d’aucune étude publiée concernant la consommation d’antibiotiques dans les services de néonatologie [9]. De ce fait, l’objectif principal de notre étude était d’évaluer la consommation des antibiotiques dans notre service de néonatalogie.
Au cours de cette période 33,9% des nouveau-nés hospitalisés ont reçu pendant au moins un jour un antibiotique. Ce chiffre est dans la fourchette de ceux publiés par d’autres équipes. Dans une enquête exhaustive menée en 2013 dans 73 hôpitaux, Versporten et al., ont rapporté que selon le niveau de l’USIN, jusqu’à 20 à 40% des nouveau-nés admis reçoivent un traitement antibiotique chaque jour, 30 à 90% d’entre eux ayant été exposés à au moins un antibiotique au cours de leur admission [10].
L’IMF documentée ou suspectée a constitué le motif principal de prescription des antibiotiques chez nos patients. Ceci est expliqué par la hantise du « Early onset sepsis », qui a impacté la plupart des recommandations et notamment celles de l’ANAES de 2002 [3]. Dans notre contexte le principe de précaution est poussé à l’extrême en raison de l’absence de politique de dépistage du Streptocoque B chez la femme enceinte en Algérie. Cette forte prescription d’antibiotiques n’est pas sans conséquences sur l’implantation de la flore néonatale dans une période critique du développement immunitaire. En effet, lorsque des antibiotiques sont administrés au cours de la période périnatale, le processus de colonisation initial est interrompu, ce qui conduit à un état de dysbiose [11]. Des études épidémiologiques ont montré une association directe entre l’utilisation d’antibiotiques au cours de la première année de vie et l’expression de l’asthme au cours de l’adolescence [12]. Des observations similaires ont été notées pour l’utilisation précoce d’antibiotiques, les maladies inflammatoires de l’intestin, le diabète de type 1 et l’obésité [13,14]. Les effets potentiellement délétères de l’antibiothérapie pendant la période périnatale à court et à long termes, imposent donc de modifier nos habitudes diagnostiques et thérapeutiques. Ainsi, dans l’objectif de minimiser l’exposition aux antibiotiques des nouveaux nés à faible risque d’infection néonatale précoce ; l’académie américaine de pédiatrie en 2012 [15], le National Institute for Health and Clinical Excellence au Royaume Uni en 2013 [16] et l’HAS en France en 2017 [17, ont successivement revu et actualisé leurs recommandations.
Par ailleurs, la gentamycine était l’antibiotique le plus prescrit dans notre service pendant cette période, ce qu’a également rapporté Versporten et al., dans leur grande enquête [10]. La bithérapie étant de règle en période néonatale, un aminoside (essentiellement la gentamycine) est systématiquement associé à une bêta-lactamine (ampicilline, pénicilline G ou céfotaxime selon le protocole utilisé). Dans notre série, une association triple a été utilisée dans 57,56%, ce qui constitue probablement une surenchère difficilement argumentable, même si les difficultés de réalisation des examens bactériologiques pourraient expliquer cette tendance. En fait, l’association triple était majoritairement prescrite au cours des gardes et l’antibiothérapie était le plus souvent réajustée le jour suivant.
Enfin, les doses et les durées d’antibiothérapies étaient dans la majorité des cas conformes avec celles préconisées dans la littérature, contrairement à Metsvaht et al., qui ont rapporté des variations extrêmement larges et non aléatoires dans la posologie des antibiotiques les plus fréquemment utilisés dans les unités de soins intensifs néonatales [18]. La méthodologie de l’auto-évaluation que nous avons adoptée dans notre étude constitue une limitation et pourrait expliquer ce résultat contradictoire.
Conclusion
Le traitement rapide de l’infection néonatale avec des antibiotiques appropriés peut sauver la vie, cependant, la disponibilité d’antibiotiques efficaces peut devenir limitée par le développement des résistances.
La rédaction des protocoles thérapeutiques et d’arbres décisionnels adaptés à notre contexte ainsi que la création d’un comité pour l’antibiothérapie à usage hospitalier devraient permettre de mieux rationaliser les prescriptions des antibiotiques.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
de Jong, P.B. van den Berg, S.T. Visser, T.W. de Vries, L.T. de Jong-van den Berg. Antibiotic usage, dosage and course length in children between 0 and 4 years. Acta Paediatr., 98 (7) (2009), pp. 1142-1148
Short MA. Guide to a systematic physical assessment in the infant with suspected infection and/or sepsis. Adv Neonatal Care. 2004 Jun. 4(3):141-53; quiz 154-7.
Diagnostic et traitement curatif de l’infection bactérienne précoce du nouveau-né. ANAES, Editor. 2002.
Yves Liem TB, Krediet TG, Andre´ Fleer, Egberts TCG, Rademaker CMA. Variation in antibiotic use in neonatal intensive care units in the Netherlands. J Antimicrob Chemother 2010;65(6):1270-5.
Young T.E. MD, Magnum B, PharmD. Neofax. 24th ed. Montvale, NJ: Thomson Reuters; 2011
Bradley JS, Nelson JD. Nelson’s pediatric antimicrobial therapy. 22. Elk Grove Village: American Academy of Pediatrics; 2016
WHO Collaborating Centre for Drug Statistics Methodology, Guidelines for ATC classification and DDD assignment 2019. Oslo, Norway, 2018.
Eili Y. Klein, Thomas P. Van Boeckel, Elena M. Martinez, Suraj Pant, Sumanth Gandra, Simon A. Levin, Herman Goossens, and Ramanan Laxminarayan. Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc Natl Acad Sci U S A. 2018 Apr 10;115(15): E3463-E3470.
Versporten A, Sharland M, Bielicki J, Drapier N, Vankerckhoven V, Goossens H. The antibiotic resistance and prescribing in European Children project: a neonatal and pediatric antimicrobial web-based point prevalence survey in 73 hospitals worldwide. Pediatr Infect Dis J. 2013;32(6):e242–53.
Zeissig S, Blumberg RS. Life at the beginning: perturbation of the microbiota by antibiotics in early life and its role in health and disease. Nature Immunol 2014;12:307–10.
Marra F, Marra CA, Richardson K et al, Antibiotic use in children is associated with increased risk of asthma. Pediatrics 2009;123:1003–1010.
Giongo A, Gano KA, Drabb DB et al,. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J 2011;5:82–91
Azad MB, Bridgman SL, Becker AB, Kozyrskyj AL. Infant antibiotic exposure and the development of childhood overweight and central adiposity. Int J Obes (Lond). 2014;38(10):1290–8.
Polin RA; Committee on Fetus and Newborn. Management of neonates with suspected or proven early-onset bacterial sepsis. Pediatrics. 2012 May;129(5):1006-15. doi: 10.1542/peds.2012-0541. Epub 2012 Apr 30.
Caffrey Osvald E, et al. Arch Dis Child Educ Pract Ed 2014;99:98–100. doi:10.1136/archdischild-2013-304629
Gras-Le Guen C., Foix-L’Hélias L., Boileau P. (2017). Infection néonatale bactérienne précoce (INBP): quel algorithme de prise en charge en 2017 ? Archives de Pédiatrie. 24.S14-S17.10.1016/S0929-693X (18)30039-3.
Metsvaht, T., Nellis, G., Varendi, H., Nunn, A. J., Graham, S., Rieutord, A., Storme, T., McElnay, J., Mulla, H., Turner, MA., Lutsar, I. (2015). High variability in the dosing of commonly used antibiotics revealed by a Europe-wide point prevalence study: implications for research and dissemination. BMC Pediatrics, 15(1), [41]. DOI: 10.1186/s12887-015-0359-y
Tableaux
Tableau I. Principales caractéristiques de la population d‘étude (n=238).
Variables
%
Moyenne±Ecart-type
Médiane
Extrêmes
Age post-conceptionnel (Jours)
2,1 ± 5,2
0
0 ; 27
0 – 7 jours
76,05
–
–
–
8 – 14 jours
10,92
–
–
–
15 – 28 jours
13,03
–
–
–
Sexe (garçons)
66,4
–
–
–
Accouchement par césarienne
26,1
–
–
–
Inborn / Outborn
65,5 / 34,5
–
–
–
Age gestationnel (Semaines)
–
37,1 ± 4,2
39
27 ; 42
< 28
3,26
–
–
–
28 – 31 + 6 j
14,61
–
–
–
33 – 36 + 6 j
26,37
–
–
–
38 – 40 + 6 j
43,34
–
–
–
> 41
2,84
–
–
–
Indéterminé
9,58
–
–
–
Poids (grammes)
–
2825 ± 887,6
2860
700 ; 5100
< 1000
4,42
–
–
–
1000 – 1499
12,30
–
–
–
1500 – 2500
34,03
–
–
–
> 2500
49,25
–
–
–
Durée d’hospitalisation (Jours)
–
11,5 ± 9,4
9
1 ; 52
Tableau II. Répartition des cas selon le type (IMF ou nosocomiale) et le site de l’infection néonatale.
Antibiothérapie non justifiéen (%)
Septicémien (%)
Méningiten (%)
Infection pulmonairen (%)
Omphaliten (%)
Infection urinairen (%)
Totaln (%)
Antibiothérapie« prophylactique »
9 (3,8)
0 (0,0)
0 (0,0)
0 (0,0)
0 (0,0)
0 (0,0)
9 (3,8)
IMF précoce
0 (0,0)
106 (44,5)
16 (6,7)
36 (15,1)
0 (0,0)
1 (0,4)
159 (66,8)
IMF tardive
0 (0,0)
27(11,3)
2(0,8)
25 (10,4)
7 (2,9)
1 (0,4)
66 (27,7)
Infection nosocomiale
0 (0,0)
2(0,8)
0(0,0)
1(0,4)
1(0,4)
0 (0,0)
8 (3,4)
Total
9 (3,8)
135(56,7)
18 (7,6)
66(27,7)
8 (3,4)
2 (0,8)
238 (100)
IMF, Infection materno-fœtale
Tableau III. Répartition des prescriptions selon les familles d’antibiotiques.
Famille d’ATB
Antibiotique prescrit*
Effectif
Pourcentage%
Pénicillines
Ampicilline Oxacilline Amoxicilline
186 10 7
29,25 1.8 1, 1
C3G
Céfotaxime
181
28,46
Aminosides
Gentamycine
220
34,6
Macrolides
Josamycine
14
2,2
Carbapénèmes
Imipénème
7
1,1
Glycopeptides
Vancomycine
1
0,15
Imidazoles
Métronidazole
5
0,78
Quinolones
Ciprofloxacine
5
0,78
Total
636
100
Tableau IV. Antibiotiques prescrits et leurs Dose Définie Journalière par 181 jours d’étude.
.
Antibiotique prescrit
Dose par unité en gramme
Nombre total d’unités
Nombre de DDJ* par unité en gramme
Nombre de DDJ total
Ampicilline
1
672,6
0,5
336,3
Céfotaxime
1
534,2
0,25
133,5
Gentamycine
0,04
9,9
0,16
39,6
Josamycine
1,5
14,5
0,75
7,25
Imipenème
0,5
22
0,25
11
Oxacilline
1
17
0,5
8,5
Amoxicilline
1
21,6
1
21,6
Métronidazole
0,5
3,2
0,33
2,11
Vancomycine
0,5
0,5
0,25
0,25
*DDJ : dose définie journalière.
Tableau V. Conformité de la prescription des antibiotiques au cours des IMF précoces
Type d’infection
Antibiotique
Effectif
(%)
Dose moyenne / Référence*(mg/kg/dose)
Intervalle moyen /Référence*(heures)
Durée moyenne / Référence*(jours)
Septicémien=106
Ampicilline Gentamycine Céfotaxime Métronidazole
95 102 95 3
89,6 96,23 89,62 2,83
35,5 / 25-50 4,2 / 4-6 38,6/ 25-50 7,4 / 7,5
11,1 / 12 23,76 / 24 11,1/ 12 11,1/ 12
10,8 / 10 2,5 / 2 10,4 / 10 9 / 10
Méningiten=16
Ampicilline Céfotaxime Gentamycine Ciprofloxacine
6 14 16 1
37,5 87,5 100 16,94
95,8 / 100 51 / 50 4,19 / 4-6 11,1 / 10
11,33 / 12 11,6 / 12 22,87/ 24 12 / 12
19,7 / 15-21 17,43 / 15-21 6 / 5-10 5/ 4-5
Infection pulmonairen=36
Ampicilline Céfotaxime Josamycine
31 27 3
86,11 75 8,34
34,5 / 25-50 30,7/ 25-50 16,8 / 15
11,5 / 12 12,2/ 12 12,6 / 12
8,6 / 7-10 8,9 / 7-10 14 / 14
Infection urinaire n=1
Céfotaxime Gentamycine
1 1
100 100
50 / 50 5 / 4-6
12,7 / 12 24 / 24
11/ 7-10 4 / 2-4
*Néofax [6], et Nelson’s Pocket Book of Paediatric Antimicrobial Therapy dans sa 22ème édition [7]
Tableau VI. Antibiotiques prescrits, coût journalier moyen et coût total.
