La maladie de Pompe apparait à tout âge, depuis la petite enfance jusqu’à l’âge adulte. Elle est de grande hétérogénéité phénotypique et de sévérité variable, corrélées en partie au degré de réduction de l’activité enzymatique.
Y.Sifi, A. Boulefkhad, Service de Neurologie, CHU Abdessalam Benbadis de Constantine, Faculté de médecine, Laboratoire de Biologie et de Génétique Moléculaire, Université Constantine 3.
Date de soumission : 09 Février 2020.
Abstract: Pompe disease occurs at all ages, from infancy to adulthood. It is phenotypically heterogeneous with a varying severity, partially correlated with the degree of reduction in enzyme activity. Patients with late onset Pompe disease begin the disease at a later age ranging from one year to 60 years or more. those are the late onset forms of Pompe disease (or LOPD), most often mimicking other neuromuscular conditions. They are usually expressed by limb and axial progressive muscular weakness frequently leading to the wheelchair; and death is often precipitated by respiratory complications. Since the advent of recombinant enzyme in the treatment of Pompe disease and in order to allow early diagnosis for early treatment, literature data have been enriched by clinical studies enlarging the spectrum of clinical manifestations of the late onset Pompe disease. The objective of our work was to report the different systems affected in the late-onset Pompe disease and their clinical expressions.
Key words: Pompe disease, type II glycogenosis, late form, acid maltase deficiency, GAA, recombinant enzyme.
Résumé : La maladie de Pompe apparait à tout âge, depuis la petite enfance jusqu’à l’âge adulte. Elle est de grande hétérogénéité phénotypique et de sévérité variable, corrélées en partie au degré de réduction de l’activité enzymatique. Les patients atteints de la forme à révélation tardive ou LOPD pour « Late onset Pompe Disease » débutent la maladie entre 1-60 ans voire plus. Elle s’exprime habituellement par une faiblesse musculaire des ceintures et axiale, d’évolution progressive conduisant fréquemment vers le fauteuil roulant. Le décès est souvent précipité par les complications respiratoires. Depuis l’avènement de l’enzymothérapie substitutive dans le traitement de la maladie de Pompe, et afin de permettre un diagnostic précoce pour un traitement précoce, les données de la littérature se sont enrichies dans la recherche clinique, élargissant ainsi le spectre des manifestations cliniques de la forme tardive de la maladie de Pompe. L’objectif de notre travail était de rapporter les différents systèmes atteints dans la forme à révélation tardive de la maladie de Pompe et leurs expressions cliniques.
Mots clés : Maladie de Pompe, glycogénose de type II, forme tardive, déficit en maltase acide, GAA, enzyme recombinante.
Introduction
La maladie de Pompe (MP) ou glycogénose de type II est une myopathie métabolique rare, de transmission autosomique récessive, causée par un déficit en α-glucosidase acide (GAA) ou maltase acide, nécessaire à la dégradation du glycogène lysosomale (1), dont le gène est localisé sur le bras long du chromosome 17 en 17q 25.3 (2).
La MP apparait à tout âge, depuis la petite enfance jusqu’à l’âge adulte. La sévérité de son phénotype est variable et est corrélée en partie au degré de réduction de l’activité enzymatique. Elle se présente principalement sous deux formes cliniques, la forme infantile (que nous n’aborderons pas dans cet article), encore appelée EOPD (pour Early onset Pompe Disease), définie par deux évènements chronologiques, un début avant l’âge d’un an, voire dès les premiers jours de la vie (nouveau-né ou nourrisson), et un décès avant l’âge de 2 ans précipité par les troubles respiratoires. Les patients atteints de cette forme précoce se présentent avec un phénotype classique, dominé par une faiblesse musculaire généralisée associée à une hypotonie et à une cardiomyopathie hypertrophique (3).
La forme à révélation tardive ou LOPD (pour Late onset Pompe Disease), débute entre 1 et 60 ans. À la différence de la forme infantile, les manifestations cliniques de la forme à révélation tardive sont polymorphes. Elles s’expriment par une faiblesse musculaire des ceintures, et axiale, d’évolution progressive conduisant fréquemment vers le fauteuil roulant. Le décès est souvent précipité par les complications respiratoires. À ce phénotype habituel, une très grande variété de symptômes additionnels et/ou révélateurs a été décrite. Actuellement la forme à révélation tardive de la MP est considérée comme une affection multisystémique, faisant intervenir l’atteinte de plusieurs tissus (4). Depuis l’avènement de l’enzymothérapie substitutive dans le traitement de la maladie de Pompe et afin de permettre un diagnostic précoce pour un traitement précoce, les données de la littérature se sont enrichies en de nombreuses études dans le domaine de la recherche clinique élargissant ainsi le spectre phénotypique de cette maladie (5). L’autopsie a été un moyen précieux pour caractériser les effets de l’accumulation de glycogène dans les différents tissus des différents organes (6). L’objectif de notre travail était de rapporter les différentes présentations cliniques de la forme tardive de la MP.