Différentes études nationales ont été réalisées, au passé, certaines multicentriques, ayant permis de mettre en évidence les différents foyers d’hémoglobinopathies
Djenouni, F. Grifi, Service d’Hématologie, CHU Annaba, Université Badji-Mokhtar.
Date de soumission : 25 Février 2019.
Abstract: Pathology at the crossroads of several disciplines, sickle cell disease poses a public health problem, pathology enamelled acute and degenerative complications at any time the vital and/or functional prognosis at stake. Various national studies have shown that the Northeast of Algeria, the border area with Tunisia is an endemic area of sickle cell disease. The objectives of our study are the identification of all the haemoglobinopathies supported by the haematology department of Annaba CHU (adult vocation); and to determine the epidemiological characteristics and the chronic complications of the major sickle cell syndromes.
Résumé : Pathologie au carrefour de plusieurs disciplines, la drépanocytose pose un problème de santé publique, pathologie émaillée de complications aiguës et dégénératives mettant à tout moment le pronostic vital et/ou fonctionnel en jeu. Diverses études nationales ont démontré que le Nord-Est Algérien, la zone frontalière avec la Tunisie constitue une zone endémique de la drépanocytose. Les objectifs de notre étude sont le recensement de toutes les hémoglobinopathies prises en charge au niveau du service d’hématologie du CHU de Annaba (à vocation adulte), et de déterminer les caractéristiques épidémiologiques et les complications chroniques des syndromes drépanocytaires majeurs.
Mots clés : Drépanocytose, vaso-occlusion, falciformation, hémoglobine Sickle, polymérisation, complications.
Introduction
Différentes études nationales ont été réalisées, au passé, certaines multicentriques, ayant permis de mettre en évidence les différents foyers d’hémoglobinopathies. Le Nord-Est du pays, essentiellement la zone frontalière avec la Tunisie constitue une zone endémique de drépanocytose, posant ainsi de nombreux problèmes de santé publique.
Objectifs
Les objectifs de notre étude sont :
Le recensement de toutes les hémoglobinopathies prises en charge au niveau du service d’hématologie CHU Annaba (à vocation adulte).
La détermination des caractéristiques épidémiologiques et des complications chroniques des syndromes drépanocytaires majeurs.
Patients et méthode
Population d’étude : La population étudiée est constituée de patients atteints de syndrome drépanocytaire majeur, dont l’âge est supérieur à 15 ans et toujours en vie.
Type d’étude : Il s’agit d’une étude transversale, analytique. La collecte des données a porté sur des informations rétrospectives complétées par un suivi prospectif.
Durée de l’étude : l’étude s’étale sur une période allant de décembre 1988 à décembre 2015.
Lieu de l’étude : Le service d’hématologie du CHU Annaba, service prenant en charge les patients du chef-lieu de la wilaya et des wilayas limitrophes.
Mode de suivi : Le suivi est régulier tous les 04 mois en dehors des complications aiguës, qui seront prises en charge en hospitalisation dans le cadre de l’urgence.
Les différentes complications dégénératives sont recherchées par un bilan annuel de dépistage.
Le suivi est fait dans le cadre d’une équipe multidisciplinaire avec un réseau de référents : cardiologues, ophtalmologues, rhumatologues, urologues, gynéco-obstétriciens, néphrologues et psychologues.
Résultats
Au terme de cette étude un total de 1.502 cas d’hémoglobinopathies est suivi et toujours en vie, dont 86% sont des syndromes drépanocytaires majeurs et 62% des formes sont homozygotes.
Graphe n°1 : Répartition des patients en fonction du type d’hémoglobinopathie
Graphe n°2 : Répartition des patients en fonction du type du syndrome drépanocytaire
Les données épidémiologiques :
L’âge moyen actuel de notre cohorte est de 30,33 ans avec des extrêmes [15-69 ans] ; le sex-ratio est de 0,84.
L’origine géographique est définie par leurs lieux de naissance, ils sont principalement originaires d’Annaba (65,1%) et d’El Tarf (21,0%).
Lieu de résidence : La grande majorité, soit 97,8% de nos sujets résident au niveau des wilayas côtières du Nord-Est du pays soit : Annaba (62,5%), El-Tarf (24,8%) et Skikda (10,5%).
Graphe n°3 : Répartition des patients en fonction de l’origine géographique
Graphe n°4 : Répartition des patients en fonction du lieu de résidence
La notion de consanguinité est retrouvée chez 35% des patients, elle est du 2ème degré dans 63% des cas.
Des cas similaires de drépanocytose dans la fratrie sont retrouvés chez 60% des sujets, avec des extrêmes de 01 et 05 cas.
Le niveau scolaire de notre cohorte reste bas, vu que seuls 13,57% ont atteints le niveau secondaire.
Sur le plan socio-professionnel ; 78,63% des patients sont sans profession et 72,36% en âge de se marier sont célibataires.
Graphe n°5 : Répartition des patients en fonction du niveau scolaire
Complications dégénératives
Durant le suivi régulier, de multiples complications dégénératives diversement associées sont colligées :
Les lithiases vésiculaires
La fréquence des lithiases vésiculaires était de 60,85%, chez les formes homozygotes ; 59,24% étaient symptomatiques (coliques hépatiques) ; et 47,13% des patients ont été cholécystectomisés. Il s’agissait dans 69% des cas de multi-microlithiases. (Voir Tableau n° 01)
Type d’hémoglobinopathieSSSBSC Pourcentage 60,85% 35% 13,6%
Tableau n°01 : prévalence de la lithiase vésiculaire
Les ostéonécroses épiphysaires
Cette complication est retrouvée chez 30% des patients homozygotes. La fréquence de l’ostéonécrose de la tête fémorale (ONTF) est de 27,98% ; découverte à un stade avancé chez 88% des patients, elle est symptomatique chez 74%, et à l’origine d’un handicap chez 28% d’entre eux. La fréquence de l’ostéonécrose de la tête humérale (ONTH) est de 06,77% chez les homozygotes, découverte à un stade avancé, chez 43,75% des patients, elle est symptomatique chez 56,27% et à l’origine d’un handicap chez 6,25% des cas. Par ailleurs, l’atteinte est bilatérale chez 43,75% des sujets. Voir Tableau n° 02 – Voir Figure 01
Tableau n°02 : prévalence des ostéonécroses épiphysaires
Type d’hémoglobinopathie
SS
SB
SC
Pourcentage
30%
15%
10%
Figure 01 : ONTF bilatérale – Droite : Stade V / Gauche : Stade IV = Patiente âgée de 34 ans « de notre série ».
L’atteinte rénale :
L’atteinte rénale est dominée par la survenue d’une protéinurie dont la fréquence chez les homozygotes est de 49,10% et 11,5% ont évolué vers une insuffisance rénale chronique. (Voir Tableau n° 03)
Tableau n°03 : prévalence de l’atteinte rénale
Type d’hémoglobinopathies
SS
SB
SC
Pourcentage
49,10%
24,75%
20,45%
Les complications cardiaques :
Elles sont survenues chez 53,28% de nos sujets et dominées par les valvulopathies (51,1%). (Voir Tableau n° 04)
Tableau n°04 : Prévalence de l’atteinte cardiaque
Type d’hémoglobinopathie
SS
SB
SC
Pourcentage
19%
18,28%
16%
Les complications oculaires
L’atteinte oculaire est fréquente chez les doubles hétérozygoties SC (20,45%), et les anomalies prolifératives sont les plus fréquentes (36,45%). (Voir Tableau n° 5)
Tableau n°05 : Prévalence de l’atteinte oculaire
Type d’hémoglobinopathie
SS
SB
SC
Pourcentage
11%
8%
20,45%
Les ulcères de jambes
La fréquence de l’ulcère de jambes dans notre série est de 06,40% dans les formes homozygotes, le caractère récidivant est retrouvé chez 40% de l’ensemble des patients atteints. (Voir Tableau n° 06- Voir Figure 02)
Tableau n°06 : Prévalence de l’ulcère de jambe
Type d’hémoglobinopathie
SS
SB
SC
Pourcentage
6,40%
1,59%
0
Figure 02 : Ulcère pré malléolaire droit. Patient âgé de 32 ans « de notre série ».
La prise en charge thérapeutique
L’éducation thérapeutique :
Information les parents et les patients sur les différents aspects de l’affection, les traitements disponibles, la nécessité d’une bonne hygiène de vie, l’éviction des facteurs déclenchants les complications aiguës, les signes cliniques alarmants et les conduites à tenir.
Le traitement de fond : supplémentation systématique en folates et l’antibioprophylaxie.
La vaccination : Les patients bénéficient tous d’une vaccination selon le calendrier vaccinal, auquel on rajoute l’anti hépatite B chez les adultes et l’antigrippal. L’anti pneumocoque n’est pas généralisé, vu sa non disponibilité régulière.
Le traitement des complications aigues : repose sur de multiples armes, diversement associées :
Les antalgiques majeurs/ mineurs
Une antibiothérapie
Un support transfusionnel : culots globulaires phénotypés (tous les patients), filtrés pour certains.
Les échanges transfusionnels manuels (absence erythrocytaphérèse)
L’hydroxyurée : son utilisation chez nos patients est large et a nettement amélioré leur qualité de vie, en réduisant de façon significative la fréquence des complications aigues.
Discussion
Les résultats colligés au terme de notre étude sont comparés avec ceux des différentes études. L’âge moyen actuel de nos patients est supérieur à celui retrouvé dans les études réalisées en Côte d’Ivoire [1] et à Dakar [2], qui trouvent respectivement des âges moyens de 21 ans et 20 ans. L’espérance de vie des drépanocytaires s’est beaucoup améliorée partout dans le monde. Aux États Unis d’Amérique, 50% des patients atteignent l’âge de 50 ans [3].
La consanguinité est retrouvée chez 35% de nos sujets, ce qui contribue à la transmission du gêne S ; ceci rejoint les résultats des études de Belhani [4] en 1981 et de Bouzid [5] en 1983, qui ont trouvé respectivement des taux de 52% et 46%.
Dans notre série, 60% des patients ont des cas similaires dans la fratrie, avec des extrêmes de 01 et 05 cas, ceci reflète le défaut d’information des parents. Ces résultats démontrent que des efforts doivent être réalisés, par la mise en place d’un programme de lutte, se basant sur le conseil génétique.
Les origines géographiques des patients sont définies par le lieu de naissance. Ils sont principalement originaires d’Annaba (65,08%) et d’El Tarf (20,95%).
Dans notre cohorte 97,78% des sujets résident au niveau des wilayas côtières du Nord-Est du pays : Annaba (62,54%), El-Tarf (24,76%) et Skikda (10,48%). Ces patients parcourent en moyenne 100 Km afin d’arriver au niveau de notre centre de soins.
Chez nos patients, la scolarisation et l’insertion socio-professionnelle ne paraissent pas satisfaisantes. 72,13% des patients avaient arrêté leur scolarité précocement, avant le secondaire, et seuls 13,57% avaient atteint le niveau universitaire ; ces résultats sont très loin de ceux de l’étude française de Bordeaux [9], qui trouve que 55,7% de ses patients avaient un niveau scolaire bac/études universitaires et 34,9% un niveau collège/lycée.
Nos résultats reflètent l’impact de la maladie sur la progression scolaire du patient d’une part et l’absence de prise en charge spécifique de ces patients au niveau scolaire.
Au terme du bilan annuel, on a colligé de multiples complications diversement associées.
La lithiase biliaire, est la principale complication abdominale de la drépanocytose ; secondaire à l’hémolyse chronique, sa fréquence dans notre étude, est de 60,85% chez les homozygotes ; 59,24% sont symptomatiques (coliques hépatiques) et 47,13% sont cholécystectomisés. Nos résultats rejoignent ceux de la littérature, où sa prévalence varie selon les séries de 30 à 70% [10,11].
La protéinurie est retrouvée dans notre cohorte avec une fréquence de 49,10% chez les homozygotes, sa prévalence est variable selon les études, certaines l’ont trouvé à 50% [12], d’autres de 15 –30% [13, 14], Guasch retrouve une prévalence de 70% [15].
La fréquence de l’insuffisance rénale est de 11,50% au sein de notre série, alors qu’elle varie dans la littérature entre 5 et 18% en Arabie Saoudite, en Amérique et au brésil [16-18].
Les complications ophtalmologiques, sont retrouvés chez nos patients avec une prévalence de 20,45%, les rétinopathies prolifératives sont les plus fréquentes (36,45%), cette fréquence est supérieure à celle retrouvée dans la littérature : Au Pas-de-Calais en France, Tran Th. [19] retrouve une prévalence des atteintes rétiniennes chez les patients homozygotes S/S de 17% et elles étaient au stade de rétinopathie proliférante. Les études faites dans la péninsule arabe ne retrouvent pas d’atteintes ophtalmologiques prolifératives ou non, ceci peut être expliqué pas le fait de l’existence dans cette région de formes bénignes de drépanocytose en rapport avec un taux d’hémoglobine F élevé [20].
Notre étude retrouve une prévalence d’ostéonécroses épiphysaires de 30% chez les homozygotes, dans la littérature elle varie en fonction des études, Adekile A.D. et Marouf R. retrouvent une prévalence de 48,6% [21,22] ; elle est de 41% dans l’étude de Ware et al. En Jamaïque, Lee et al. retrouvent une prévalence de 82% [24], et dans l’étude de Lionnet elle est de 26% [25].
La fréquence de l’ulcère de jambe dans notre étude est de 6,40% chez les homozygotes. Dans la littérature, elle varie en fonction des pays et des études, en France [26] on retrouve une prévalence de 5,5%, aux USA [27] elle est de 2,5% ; entre 1,5 et 13,5% en Afrique [28,29] et > 40% en Jamaïque [30]. L’origine géographique et le niveau socioéconomique influencent la prévalence de survenue d’ulcère de jambes.
Conclusion : Vu la forte prévalence de la drépanocytose dans le Nord-Est du pays, la gravité de cette pathologie par ses complications aigues et dégénératives, pouvant mettre en jeu le pronostic vital et/ou fonctionnel ; des efforts doivent être fournis afin de réduire sa prévalence dans notre région et de prendre en charge précocement et correctement ces patients ; en mettant en place des programmes nationaux d’information, de prévention et de prise en charge.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
Aissata Tolo-Diebkilé et al. Drépanocytose homozygote chez l’adulte ivoirien de plus de 21 ans. Cahiers santé. 2010; Volume 20, n°2, avril, mai, juin.
Diop S. et al. La drépanocytose homozygote après l’âge de 20 ans. La revue de médecine interne. 2003; 24: 711-5.
Koduri PR. Acute splenic sequestration crisis in adults with sickle cell anaemia. J. Hematol. 2007; 82: 174-5.
Belhani M, Bouzid K, Henni T, Colona P. La drépanocytose homozygote en Algérie. Méd Afr Noire. 1981;28 5,337-8.
Bouzid K. Syndromes drépanocytaires : Aspects épidémiologiques, génétiques, cliniques, biologiques et hématologiques. Thèse de diplôme de Doctorat d’état en sciences médicales Alger, 1987.
Malou M., Grifi F. Profil des Anémies hémolytiques congénitales dans le Nord-Est 1994; IV N°4 Juillet-Aout.
MT. Les Syndromes drépanocytaires en Algérie : Épidémiologie nationale 1996- 2005. Rev. Alg. d’Hématologie. 2009; 1: 29-31.
GRIFI F. Étude phénotypique et génotypique de la drépanocytose dans la région d’Annaba. Thèse de diplôme de Doctorat d’état en sciences médicales Annaba, 2006.
BOURDEAU D., Caractéristiques sociologiques des drépanocytaires adultes vivant en France : étude multicentrique sur 369 patients, in : Rev. Hématologie, vol. 19,supl 2, mars 2013, Paris, pp. 36.
Vichinsky E., Johnson J et al. Blood Alloimmunization in sickle cell anaemia and transfusion of racially unmatched. New Engl. J. Med. 1990; 322: 1617.
F BD, Nenert .M et al. Vascular lesions of the liver in sickle cell disease: a clinicopathological study in 26 living patients. Archives of Pathology and Laboratory Medicine. 1995; vol. 119 no. 1:46-52.
R, Kuti.M. A, Olawale.O, Akinola.N. Renal Disease in Adult Nigerians with Sickle Cell Anemia: A Report of prevalence, Clinical Features and Risk Factors. Saudi Journal of Kidney Diseases and Transplantation. 2012; Vol. 23, 171-175.
Ataga K, Orringer E. Renal abnormalities in sickle cell disease. American Journal of Haematology. 2000; vol. 63, no. 4:205-11.
Wesson D.E. The initiation and progression of sickle cell nephropathy, Kidney International, 2002; vol. 61, no. 6:2277- 86.
Guasch A, Nass K, Zayas CF. Glomerular involvement in adults with sickle cell hemoglobinopathies: Prevalence and clinical correlates of progressive renal failure. J Am Soc 2006;17(8):2228-35.
Aleem A. Proteinuria in adult Saudi patients with sickle cell disease is not associated with identifiable risk factors. Saudi J Kidney Dis Transpl.2010.21:903–908
Powars DR, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anaemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore). 2005; vol.84: 363-76.
Silva Junior G.B., Vieira A.P. Evaluation of renal function in sickle cell disease patients in Brazil. Braz J Med Biol Res. 2012; 45 (7).
Tran T.H.C MA, Godinaud M, Rose C. Rétinopathie drépanocytaire chez les adultes de la région Nord-Pas-de-Calais. J Fr Ophtalmol. 2008; 31: 987-92.
Al-Salem M. Benign ocular manifestations of sickle cell anaemia in Arabs. Indian J Ophtalmol. 1991; 39:9-11.
Adekile AD. Limitations of Hb F as a phenotypic modifier in sickle cell disease: study of Kuwaiti Arab patients. Hemoglobin. 2011;35(5-6):607-17.
Marouf R. Blood transfusion in sickle cell disease. Hemoglobin. 2011;35(5-6): 495-502.
Ware H E., Toye R., Berney S. I. Sickle cell disease and silent avascular necrosis of the hip. Journal of Bone and Joint Surgery-Series A. 1991; vol. 73, no. 6:947-9.
Lee JS. Serjeant G. R. The radiological features of avascular necrosis of the femoral head in homozygous sicke cell disease, Clinical Radiology. 1981; vol. 32 no. 2:205-14
Lionnet F, Katia Stankovic Stojanovic, Virginie Avellino, Gilles Grateau, Robert Girot, Jean-Philippe Haymann. Haemoglobin sickle cell disease complications: a clinical study of 179 cases. 2012;97(8):1137.
Halabi-Tawil M, Girot R, Bachmeyer C, Levy PP, Aractingi S. Sickle cell leg ulcers: A frequently disabling complication and a marker of severity. Br J Dermatol. 2008;158: 339-44.
Koshy M, Koranda A. Leg ulcers in patients with sickle cell disease, Blood, 1989; vol. 74, no. 4:1403-8.
Elira Dokekias. A. Étude analytique des facteurs d’aggravation de la maladie drépanocytaire au Congo. Pub. Med. Afr. 2001; 131 :12-6.
Eckman J. R. Leg ulcers in sickle cell disease. Haematology/ Oncology Clinics of North America, 1996; vol. 10, no. 6:1333-44.
Catherina P, Minniti JE, SamirK.Ballas and al. Leg Ulcers cell disease. Am J Hematol. 2010 85(10) October: :831-3.
La qualité et l’espérance de vie des patients thalassémiques se sont nettement améliorées ces dernières années
Djenouni, F. Grifi, Université Badji-Mokhtar Faculté de Annaba, Algérie.
Date de soumission : 22 Février 2019.
Abstract: Β-Thalassemia major is a heterogeneous genetic disorder, characterized by abnormalities of the β gene of haemoglobin, currently more than 130 mutations have been identified. Its conventional treatment uses two main therapeutic modalities: 1. The transfusion program: which induces long-term iron overload, affecting various organs and constituting the leading cause of mortality and morbidity. 2. The iron chelation: the quality of which remains the major element of the vital prognosis of the disease via the prevention of the haemochromatosic organic lesions. The objective of our study is to determine the epidemiological, clinical and evolutionary characteristics of major thalassaemic patients followed in the haematology department at Annaba Hospital.
Résumé : La β thalassémie majeure est une affection génétique hétérogène, caractérisée par des anomalies du gène β de l’hémoglobine, actuellement plus de 130 mutations ont été identifiées. Son traitement conventionnel fait appel à deux modalités thérapeutiques principales : 1. le programme transfusionnel : qui induit à long terme une surcharge en fer, affectant différents organes et constituant la première cause de mortalité et de morbidité ; 2. La chélation du fer : dont la qualité reste l’élément majeur du pronostic vital de la maladie par la prévention des atteintes organiques hémochromatosiques. L’objectif de notre étude est de déterminer les caractéristiques épidémiologiques, cliniques et évolutives des patients thalassémiques majeurs suivis au niveau du service d’hématologie CHU Annaba.
Mots clés : ß thalassémie, hémochromatose, transfusion.
Introduction : La qualité et l’espérance de vie des patients thalassémiques se sont nettement améliorées ces dernières années ; mais l’évolution reste émaillée de nombreuses complications en rapport, dans la grande majorité des cas, avec la surcharge en fer dont les moyens d’évaluation restent insuffisants dans notre pays.
Objectif : Déterminer les caractéristiques épidémiologiques, cliniques et évolutives de nos patients thalassémiques majeurs.
Matériel et méthode : Étude rétrospective, descriptive, ayant inclus 68 patients atteints de β thalassémie majeure ; s’étalant de Décembre 2015 à Janvier 2017.
La surveillance des patients est régulière : clinique, biologique et radiologique, à la recherche des complications liées au traitement ou en rapport avec la maladie.
Résultats et discussion : Au terme de notre travail, certaines caractéristiques sont colligées.
L’âge moyen de notre cohorte est de 23 ans, avec des extrêmes de [15 – 37 ans], cet âge est supérieur à celui de l’étude tunisienne de 20071 (10,7ans) et marocaines2 de 2014 (11ans). Le sex-ratio est de 1,10.
Une consanguinité est notée chez les parents dans 43% des cas, ce taux rejoint celui des études nationales de 20093 (40%) et marocaines2 de 2014 (45%), mais nettement inférieur à celui de l’étude tunisienne1 réalisée en 2007 (75,3%).
Des cas similaires dans la fratrie sont retrouvés chez 40% de nos patients, ce taux est proche de celui de l’étude nationale de 20093 (39,4%) et il est supérieur à celui de l’étude tunisienne de 20071 (34,3%) ; ceci reflète l’absence à l’heure actuelle d’un programme national de prévention.
Sur le plan scolaire : 40% de nos sujets ont un niveau moyen, 25% ont atteint le secondaire et 16,39% ont fait des études universitaires (médecine, droit et informatique). Le niveau d’instruction de nos patients est inférieur à celui retrouvé dans les études internationales notamment à chypre4, où 83% des patients ont un niveau universitaire, ceci serait expliqué par la fréquence de l’absentéisme, des complications imprévisibles ainsi que les troubles psychologiques qui peuvent survenir chez ces patients.
Le traitement est toujours conventionnel se basant sur :
Un programme transfusionnel régulier chaque 3 à 4 semaines, avec des culots globulaires phénotypés ; filtrés seulement s’il y a une allo immunisation leuco-plaquettaire et lavés si réaction allergique. Le taux d’Hb pré-transfusionnel moyen est de 08,5g/dl +/- 1,21, avec des extrêmes de [7-10,5g/dl], les recommandations actuelles visent un taux d’hémoglobine pré transfusionnel > 9,5g/dl.
Le traitement chélateur du fer : Deux types de chélateurs sont actuellement disponibles en Algérie (deferoxamine/deferasirox) et sont utilisés avec la même fréquence en monothérapie ou en association. La deferoxamine est utilisée par voie sous-cutanée en bolus chez la grande majorité des patients, vu la non disponibilité des micro-pompes, ceci est à l’origine d’une non observance du traitement.
Tableau n°1 : Le type de chélation
Molécule
Type
Fréquence(%)
Dose
Monothérapie
Deferoxamine (DFO)
27
50mg/Kg/j
Deferasirox (DFX)
50
30-40mg/Kg/j
Association
DFO+DFX
23
50mg/Kg/j +30mg/Kg/j
Tableau n°1 : le type de chélation
La vaccination : Les patients bénéficient tous d’une vaccination selon le calendrier vaccinal auquel on rajoute l’anti hépatite B chez les adultes et l’antigrippal. L’anti pneumocoque n’est pas disponible de façon régulière.
À notre niveau, la surveillance de la surcharge en fer, se base essentiellement sur la ferritinémie, dont le taux moyen est de 3.558 ng/ml avec des extrêmes de [521-8.220 ng/ml], le seuil critique de 2.500 ng/ml est dépassé chez 64% des patients, ce qui expose à de nombreuses complications. Ce taux est supérieur à celui de l’étude tunisienne1 de 2007 (26,50%).
Depuis leur recrutement, de nombreuses complications, diversement associées ont été colligées chez nos patients :
Les complications endocriniennes :
Tableau n°2 : Les complications endocriniennes
Type
Fréquence (%)
Age moyen de découverte (ans)
Retard staturo-pondéral
36
18
Retard pubertaire
37
18
Diabète insulinodépendant
16
19
Hypothyroïdie
9
19
Hypoparathyroïdie
5
24
L’hypogonadisme est la plus fréquente et la plus précoce des complications endocriniennes de la surcharge en fer5. Dans notre étude, elle est retrouvée avec une fréquence de 37%, taux supérieur à celui de l’étude française6 de 2010 (15%) ; mais inférieur à ceux des études Iranienne7 de 2016 (42,3%) et tunisienne1 de 2007 (51%).
Le retard de croissance est retrouvé dans notre série avec une fréquence de 36%, ce taux est supérieur à celui de l’étude tunisienne1 de 2007 (26,7%).
L’hypothyroïdie et l’hypoparathyroïdie sont liées à l’hémochromatose et sont souvent associées à un hypogonadisme ; dans notre étude elles sont retrouvées avec des fréquences respectives de 09% et 05%, ces taux rejoignent ceux des différentes études retrouvées dans la littérature.
Le diabète insulinodépendant est retrouvé avec une fréquence de 16%, ce qui est supérieur à celle retrouvée dans la littérature.
Tableau n° 3 : Les complications endocriniennes dans la littérature
Etude
Hypothyroïdie Fréquence(%)
Hypoparathyroïdie Fréquence (%)
DID Fréquence(%)
France6 2010
10
——
08
Iran82016
9.8
——
9.5
Tunisie1 2014
6.87
——
4.7
Arabie Saoudite9
——-
10
—————–
Oman10
——-
10
—————–
Italie11
——-
3.6
—————–
Les complications cardiaques : 18 cas (26,50%)
Tableau n°5 : Les complications cardiaque
Type
Effectif (n)
Age moyen de découverte (ans)
Cardiomyopathie
10
19
Trouble du rythme
02
22
HTAP
02
22
Thromboses
04
25
L’hémosidérose cardiaque constitue la 1ère cause de mortalité12 chez les patients thalassémiques majeurs, elle est secondaire à un dépôt de fer au niveau des cardiomyocytes aboutissant à une dysfonction cardiaque et à une arythmie ; si cette surcharge se poursuit une fibrose s’installera.
Ces complications même sévères peuvent être réversibles par une intensification du traitement chélateur. L’IRMT2* constitue l’examen de référence pour détecter une surcharge cardiaque à un stade pré symptomatique, infra clinique.
Chez nos patients, cette atteinte a été retrouvée avec une fréquence de 26,50%, ce taux est supérieur à celui des séries françaises6 (10%) et tunisiennes1 (17,52%).
Les complications hépatiques : 32 cas (47%)
Tableau n°5 : Les complications hépatiques
Complications
Effectif (n)
Age de découverte (ans)
Cirrhose
04
25
Lithiase vésiculaire
11
22
Hépatite virale
08
19
La surcharge en fer est la 1ère cause d’atteinte hépatique13, s’exprimant sous forme de fibrose pouvant évoluer vers une cirrhose et/ou un carcinome hépatique, elle constitue la 2ème cause de mortalité chez les thalassémiques majeurs13. L’IRMT2* est à ce jour la méthode non invasive de choix pour évaluer la surcharge hépatique en fer. Dans notre série, cette atteinte est retrouvée dans 47% des cas, ce taux est supérieur à celui des études européennes14 (10%) et même celui de l’étude nationale15 de 2014 (36%).
Les complications osseuses : 36 cas (52%)
Tableau n°6 : Les complications osseuses
Complications
Effectif (n)
Age de découverte (ans)
Ostéopénie
18
24
Tassements vertébraux
05
22
Fractures pathologiques
07
22
Déformations : Genu-valgus
04
19
Foyers ectopiques hématopoïèse
02
28
L’atteinte osseuse est complexe et multifactorielle, en rapport avec l’expansion médullaire, les troubles endocriniens, principalement l’hypogonadisme et la surcharge en fer. La deferoxamine joue également un rôle dans la genèse de l’ostéopénie.
Conclusion
La prise en charge de nos patients thalassémiques s’est nettement améliorée, mais il est nécessaire qu’elle soit uniformisée chez l’enfant et l’adulte et il est important d’avoir des plateaux techniques régulièrement fonctionnels au niveau du secteur public pour la réalisation des différents bilans annuels et de surveillance .
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Bibliographie
Bejaoui M. Thalassemia in Tunisia-the first Maghreb Summit 2014
Khatab MD: Thalassemia in Morocco-the first Maghreb Summit 20
Belhani M. Epidémiologie de la B thalassémie homozygote en Algérie. Revue Algérienne d’hématologie n°1-Sept 2009
Eleftheriou A, à propos de la thalassémie, TIF ISBN : 9963-623-40-9
Léger J. Girot R, Normal Growth hormone (GH) response to GH-releasing hormone in children with thalassemia major before puberty: a possible age-related effect. J Clin Endocrionl Metab 1989;69:453-6
Thuret I & coll. Complications and treatment of patients with beta-thalassemia; result of the national registry, Hematologica 2010;95:724-9
Sayermiri K et coll. The prévalence of hypogonadism in patients with thalassemia major in Iran-a systematic meta-analysis study. J Shahrekord univ med sci 2016.
Milad et coll. Prevalence of Hypothyroïdism and hypoparathyroïdism. Iran journal Ped Hematol Oncol. 2016,vol 6 N° 3,261-276.
Habeb et coll. Endocrinopathies in beta-thalassemia major in northwest Saudi Arabia. Saudi Arabia journal 2013; 34(1):67-73.
Mula-Abed et coll. Prevalence of endocrinopathies in patients with beta thalassemia in Oman; Oman medical journal2008;23(4):257-62
Multicentre study on prevalence of endocrine complications in thalassemia major. Italian Working Group on Endocrine complications in non-endocrine disease. Clinical endocrinology 1995;42(6):581-6
Rose C et al, Hemochromatose post-transfusionnelle, EMC Hematologie,2008,13-038-A-10
Girot R et al. Traitement de la surcharge en fer dans les maladies hématologiques. Revue hématologie 2006;12(3):181-93
Mirault t. Non-invasive assessment of liver fibrosis by transient elastography in post transfusionnal iron averload . Eur J Haematol 2008; 80:337-40
Belhani et al, Situation de la B thalassémie en Algérie, First Maghreb Summit 2014
L’anémie de Fanconi (AF) décrite la première fois en 1927 par Guido Fanconi est une maladie génétique héréditaire rare, le plus souvent à transmission autosomale récessive
Benakli, R. Ahmed-Nacer, R.M. Hamladji, Service d’hématologie-greffe de moelle osseuse, Centre Pierre et Marie Curie, Alger
Date de soumission : 23 Février 2019.
Abstract: Fanconi anaemia (FA) is a rare hereditary genetic disease most often with autosomal recessive inheritance. It carries out a syndrome of chromosomal instability whose clinical presentation is variable including congenital anomalies, a progressive pancytopenia and a particular susceptibility to cancer. FA is the most common constitutional medullary aplasia. The diagnosis of certainty is based on the existence of chromosomal breaks in response to desoxyribonucleic acid bridging agents, mitomycin C or diepoxybutane. The only cure for haematological manifestations is allogeneic hematopoietic stem cell transplantation, which prevents progression to acute leukaemia.
Résumé : L’anémie de Fanconi (AF) est une maladie génétique héréditaire rare le plus souvent à transmission autosomale récessive. Elle réalise un syndrome d’instabilité chromosomique dont la présentation clinique est variable incluant des anomalies congénitales, une pancytopénie progressive et une susceptibilité particulière au cancer. L’AF est la plus fréquente des aplasies médullaires constitutionnelles. Le diagnostic de certitude repose sur l’existence de cassures chromosomiques en réponse à des agents pontants de l’acide désoxyribonucléique, la mitomycine C ou le diepoxybutane. Le seul traitement curatif des manifestations hématologiques est l’allogreffe de cellules souches hématopoïétiques qui permet de prévenir l’évolution vers une leucémie aiguë (LA).
Mots clés : Fanconi, aplasie constitutionnelle, anomalies congénitales, allogreffe de CSH.
Introduction
L’anémie de Fanconi (AF) décrite la première fois en 1927 par Guido Fanconi est une maladie génétique héréditaire rare, le plus souvent à transmission autosomale récessive. Elle réalise un syndrome d’instabilité chromosomique dont la présentation clinique est variable, incluant des anomalies congénitales, une pancytopénie progressive et une susceptibilité particulière au cancer. L’AF est la plus fréquente des aplasies médullaires constitutionnelles. Le diagnostic de certitude repose sur l’existence de cassures chromosomiques en réponse à des agents pontants de l’acide désoxyribonucléique (ADN), la mitomycine C (MMC) ou le diepoxybutane (DEB). Le seul traitement curatif des manifestations hématologiques est l’allogreffe de cellules souches hématopoïétiques qui permet de prévenir l’évolution vers une leucémie aiguë (LA).
Épidémiologie
La fréquence des sujets hétérozygotes a été estimée à 1/300 aux États Unis et en Europe. L’AF est observée dans tous les groupes ethniques, avec toutefois une fréquence plus élevée dans 2 ethnies : les juifs ashkénazes et les afrikaners (1/22.000 naissances dans la région du Cap). La transmission autosomique récessive favorise la survenue de cas dans groupes ethniques avec forte consanguinité (1). En Algérie, 13% des aplasies médullaires sont des AF selon une étude réalisée entre 1994 et 2004 (2).
Génétique de l’anémie de Fanconi
L’AF est marquée par une hétérogénéité phénotypique reflétant en partie son hétérogénéité (3,4). Il a été mis en évidence une vingtaine de gènes dont la mutation peut être responsable d’une AF (Tableau 1). Ces gènes sont impliqués dans une voie de réparation des lésions acquises de l’ADN, la voie FANC/BRCA et sont également à l’origine de l’apparition de tumeurs (5). L’instabilité génétique décrite dans l’AF résulte d’une atteinte des mécanismes de réparation de l’ADN.
Les gènes les plus souvent impliqués sont FANCA (60% environ), FANCC (14%) et FANCG (10%). La maladie se transmet selon un mode autosomique récessif sauf en cas de FANCB qui est situé sur le chromosome X, se transmettant alors selon le mode récessif lié à l’X.
Les protéines codées par les gènes FANC agissent de concert au sein d’une voie métabolique activée dans la cellule après survenue d’une lésion de l’ADN. Cette voie métabolique a pour cette raison été dénommée voie AF/BRCA. Il se forme alors un complexe constitué de 8 protéines FANC (FANCA/B/C/E/F/G/L/M) qui va activer la voie AF/BRCA en mono-ubiquitinant 2 autres protéines : FANCD2 et FANCI qui vont former un dimère. Ce dimère va stabiliser la fourche de réplication et interagir, au niveau des foyers de réparation nucléaires, avec des protéines dites d’aval qui coopèrent dans la réparation des lésions de l’ADN. L’atteinte des protéines intervenant dans cette voie de réparation de l’ADN est à l’origine de cassures chromosomiques, spontanées ou induites par les agents pontants (comme les agents alkylants) ou les radiations ionisantes. Certains patients peuvent présenter un mosaïcisme génétique qui peut aboutir à un développement clonal et sont donc aussi prédisposés au développement de tumeurs solides et de l’évolution vers une LA (6,7).
Tableau 1 : Mutations génétiques au cours de l’AF
Gène
Transmission
FANCA
Autosomale Récessive (AR)
FANCB
Récessive liée à l’X
FANCC
AR
FANCD1/BRCA2
AR
FANCD2
AR
FANCE
AR
FANCF
AR
FANCG/XRCC9
AR
FANCI/KIAA1794
AR
FANCJ/BRIP1/BACH1
AR
FANCL
AR
FANCM
AR
FANCN/PALB2
AR
FANCO/RAD51C
AR
FANCP/SLX4
AR
FANCQ/ERCC4
AR
FANCR/RAD51
Autosomale Dominante
FANCS/BRCA1
AR
FANCT/UBE2T
AR
FANCU/XRCC2
AR
FANCV/REV7/MAD2L2
AR
Présentation clinique
L’expression clinique de la maladie est marquée par une grande variabilité phénotypique, y compris au sein d’une même famille, reflet d’une hétérogénéité génétique. Certains patients ne présentent aucune anomalie ou malformation associée. Le tableau clinique associe classiquement une petite taille, un syndrome malformatif variable, et une insuffisance médullaire d’apparition secondaire s’aggravant avec l’âge.
La majorité des patients ont des malformations congénitales, les plus fréquentes étant les malformations squelettiques présentes chez 2/3 d’entre eux. Si un tiers des patients ne présentent aucune malformation congénitale, il existe le plus souvent un visage particulier, un retard staturo-pondéral et des signes cutanés (3,4,8).
Manifestations hématologiques
L’atteinte hématologique est le plus souvent absente à la naissance. Progressivement se développe une anémie macrocytaire, suivie d’une thrombopénie et d’une neutropénie. Dans une étude de l’IFAR (International Fanconi Anemia Registry) portant sur 388 sujets, 85% présentent des anomalies de la numération. L’âge médian de l’apparition de ces anomalies est de 7 ans (0 à 36 ans). L’évolution se fait progressivement vers un tableau d’insuffisance médullaire sévère. Il existe un risque d’évolution vers un syndrome myélodysplasique ou une leucémie aiguë. Le risque pour un patient atteint d’AF de développer une leucémie aiguë myéloïde est environ 500 fois supérieur à celui de la population générale. Ce risque augmente avec l’âge : 7% à 10 ans, 27% à 20 ans, 43% à 30 ans.
Le myélogramme montre une moelle pauvre, érythroblastique ou franchement hypoplasique. Un aspect dysplasique d’une ou plusieurs lignées est fréquent.
La biopsie ostéo-médullaire montre une hypoplasie médullaire, voire une moelle désertique.
Manifestations extra-hématologiques (Tableau 2)
Le retard staturo-pondéral est pratiquement constant, débutant dès la vie intra-utérine par un retard de croissance harmonieux.
La dysmorphie faciale est caractérisée par un visage triangulaire, une macrognathie et une microphtalmie responsable d’un pseudo-hypertélorisme. Une microcéphalie inférieure au 5ème percentile est présente dans 30 à 40% des cas.
L’atteinte cutanée est très fréquente, souvent très évocatrice. L’expression clinique est variée et évolutive dans le temps. On note une association de tâches pigmentées (dites «café-au-lait»), de tâches achromiques et d’une mélanodermie s’accentuant avec l’âge, siégeant préférentiellement au tronc et au cou.
Des malformations squelettiques peuvent exister : pouce absent, hypoplasique, bifide, dupliqué ou triphalangé ; hypoplasie de l’éminence thénar, absence du 1er métacarpien; radius absent ou hypoplasique, syndactylie, malformations des orteils, luxation congénitale des hanches, spina bifida, scoliose et aplasie coccygienne.
Des malformations rénales et de l’appareil urinaire (chez environ 20%) : reins uniques, en fer à cheval, ectopiques ou pelviens, hypoplasiques, dysplasiques ou encore d’une urétéro-hydronéphrose.
Des anomalies oto-rhino-laryngologiques (environ 10%) : malformation de l’oreille externe (malformation du pavillon de l’oreille, oreilles implantées basses, dysplasie ou atrésie du conduit auditif externe), ou surdité, le plus souvent de transmission, secondaire à des malformations des osselets de l’oreille moyenne.
Les anomalies du tube digestif (environ 7%) : atrésie de l’œsophage, avec ou sans fistule trachéo-oesophagienne, atrésie duodénale, imperforation anale, et l’anus ectopique.
Ces anomalies peuvent être isolées ou s’intégrer dans une association de type VACTERL (V: anomalies vertébrales, A: atrésie anale, C : cardiopathie, TE : fistule trachéo-œsophagienne avec atrésie de l’œsophage, R : atteinte rénale, L (limb) : anomalies des membres) qui doit faire systématiquement évoquer une AF (9,10,11).
Tableau 2 : manifestations extra-hématologiques au cours de l’AF
Peau
Taches café-au-lait Anomalies de pigmentation (mélanodermie, taches ou zones achromiques)
Membres supérieurs
Radius: absent, hypoplasique, absence de pouls ou pouls faible Pouce: absent, hypoplasique, bifide, dupliqué, non articulé, triphalangé, long, implantation basse Éminence thénar: plate, absente Main et doigts: absence du premier métacarpien, clinodactylie, polydactylie Cubitus: court, dysplastique
Surdité : de transmission ou neurosensorielle Forme: anomalies du pavillon, aspect dysplastique ou atrésique, canal auditif étroit, anomalies de l’oreille moyenne Langage: retardé, dyslexie
Cœur et poumons
Malformations cardiaques congénitales: persistance du canal artériel, communication inter-auriculaire, communication inter-ventriculaire, coarctation de l’aorte, situs inversus
Retard de croissance intra-utérin
Membres inférieurs
Hanches: luxation congénitale Pieds: syndactylie des orteils, anomalies des orteils
Hypophyse: petite, syndrome d’interruption de la tige pituitaire Cerveau: agénésie du corps calleux, hydrocéphalie
Anémie de Fanconi et néoplasies
En ce qui concerne le risque de tumeurs solides, les plus fréquentes chez les patients AF sont des carcinomes épidermoïdes de la tête et du cou et des muqueuses vulvaire et vaginale. Ces tumeurs semblent être plus fréquentes chez les patients survivant aux complications hématologiques (aplasie médullaire, LAM) mais ceci peut être lié simplement au fait que les patients aient vécu assez longtemps pour que survienne ce type de tumeur. D’autres tumeurs solides spécifiques, le plus souvent médulloblastome et néphroblastome sont associées aux mutations des gènes d’aval FANCD1/BRCA2 et FANCN/PALB2, pour lesquels le risque de cancer et de LAM dans la petite enfance est très élevé. L’allogreffe de CSH surtout suivie de GVH chronique augmente le risque de survenue d’un cancer.
La connaissance de la mutation peut dans ces cas avoir un impact sur la prise en charge (12).
Diagnostic biologique
Examens d’orientation
Alpha-foeto protéine : son élévation a été identifiée comme un marqueur biologique sensible et spécifique.
Hémoglobine F : moins spécifique, son élévation oriente vers l’origine constitutionnelle de l’aplasie.
Examens de certitude
Étude des cassures chromosomiques
L’étude cytogénétique réalisée sur les lymphocytes du sang périphérique est la méthode diagnostique de référence. Elle permet d’observer une augmentation du nombre de cassures spontanées, mais surtout une augmentation significative du nombre de cassures chromosomiques et des figures anomalies de type tri ou quadri-radiales en présence de diepoxybutane (DEB) ou de mitomycine C (MMC), 2 agents cassants (ou pontants), créant des liaisons entre les 2 brins de l’ADN. Sont très évocateurs :
Un pourcentage de métaphases avec cassures (figures radiales incluses) > 20%
Un nombre moyen de cassures par métaphase >2 (x10 par rapport à la culture sans MMC)
Un nombre moyen de cassures par métaphase avec cassures > 4
Un pourcentage de métaphases avec figure radiale >10% (plusieurs figures /métaphase).
Ce test est considéré comme pathognomonique de l’AF. Il existe néanmoins des faux positifs qui peuvent se voir en cas de problème technique ou si le test est réalisé dans les semaines ou mois qui suivent une chimiothérapie. De faux négatifs sont également possibles en raison, ici aussi, de problèmes techniques lors de la réalisation du test ou en cas de mosaïcisme somatique. Dans ce cas, c’est l’étude de fibroblastes (test FANCD2, test de viabilité après exposition à la mitomycine C), obtenus par culture à partir d’une biopsie cutanée, qui permet de trouver ces anomalies et d’affirmer le diagnostic. Les tests sur les fibroblastes sont utiles dans les cas où le caryotype effectué sur le sang est normal alors que le phénotype clinique est très évocateur. Il permet également d’exclure formellement le diagnostic d’AF chez un membre de la fratrie avant un don de cellules souches hématopoïétiques, cependant ce test n’est pas de pratique courante.
Cytométrie de flux : l’étude du cycle cellulaire, faite sur sang périphérique, permet de montrer une augmentation significative du taux de cellules bloquées en phase G2/M après adjonction de MMC ou de DEB.
Le diagnostic moléculaire
Il se fait par séquençage pour l’identification du gène, et n’est pas de pratique courante. Il permet de préciser si une sœur ou un frère est atteint, aide au conseil génétique familial et au diagnostic anténatal. Afin d’exclure un mosaïcisme, cette recherche doit, à l’idéal, être faite sur 2 sources d’ADN différentes (sang et autre tissu).
Par ailleurs, il existe quelques corrélations génotype–phénotype : la mutation de FANCA induit souvent un début plus précoce de la maladie et les sujets sont plus à risque de leucémie aiguë myéloïde (LAM). Les mutations FANCA et FANCC sont associées à moins de malformations congénitales. Le développement d’une aplasie médullaire est plus fréquent pour les patients avec mutation nulle de FANCA, et aussi pour les mutations de FANCB, FANCC, FANCF, FANCG et FANCD2.
Autre test
Le caryotype médullaire
Il peut révéler la présence d’anomalies clonales. Les anomalies cytogénétiques décrites sont variées, touchant le plus souvent les chromosomes 1, 3, ou 7. Leur présence doit faire craindre une évolution leucémique et le caractère péjoratif de ces anomalies est bien établi. L’estimation de la survie à 5 ans des patients avec clone cytogénétique est de 40% versus 94% en l’absence de clone.
Diagnostic anténatal
La mise en évidence de l’augmentation du nombre de cassures chromosomiques induites par les alkylants au caryotype sur prélèvement de sang fœtal, de liquide amniotique ou de villosités choriales est possible mais difficile à réaliser. Actuellement, la meilleure approche est l’identification de la mutation d’un gène sur ADN extrait de biopsie de trophoblastes (possible dès la 14ème semaine d’aménorrhée). Ceci implique néanmoins que le gène en cause et la mutation responsables soient déjà connus pour la famille étudiée.
Diagnostic différentiel (tableau 3)
Tableau 3 : Autres aplasies médullaires constitutionnelles
La prise en charge des patients atteints d’AF doit être pluridisciplinaire. Une surveillance régulière doit être mise en place, avec en particulier la réalisation annuelle d’un myélogramme avec caryotype afin de dépister les évolutions clonales.
Transfusions de culots globulaires : elles doivent être limitées au maximum afin d’éviter toute allo-immunisation en pré-greffe. Le seuil transfusionnel est fixé à 7 ou 8 g/dL d’hémoglobine avec des signes cliniques. Une surveillance de la surcharge en fer s’impose, par le dosage de la ferritine, voire par la réalisation d’une imagerie par résonance magnétique cardiaque ou hépatique. Un traitement par chélateur du fer doit être envisagé rapidement en cas de surcharge importante (ferritinémie >1.000 mg/L) pour éviter l’évolution vers la fibrose voire la cirrhose hépatique, l’insuffisance cardiaque.
Les facteurs de croissance hématopoïétiques : ne sont pas indiqués en dehors du G-CSF qui peut être prescrit ponctuellement lors d’un état infectieux sévère. Son usage peut favoriser l’évolution clonale.
Les androgènes : permettent une élévation du taux d’hémoglobine chez 50% des patients et donc réduisent le recours aux transfusions. Un effet sur les plaquettes et les polynucléaires neutrophiles a également été mis en évidence mais dans une moindre mesure. Les plus utilisés sont : le Danazol (0,1 mg/kg/j) a permis une réponse hématologique dans 78% des cas dans un essai récent. La Norethandrolole (Nilevar®) à la dose initiale de 0,5 mg/kg/j, la réponse apparaît généralement dans les 1ers mois. Les effets secondaires sont nombreux et peuvent être graves : virilisation avec acné sévère, avance staturale avec risque de fusion précoce des cartilages de croissance, hyperactivité ou changements de comportement, toxicité hépatique (cytolyse, cholestase, risque d’adénome ou carcinome hépatiques), et surtout le Danazol dont les effets secondaires sont moindres. Cette option ne prévient pas l’évolution vers une LAM, elle est réservée aux patients chez lesquels une allogreffe de CSH ne peut être envisagée. La réponse au long terme est observée chez 10 à 20% seulement (13,14).
Allogreffe de cellules souches hématopoïétiques
C’est le seul traitement curatif qui empêche l’évolution vers une LA. Elle modifie uniquement l’atteinte médullaire mais ne corrige pas les autres anomalies ou malformations. Elle est indiquée devant :
Une pancytopénie persistante et modérément sévère : taux d’hémoglobine inférieur à 8 g/dL, taux de polynucléaires neutrophiles inferieur à 500/mm3 ou taux de plaquettes inferieur à 20.000/mm3.
Évolution clonale (apparition d’un syndrome myélodysplasique ou d’une LAM). L’âge médian de survenue de cette atteinte hématologique est de 7 ans.
L’intensité du conditionnement pré-greffe est considérablement réduite chez les patients atteints d’AF. Ceci est dû à leur hypersensibilité unique aux agents alkylants et à l’irradiation, à la suite du défaut des mécanismes de réparation d’ADN. Il y a actuellement un consensus sur l’abandon des agents alkylants et la radiothérapie dans le conditionnement de ces greffes. Les régimes d’intensité réduite contenant de la fludarabine font maintenant figure de référence. Cette molécule, beaucoup moins toxique, entraine une importante T-déplétion, diminuant le risque de GvHD et morbi-mortalité.
Les facteurs de mauvais pronostic identifiés sont : un conditionnement sans fludarabine, un âge supérieur à 10 ans au moment de la greffe, une antériorité de plus de 20 transfusions en pré-greffe, le recours à un greffon alternatif (donneur non apparenté ou sang placentaire) et un traitement préalable par androgènes.
Le suivi au long cours est indispensable en raison de l’existence d’anomalies d’organe, ainsi que la susceptibilité aux cancers en particulier ORL (incidence cumulative à 10 ans 28% en cas de survenue d’une GvHD). A âge égal, le risque de développer un carcinome épidermoïde est 4 fois plus élevé chez les patients greffés (15,16,17).
Traitement de la leucémie aiguë secondaire
Les patients atteints d’AF qui développent une LA ne sont pas faciles à prendre en charge car la sensibilité aux agents endommageant l’ADN limite l’utilisation classique de la chimiothérapie qui peut entrainer des aplasies sévères et prolongées, voire irréversibles. L’intérêt potentiel d’une chimiothérapie pré-greffe n’est à ce jour pas démontré. Certains utilisent des chimiothérapies à faible doses, de type mini-FLAG en attendant la greffe (18).
Traitement des néoplasies secondaires
En raison de la chimio et radiosensibilité, le traitement chirurgical est recommandé si toutefois le diagnostic est précoce, sans signes de dissémination. Les patients doivent donc être soumis à un programme de dépistage régulier.
Conclusion
Les patients atteints d’AF doivent bénéficier d’une prise en charge multidisciplinaire en raison de la variété des manifestations de la maladie, mais surtout du risque néoplasique. Le diagnostic doit être le plus précoce possible, ce qui permettra de dépister et de prendre en charge au mieux les malformations ou déficits d’organes existants et des lésions cancéreuses. Le seul traitement curatif actuel de l’atteinte hématologique est l’allogreffe de CSH, en attendant les résultats prometteurs de la thérapie génique.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
Soulier J. Fanconi anemia. Hematology Am Soc Hematol Educ Program Book 2011;492–7.
Djouadi K. Approche épidémiologique des aplasies médullaires en Algérie. Revue Algérienne d’Hématologie, N°00, Mars 2009; 24-27.
Giampietro PF, Verlander PC, Davis JG, Auerbach AD. Diagnosis of Fanconi anaemia in patients without congenital malformations: an international Fanconi Anaemia Registry Study. Am J Med Genet. 1997 Jan 10;68(1):58-61.
Butturini A, Gale RP, Verlander PC, Adler-Brecher B, Gillio AP, Auerbach AD. Hematologic abnormalities in Fanconi anaemia: An International Fanconi Anaemia Registry study. Blood. 1994 Sep 1;84(5):1650-5.
Kee Y, D’Andrea AD (2012) Molecular pathogenesis and clinical management of Fanconi anaemia. J Clin Invest. 122(11):3799-3806.
Moldovan GL, D’Andrea AD. How the Fanconi anaemia pathway guards the genome. Ann. Rev Genet 2009; 43:223–49.
Mehta PA, Tolar J. Fanconi anaemia. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. Gene Reviews. Seattle, WA: University of Washington, Seattle; 1993-2017. (updated 23 February 2017).
Kutler DI, Singh B, Satagopan J, et al. A 20-year perspective on the International Fanconi Anaemia Registry (IFAR). Blood 2003; 101:1249–56.
Shaw-Smith C. Oesophageal atresia, tracheo-oesophageal fistula, and the VACTERL association: review of genetics and epidemiology. J Med Genet. 2006 Jul;43(7):545-54.
Mialou V, Leblanc T, Peffault de Latour R, Dalle JH, Socie G. Aplasies médullaires constitutionnelles. EMC Hématologie, 13-008-C10, 2011
Lanneaux J, Poidvin A, Soole F, Leclerc G, Grimaud M, Dalle JH. L’anémie de Fanconi en 2012: diagnostic, suivi pédiatrique, traitement. Archives de Pédiatrie 2012; 19:1100-1109
Soulier J. Fanconi anemia. Hematology Am Soc Hematol Educ Program Book 2011;492–7.
Calado RT, Clé DV. Treatment of inherited bone marrow failure syndromes beyond transplantation. ASH Education Program, December 2017
Chaudhury S, Auerbach AD, Kernan NA, et al. Fludarabine based cytoreductive regimen and T-cell-depleted grafts from alternative donors for the treatment of high-risk patients with Fanconi anaemia. Br J Haematol 2008; 140:644–55.
Fanconi anaemia: Guidelines for diagnosis and management, 4th ed, Eugene OR : Fanconi Anaemia Research Fund, Inc; 2014
Peffault de Latour R, Peters C, Gibson B, Strahm B, Lankester A, de Heredia CD, Longoni D, Fioredda F, Locatelli F, Yaniv I, WachowiakJ, Donadieu J, Lawitschka ABierings M, Wlodarski M, Corbacioglu S, Bonanomi S, Samarasinghe S, Leblanc T, Dufour C and Dalle JH. Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Bone Marrow Transplantation (2015) 50, 1168–1172
Peffault de Latour R, Soulier J. How I treat MDS and AML in Fanconi anaemia. Blood 2016; 127: 2971-2979.
L’hémoglobinurie paroxystique nocturne (HPN), ou maladie de Marchiafava-Micheli est une hémolyse corpusculaire acquise, encore appelée hémoglobinurie paroxystique du fait des crises hémolytiques survenant en deuxième moitié de la nuit
A.F. Bendahmane(1)(2), S. YADI (1), N. MESLI (1)(2)
(1) Service d’hématologie – CHU Tidjani Damerdji de Tlemcen
(1)(2) Université de Tlemcen Abou Bakr Belkaïd – Département de Médecine
Date de soumission : 28 Février 2019.
Abstract: Paroxysmal nocturnal haemoglobinuria, or Marchiafava-Micheli disease, is a rare disease. It is a disease related to the clonal expansion of one or more hematopoietic stem cells that have acquired a somatic mutation gene of the class A Phosphatidylinositol Glycan. The PNH is a clinical diagnosis that must be confirmed by peripheral blood flow cytometry to detect the absence or severe deficiency of GPI on at least two lines. It is a very polymorphic clinical-biological expression disorder. The progress of flow cytometry and, more recently, of molecular biology have led to a real advance in the knowledge of the pathophysiology and diagnosis of this disease.
Keywords: Haemoglobinuria, corpus haemolysis, flow cytometry.
Résumé : L’hémoglobinurie paroxystique nocturne (HPN), ou maladie de Marchiafava-Micheli, est une maladie rare. C’est une maladie liée à l’expansion clonale d’une ou de plusieurs cellules souches hématopoïétiques ayant acquis une mutation somatique du gène Phosphatidylinositol Glycane de classe A. L’HPN est un diagnostic clinique qui doit être confirmé par cytométrie de flux sanguin périphérique pour détecter l’absence ou le déficit grave en GPI sur au moins deux lignées. C’est une affection d’expression clinico-biologique très polymorphe. Grace au progrès de la cytométrie en flux et, plus récemment de la biologie moléculaire ont conduit à une réelle avancée dans la connaissance de la physiopathologie et diagnostic de cette maladie.
Mots clés : Hémoglobinurie, hémolyse corpusculaire, cytométrie en flux.
Introduction :
L’hémoglobinurie paroxystique nocturne (HPN), ou maladie de Marchiafava-Micheli est une hémolyse corpusculaire acquise, encore appelée hémoglobinurie paroxystique du fait des crises hémolytiques survenant en deuxième moitié de la nuit (sensibilité des globules rouges à l’activation complémentaire liée à l’acidification physiologique sérique de la 2ème moitié de la nuit) et responsable d’une hémoglobinurie matinale du primo lever.
L’HPN est une anémie hémolytique particulière par son association fréquente avec une leucopénie et une thrombopénie et par la survenue de thromboses veineuses.
Depuis le début des années 1980, les progrès de la cytométrie en flux puis, plus récemment, de la biologie moléculaire ont conduit à une réelle avancée dans la connaissance de la physiopathologie de cette maladie rare.
L’HPN est aujourd’hui considérée comme une maladie liée à l’expansion clonale d’une ou de plusieurs cellules souches hématopoïétiques ayant acquis une mutation somatique du gène Phosphatidylinositol Glycane de classe A (PIG-A).
Le diagnostic précoce est essentiel pour améliorer la prise en charge et le pronostic des patients. L’HPN peut survenir de novo, mais aussi fréquemment dans le cadre d’une insuffisance médullaire comme une aplasie médullaire.
Historique
Parmi les premières descriptions de l’HPN réalisées vers la fin du 19ème siècle, le Dr Paul Strübing a décrit en 1882 un cas chez un jeune homme qui souffre d’une fatigue, de douleurs abdominales et de grave hémoglobinurie intermittente nocturne.
Marchiafava et Nazani en 1911, puis Micheli en 1931, ont établi le tableau clinique classique de la maladie, mais c’est Enneking, en 1925, qui aurait introduit le terme d’hémoglobinurie paroxystique nocturne.
En 1937, Thomas Ham signala que les érythrocytes de l’HPN étaient hémolysés lorsqu’ils étaient incubés avec un sérum acidifié. Cette découverte majeure a donné lieu au premier test de diagnostic de l’HPN, le test de sérum acidifié (Ham).
Ce n’est qu’en 1954, avec la découverte de la voie alternative de l’activation du complément, qu’il a été officiellement démontré que le complément provoquait l’hémolyse des globules rouges de l’HPN.
Dans les années 1980, il a été découvert que les cellules HPN présentaient un déficit global dans un groupe de protéines fixées à la surface de la cellule par une protéine d’ancrage.
Quelques années plus tard, la mutation génétique responsable du déficit en protéine d’ancrage a été découverte et, plus récemment, un anticorps monoclonal humanisé qui inhibe l’activation terminale du complément.
Épidémiologie
L’HPN est une maladie rare ou orpheline. Elle est observée surtout chez l’adulte. Elle est exceptionnelle chez l’enfant. Dans une série de la Société Française d’Hématologie, la moyenne d’âge était 33 ans mais près de 15% des patients étaient des enfants (moins de 16 ans). Elle touche les 2 sexes avec une fréquence variable selon la population.
Physiopathologie
L’HPN est liée à un défaut de l’expression des protéines membranaires inhibitrices du complément, les protéines CD55 ou DAF (decay accelerating factor, ou inhibiteur de la C3 convertase) et CD59 ou MIRL (membrane inhibitor of reactive lysis, ou inhibiteur du complexe C5-C9). Ces protéines sont fixées sur la membrane via une molécule de glycosylphosphate inositol (GPI). Ce déficit s’exprime sur une cellule souche hématopoïétique totipotente, expliquant l’atteinte de l’ensemble des lignées myéloïdes et lymphoïdes quand la maladie évolue. Donc il s’agit d’une pathologie clonale avec déficits membranaires des protéines d’ancrage.
Il existe une hémolyse intra vasculaire en raison de la sensibilité excessive des globules rouges au complément activé. La baisse nocturne physiologique du pH activant le complément expliquerait les hémoglobinuries nocturnes révélées au réveil par une hémoglobinurie matinale (1ère description de la maladie).
· Sensibilité anormale des cellules à l’action du complément
La sensibilité anormale des globules rouges à l’action lytique du complément a été considérée comme la caractéristique princeps de la maladie.
Elle est encore de nos jours à la base des tests diagnostiques de Ham et Dacie et du test au sucrose. Dans ces tests, les globules rouges atteints sont identifiés, indirectement, par leur hémolyse sélective en présence de complément activé en milieu acide ou en présence de sucrose. Cette sensibilité anormale des globules rouges à l’action du complément a très vite fait évoquer un déficit membranaire impliquant un système de régulation de la cascade d’activation du complément.
· Défaut de molécules régulatrices de l’action du complément
Les deux voies d’activation du complément (classique et alterne) (figure 1) se rejoignent en une voie effectrice commune conduisant à la formation d’un complexe multimoléculaire appelé complexe lytique ou complexe d’attaque membranaire. L’étape centrale d’activation est la formation de fragments activés de la fraction C3 (C3b) par les C3 convertases des deux voies d’activations. Deux protéines, dont le rôle est d’inhiber l’action du complément, ne sont pas exprimées à la surface des globules rouges de patients atteints d’HPN. Ces 2 molécules sont le DAF, ou CD55, qui agit au niveau des C3 convertases, et le MIRL, ou CD59, qui agit en inhibant la formation du complexe d’attaque membranaire.
Figure 1 : voies d’activation du complément : classique et alterne
· Défaut du système d’ancrage
En plus du défaut d’expression du CD55 et du CD59, d’autres déficits moléculaires ont été identifiés sur les cellules de patients atteints d’HPN (Tableau I). Toutes ces molécules ont un élément structurel commun : elles sont attachées à la membrane par une ancre (système d’ancrage) : le glycosyl-phosphatidylinositol (GPI). Ceci peut expliquer le fait que des molécules dont des fonctions sont apparemment aussi éloignées comme par exemple, le DAF et la molécule d’adhésion LFA3 (CD58) soient manquantes dans l’HPN.
Tableau I. – Principales protéines glycosyl-phosphatidylinositol chez l’homme
Protéines du complément DAF (CD55) MIRL (CD59) Protéine porteuse du C8 Enzymes Acétylcholinestérase (érythrocyte) Phosphatase alcaline (leucocyte) 5′-exonucléotidase (lymphocyte) CD73
Génétique
Le déficit en GPI est dû à une atteinte de la voie de biosynthèse du gène responsable situé sur le chromosome X. Il s’agit du gène Phosphatidylinositol Glycane de classe A (PIG-A).
· Mutations PIG- A
Le produit du gène PIG-A est l’une des sept protéines impliquées dans la première étape de la biosynthèse des ancres GPI. Théoriquement, une mutation de n’importe quel gène de la voie pourrait conduire à l’HPN. Cependant, jusqu’à maintenant, PIG-A était le seul gène muté trouvé chez les patients atteints d’HPN. Une seule mutation somatique dans une cellule souche hématopoïétique est suffisante pour produire un phénotype HPN.
· Autres mutations
L’absence de CD59 est le principal responsable des manifestations cliniques de l’HPN. En conséquence, de rares cas de mutations héréditaires dans CD59 conduisant à une perte de CD59 à la surface des cellules ont été bien documentés. Le phénotype de ces patients imite l’HPN en ce sens qu’ils se manifestent par une hémolyse intravasculaire chronique accompagnée de poussées d’hémolyse paroxystiques et d’une propension à la thrombose. Contrairement aux patients atteints d’HPN, les patients présentant un déficit héréditaire en CD59 présentent également une neuropathie périphérique.
Dans l’HPN classique, le déficit en CD59 ne se trouve que sur les cellules sanguines ; chez les patients présentant des mutations de la lignée germinale CD59 ; CD59 est déficient dans toutes les cellules du corps.
Clinique
Le type et la sévérité des symptômes peuvent varier considérablement en fonction des patients. Généralement, le début de la maladie est souvent silencieux (asymptomatique), ce qui rend le diagnostic très difficile. Certains patients peuvent donc attendre plusieurs années avant que le diagnostic d’HPN soit réellement posé.
La forme la plus classique de la maladie, est une anémie hémolytique acquise d’origine corpusculaire, apparaissant généralement chez l’adulte jeune, associée à des urines foncées le matin (hémoglobinurie) et parfois d’un ictère modéré. Les principaux symptômes (Figure 2) sont :
Figure 2 : Symptômes et fréquences de l’HPN
Anémie: accompagnée d’une réticulocytose et souvent d’une leucopénie et/ou d’une thrombopénie modérée. L’anémie est secondaire soit à l’hémoglobinurie paroxystique matinale avec hémosidérose rénale chronique (hémosidérinurie), responsable de carence martiale et de microcytose, soit à l’hémolyse chronique avec l’ictère.
Insuffisance médullaire : l’HPN peut se révéler par une pancytopénie révélant une aplasie médullaire ou un syndrome myélodysplasique à moelle riche mais dysplasique. On peut voir des aplasies médullaires en apparence idiopathiques après traitement immunosuppresseur, se compliquer d’HPN.
Thromboses veineuses: le plus souvent profondes et récidivantes qui touchent les veines sus-hépatiques (syndrome de Budd-Chiari), le système porte ou les sinus veineux crâniens. Ils sont secondaires à l’atteinte des plaquettes par le complément qui libèrent des substances pro-coagulantes, en plus le récepteur du plasminogène est ancré par le GPI (Figure 3).
Figure 3 : Mécanisme des thromboses dans l’HPN
Dystonie des muscles lisses: les crises douloureuses abdominales, les spasmes œsophagiens, la dysphagie et le dysfonctionnement érectile sont des symptômes courants associés à l’HPN classique et sont une conséquence directe de l’hémolyse intravasculaire et de la libération d’hémoglobine libre. Les taux élevés d’hémoglobine libre dans le plasma conduit à l’épuisement du NO. Le NO est synthétisé par les cellules endothéliales et dont le rôle est de maintenir la relaxation des muscles lisses et d’inhiber l’activation et l’agrégation plaquettaires.
Autres manifestations :
Asthénie : la fatigue est l’un des symptômes les plus fréquents de l’HPN. Elle est souvent intense et provoque des épisodes de somnolence dans la journée.
Fièvre due à des infections.
Atteinte rénale : Les lésions tubulaires rénales sont causées par une thrombose micro vasculaire et une accumulation de dépôts de fer.
Une hypertension pulmonaire (légère à modérée) : l’augmentation des pressions pulmonaires et la réduction de la fonction ventriculaire droite provoquée par les microthrombi subcliniques et le balayage de NO contribuent aux symptômes de fatigue et de dyspnée.
Biologie :
Hémogramme :
L’anémie est normocytaire normochrome ; plus rarement macro ou microcytaire hypochrome. La réticulocytose est élevée.
Une leucopénie avec neutropénie est présente dans la moitié des cas.
Une thrombopénie modérée est habituelle. Elle peut être importante dans 50% des cas.
Une pancytopénie peut ainsi être constaté dans 1/3 des cas.
Sans anomalie morphologique des globules rouges sur le frottis sanguin
Bilan d’hémolyse :
Les taux de bilirubine totale et libre sont augmentés
Le taux d’haptoglobine plasmatique est abaissé
Les lactico-deshydrogénases (LDH) sont augmentés
Autres données biologiques :
Une hémoglobinurie ou une hémosidérinurie positive
Le test de Coombs direct est négatif.
Tests spécifique classiques
Le test de Ham et Dacie : il consiste à mesurer l’hémolyse des globules rouges in vitro en présence de complément. Il permet d’observer l’hémolyse en sérum frais normal (pH entre 6,4 et 7). Il est positif avec le sérum acidifié du malade mais négatif si le sérum est inactivé par la chaleur ou si le sérum du malade est utilisé sur des hématies normales.
Test au sucrose : qui favorise la fixation du complément, est un test très sensible positif avec un sérum normal ou le sérum du malade.
Cependant ces tests, utilisés depuis de nombreuses années afin de poser le diagnostic, tend aujourd’hui à être remplacé par l’étude des molécules GPI grâce à l’étude par cytométrie en flux qui reste une méthode plus sensible et spécifique que les tests classiques.
Étude immunocytologique par cytométrie en flux
Les tests actuels : étude de l’expression en cytométrie de flux des protéines CD55 et CD59 à la surface des hématies et des leucocytes : mise en évidence de l’anomalie membranaire du déficit en protéines CD55 et CD59.
Cette technique permet l’analyse rapide des différentes populations leucocytaires et lymphocytaires à partir de sang total. Les résultats obtenus peuvent être exprimés en pourcentage de cellules négatives et/ou en moyenne d’intensité de fluorescence.
Cette dernière manière est utile d’une part, pour l’étude de certaines molécules comme le CD16 ou le CD58 dont l’expression physiologique est faible ; et d’autre part, dans la mise en évidence de populations dites “intermédiaires” ou PNH II (populations cellulaires exprimant à leur surface un niveau faible, mais non nul d’une molécule GPI donnée) (Figure 4).
La perte de GPI est détectée après coloration de cellules avec des anticorps monoclonaux et un réactif appelé aérolysine fluorescente (FLAER). FLAER est un variant de la proaérolysine marqué à la fluorescéine qui se lie à la partie glycane de l’ancre GPI.
La sensibilité de la cytométrie en flux dans le diagnostic d’un déficit des molécules GPI permet de détecter des clones dont l’importance n’excède pas quelques pourcents. En pratique pour la majorité des molécules étudiées, un déficit peut être considéré comme significatif lorsque le pourcentage de cellules négatives est supérieur à 5%.
Figure 4 : Différents marqueurs évaluer par la CMF au cours d’HPN
Classifications
Le test de Ham et Dacie permet de classer (Figure 5) certains patients en trois populations cellulaires :
Type I : hématies de sensibilité normale au complément.
Type II : sensibilité intermédiaire.
Type III : hématies très sensibles.
La CMF permet de distinguer trois populations cellulaires :
L’HPN classique, qui inclut les patients hémolytiques et thrombotiques.
L’HPN dans le contexte d’autres troubles primaires de la moelle osseuse, tels que l’aplasie médullaire ou le syndrome myélodysplasique.
L’HPN sous-clinique, dans laquelle les patients ont de petits clones d’HPN, mais aucune preuve clinique ou biologique d’hémolyse ou de thrombose.
Figure 5 : Classification selon le groupe international d’HPN
Évolution et complications
L’évolution de la maladie est variable et dépend du degré d’atteinte des cellules sanguines. La médiane de survie des patients est de 10 à 15 ans. Le problème majeur qui vient grever l’évolution des patients atteints d’HPN est représenté par les thromboses (qui peuvent être inaugurales). L’incidence des thromboses dans les séries françaises est de 25% à 5 ans.
Deux autres complications sont aussi fréquemment rencontrées (chacune chez 20% des malades) : des crises douloureuses abdominales et des infections récurrentes de la sphère ORL et pulmonaire, en particulier.
Chez les patients atteints d’HPN de novo la probabilité de développer une pancytopénie durant l’évolution est estimée à 15% à 8 ans.
Le risque de développement d’un syndrome myélodysplasique ou une leucémie aiguë est classique mais rare (5 et 1% à 8 ans, respectivement).
Chez les patients positifs à l’HPN ayant de petits clones HPN, la surveillance est essentielle car la taille du clone peut rapidement augmenter en quelques mois. Les patients à haut risque doivent être dépistés et surveillés en continu par cytométrie en flux de haute sensibilité effectuée sur sang périphérique.
Une étude multifactorielle des facteurs de risque de décès a pu être réalisée pour la première fois dans cette maladie rare. Les facteurs affectant de manière indépendante la survie de ces patients sont :
Le développement d’une thrombose ;
La progression d’une HPN de novo vers un tableau de pancytopénie ;
Le développement d’un syndrome myélodysplasique ou une leucémie aiguë ;
L’âge (plus de 54 ans) ;
La thrombopénie au diagnostic.
Traitement
Traitement symptomatique
Les transfusions par des concentrés globulaires sont indispensables en cas d’anémie sévère ou mal tolérée : l’indication de l’utilisation de culots globulaires déplasmatisées et de globules rouges lavés (décomplémentés) est classique mais d’efficacité et de protection contre l’hémolyse induite discutée. Il faut utiliser des culots globulaires « frais » (car le complément s’active progressivement dans les poches de sang)
Traitement martial : hormis les transfusions, certains patients peuvent bénéficier de supplémentation en fer, car perte urinaire journalière de 20mg de fer sous forme d’hémosidérine. Mais la prise de fer peut provoquer des douleurs abdominales. La compensation martiale doit être prudente car le fer peut accentuer l’hémolyse.
Acide folique : tous les patients présentant une anémie hémolytique chronique doivent être supplémentés du fait de l’hyperproduction médullaire compensatrice.
Facteurs de croissance : le G-CSF peut être prescrit dans les formes aplasiques, on observe des résultats mitigés. Certaines équipes médicales ont essayé l’érythropoïétine (EPO) qui peut aider à limiter les transfusions.
Anticoagulants, thrombolytiques et antiagrégants plaquettaires : La prophylaxie contre les événements thromboemboliques chez les patients atteints d’HPN est un sujet de débat. Le risque thrombotique peut être corrélé à la taille du clone PNH, ce qui a conduit de proposer une anticoagulation prophylactique aux patients présentant un déficit en GPI-AP supérieur à 50% à 60%.
Antalgiques : en cas de douleurs abdominales, de céphalées provoquées par les crises hémolytiques, des antalgiques plus ou moins puissants sont prescrits.
Antibiothérapie : Les infections sont souvent récurrentes (sphère ORL et pulmonaire), et traitées par antibiotiques.
Traitement de fond
Les corticoïdes sont parfois efficaces. Ils auraient un intérêt dans les formes hémolytiques mais n’auraient pas d’effet sur l’hématopoïèse. Un traitement de 6 semaines inefficace doit être interrompu.
Les immunosuppresseurs sont potentiellement utiles : La cyclosporine et le sérum anti-lymphocytaire ont été employés avec succès dans certaines formes très pancytopéniques.
Les androgènes (Danazol, Danatrol®) semblent avoir une certaine efficacité, bien que leur mécanisme d’action ne soit pas très bien connu (augmentation de l’inhibiteur de la C1 estérase).
L’eculizumab est un anticorps monoclonal humanisé inhibant la partie C5 du complément ce qui permet d’obtenir une inhibition de la voie d’activation impliquée, et d’autre part, l’inhibition de son clivage. Ce traitement s’est révélé améliorer l’hémolyse et la thrombose et améliorer la qualité de vie des patients atteints d’HPN. Le traitement par eculizumab est administré par voie intraveineuse selon le schéma posologique suivant :
Phase initiale avec des injections hebdomadaires de 600 mg pendant quatre semaines puis de 900 mg à la cinquième semaine.
Phase d’entretien avec des injections de 900 mg toutes les deux semaines.
Nb : une vaccination contre le méningocoque, au moins deux semaines avant la première dose d’eculizumab doit être faite.
Greffe de moelle osseuse allogénique reste le seul traitement curateur de l’HPN.
Attitude thérapeutique
Formes graves
Les formes pancytopéniques et aplasiques, avec complications thrombotiques sévères ou avec hémolyse majeure doivent bénéficier – s’il existe un donneur HLA identique – d’une greffe de moelle allogénique.
Dans les formes aplasiques sans donneur HLA identique, un traitement immunosuppresseur ± androgènes est réalisé.
Formes modérées
Eculizumab
Androgènes
Traitement symptomatique
Étude nationale sur l’HPN du 2014
Sur une période de plus de dix ans (2003-2014), 67 patients adultes atteint d’HPN ont été colligés et examinés rétrospectivement. Le diagnostic était établi devant la présentation clinique et les résultats de la cytométrie en flux.
Le recueil des données a été réalisé à l’aide d’une fiche d’enquête diffusée dans tous les services d’hématologie de l’Algérie. 15 services d’hématologies ont participé.
Les patients atteints HPN sont principalement des jeunes adultes de sexe masculin. Au moment du diagnostic, l’âge médian est de 38 ans. La fréquence de l’HPN reste sous-estimée.
Le tableau clinique est dominé par l’asthénie et le syndrome anémique. La CMF est pratiquée chez la majorité des patients suivis. Le traitement est basé essentiellement sur le traitement symptomatique.
Les centres de recrutement
Répartition des nouveaux cas par année
Répartition selon les tranches d’âges : Les extrêmes d’âge sont de 16 à 74 ans avec une médiane d’âge à 37,7
Répartition selon le sexe : sur la population étudiée il y a une prédominance masculine avec un sexe ratio 1,72.
Circonstances de découverte : elles sont dominées par le syndrome anémique.
Tableau clinique à l’admission : marqué par l’asthénie dans la majorité des cas, suivi par l’anémie. Les thromboses sont présentes chez moins d’un tiers des patients.
Siège de la thrombose : le siège cérébral et du système porte dans l’ensemble des thromboses veineuses.
Tests au diagnostic : dans l’ensemble le diagnostic est posé par CMF
Thérapeutiques utilisés
Présentation d’une observation
Monsieur R.B. âgé de 55 ans, originaire et demeurant à Tlemcen (Algérie), aux antécédents de diabète sous insulinothérapie, consulté en 2013 en urologie pour la prise en charge d’une pyélonéphrite aiguë sur macro lithiase, découverte d’une anémie avec une notion d’urine foncée puis le patient fut orienté en consultation d’hématologie.
A l’admission, sur le plan général le patient était conscient coopérant, asthénique, normo tendu. Il présentait une pâleur cutanéomuqueuse avec un subictère conjonctival, une hémoglobinurie, sans syndrome infectieux ni tumoral.
L’hémogramme a retrouvé une anémie à 7,4 g/dl normochrome normocytaire régénérative à 170.000/mm3 de réticulocytes, un taux de leucocyte normal à 4.140/mm3 avec une répartition correcte, un taux de plaquette normal à 185.000/mm3 et absence d’anomalie notable sur le frottis de sang.
Le bilan d’hémolyse a montré une bilirubine totale augmentée à 20mg/l avec une bilirubine libre à 18mg/l, les lactico-deshydrogénases (LDH) très élevés à 3.320 UI/l et une Haptoglobine effondrées. Le test de Coombs directe (TCD) était négatif.
Devant ce tableau une cytométrie en flux (CMF) a été demandé à la recherche d’un clone HPN objectivant :
Déficit FLEAR/CD 24 sur les granulocytes à 84%
Déficit FLEAR/CD 14 sur les monocytes à 85%
Déficit en CD 59 sur les GR : type II 6,7%, type III 23.6%, type II + lll 30,3%
Devant ces résultats le diagnostic d’HPN a été posé et traité par des traitements symptomatiques.
En 2016 décision de mettre le patient sous anticorps monoclonal Eculizumab et ceci après vaccination systématique par l’antiméningococcique, associé à un antiagrégant plaquettaire et de l’acide folique. Une nette amélioration clinique et biologique a été noté au bout d’un mois de traitement :
Disparition de l’asthénie et de l’hémoglobinurie.
Le taux d’hémoglobine à 11g/dl
La diminution des LDH à 800 UI/l.
Au bout de 6 mois et faute de disponibilité du produit (Eculizumab), un phénomène de rebond important a été déclenché après l’arrêt du traitement avec réapparition de l’asthénie, l’hémoglobinurie, une augmentation des LDH, et une importante atteinte de la fonction rénale.
Une CMF de contrôle a été réalisée en 2017 objectivant :
Déficit FLEAR/CD 24 sur les granulocytes a 77,8%
Déficit FLEAR /CD 14 sur les monocytes 84,1%
Déficit en CD 59 sur les GR : type ll 3,7%, type III 70,2%
Le patient a été remis sous Eculizumab, cela depuis 4 mois, avec une bonne réponse clinique et biologie (augmentation de l’hémoglobine de 9 à 12,8 g/dl et une diminution de LDH de 1834 vs 830 UI/l).
Conclusion
Une meilleure connaissance des bases moléculaires et cellulaires de l’HPN au cours des deux dernières décennies a permis de mieux comprendre la biologie et l’histoire naturelle de l’HPN.
Les progrès de la cytométrie en flux ont conduit à des avancées dans le diagnostic de cette pathologie rare. La rareté de cette maladie a conduit à l’emploi de thérapeutiques variées dont l’intérêt potentiel est difficilement évaluable.
L’utilisation de l’anticorps monoclonal, l’éculizumab, démontrent que l’inhibition du complément terminal contrôle les symptômes et des complications potentiellement mortelles de l’HPN.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
Peffaut de Latour R et al. L’hémoglobinurie paroxystique nocturne. Rev Med Int 2010 ;31:200-207.
Richards SJ et al. Application of FCM to the diagnosis of PNH. Cytometry 2000;42:223-233.
Régis Peffault De La Tour – Dr Flore Sicre De Fontbrune – Pr Gérard Socie – Dernière mise à jour : Octobre 2017
Arruda MMAS, Rodrigues CA, Yamamoto M, et al. Hemoglobinúria paroxística noturna: da fisiopatologia ao tratamento. Rev Assoc Med Bras. 2010; 56(2): 214-21.
Brodsky RA. Paroxysmal nocturnal hemoglobinuria: stem cells and clonality. Hematology Am Soc Hematol Educ Program. 2008; 111-5.
Nomura ML, Surita FGC, Parpinelli MA, et al. Hemoglobinúria paroxística noturna e gravidez. Rev Bras Ginecol Obstet. 2004; 26(7): 579-82.
Brito Junior LC, Cardoso MS, Rocha EG, et al. Frequency of paroxysmal nocturnal hemoglobinuria in patients attended in Belém, Pará, Brazil. Rev Bras Hematol Hemoter. 2011; 33(1): 35-7.
Conférence HPN par Pr HAMLADJI R.M novembre 2007.
HPN PAR Dr Gérard Socié service d’hématologie-greffe de moelle hôpital Saint-Louis-Paris.
Encyclopédie Orphanet Grand Public; Maladies Rares; mai 2007


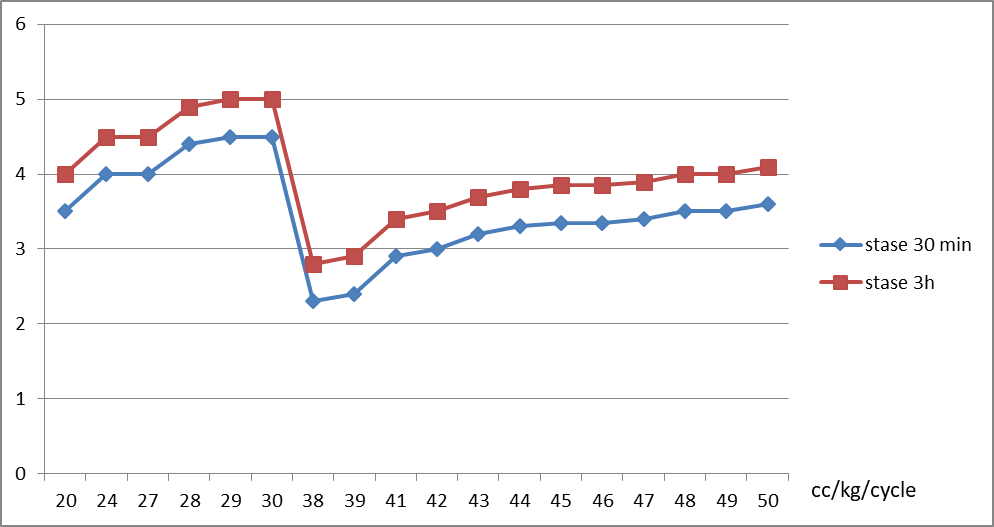
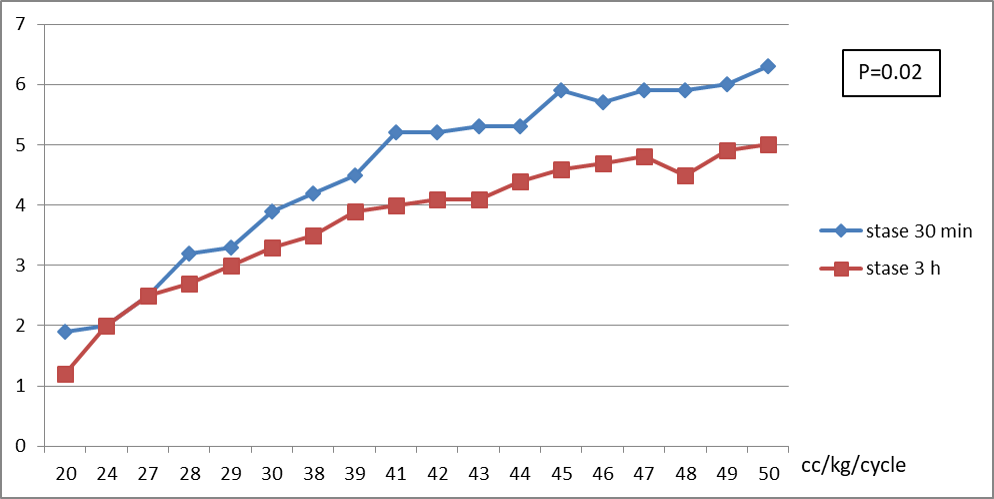
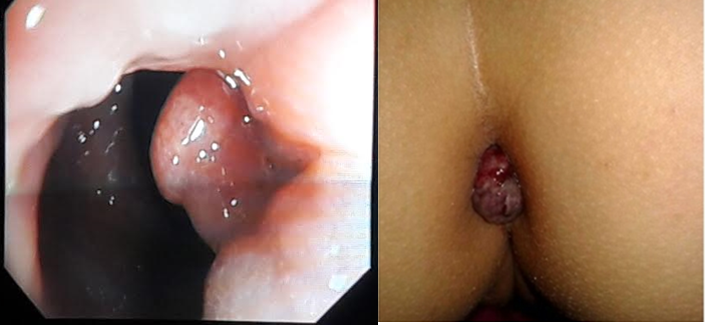
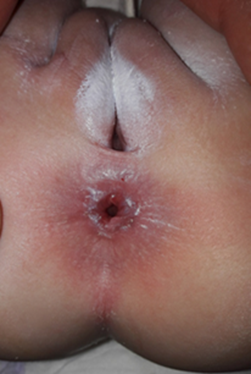 Figure 2: Fissure anale chez un nourrison de 6 mois secondaire à la constipation.
Figure 2: Fissure anale chez un nourrison de 6 mois secondaire à la constipation.