Manifestations musculaires
Le déficit de la musculature squelettique est d’évolution progressive et constitue le maitre symptôme de la forme à révélation tardive de la maladie de Pompe (7). Il apparait à tout âge, de la petite enfance à l’âge adulte, souvent précédé par une fatigue, des myalgies, une intolérance à l’effort ou une hyperCKémie[1] longtemps isolée. Il n’a souvent rien de spécifique, proximal des ceintures pelvienne et scapulaire mimant d’autres affections neuro musculaires (une LGMD[2], une amyotrophie spinale de type 3 ou 4, ou une myopathie inflammatoire et traitée inutilement). Cependant il est parfois distal, pouvant évoquer une myopathie distale ou une neuropathie héréditaire.
Devant toute suspicion de MP, l’examen neurologique fin recherchera particulièrement : une amyotrophie et une faiblesse précoce de la musculature axiale atteignant les muscles fléchisseurs de la nuque, spinaux, et/ou abdominaux. L’atteinte diaphragmatique et des muscles intercostaux serait responsable dans tous les cas d’une insuffisance respiratoire. L’atteinte des muscles bulbaires s’exprime par une faiblesse des muscles de la langue avec dysphagie et dysarthrie (8). Les muscles de la jambe et des pieds sont épargnés voire rarement atteints. L’évolution se fait vers l’extension du déficit moteur aux muscles proximaux de la ceinture scapulaire. Le taux des CK[3] au cours de la maladie de Pompe est généralement assez élevé, atteignant souvent plus de 2.000 U/L, mais dans les formes à révélation tardive le taux le plus élevé atteint rarement 1.000 à 1.500 U/L. Il peut par contre être normal (4).
Atteinte cardiaque
Contrairement à la forme à début précoce, l’atteinte cardiaque dans la forme à début tardif est rare (<5%) et modérée. Elle est secondaire à une accumulation massive de glycogène dans le tissu cardiaque, il s’agit de cardiomyopathie hypertrophique (hypertrophie ventriculaire gauche concentrique modérée, non obstructive) de troubles conductifs le plus souvent modérés, d’arythmies sinusales, de tachycardie supraventriculaire, de syndrome de Wolff Parkinson-White ou de bloc auriculo-ventriculaire nécessitant l’implantation d’un stimulateur cardiaque (9, 10).
Atteinte respiratoire
L’atteinte respiratoire est fréquente, elle se voit pratiquement chez 80 % des patients (11). Elle met souvent en jeu le pronostic vital, et est secondaire en grande partie à une atteinte du diaphragme mais aussi des muscles thoraco-abdominaux. Elle peut par ailleurs révéler la maladie même en l’absence de toute atteinte musculaire cliniquement patente sous forme d’insuffisance respiratoire restrictive, parfois aigüe. Le mécanisme physiopathologique de ces troubles respiratoires est double, lié à l’accumulation de glycogène dans la région ventrale de la moelle épinière où se trouvent les motoneurones phréniques innervant le diaphragme, et au niveau des fibres musculaires lisses (trachée et bronches).
Les premiers signes de détresse respiratoire se manifestent cliniquement par une altération de la qualité du sommeil, fatigue, somnolence diurne et céphalées. L’atteinte du diaphragme est mise en évidence par l’exploration respiratoire fonctionnelle (EFR) mettant en évidence une diminution de la capacité vitale forcée (CVF), cette dernière doit être réalisée en position assise et couchée.
Manifestations musculo-squelettiques
Elles s’observent chez les patients ayant perdu la marche et placés sur fauteuil roulant. On note des scolioses, des cyphoses, des hyperlordoses et un syndrome de la colonne raide (4).
Par ailleurs, l’ostéoporose est fréquente, elle est habituellement accompagnée de fractures spontanées répétées (4).
Manifestations neurologiques
Les manifestations neurologiques sont de type central et périphérique. Elles sont secondaires à l’accumulation de glycogène lysosomal dans les régions du système nerveux central (SNC) et du système nerveux périphérique (SNP).
Atteinte du système nerveux périphérique
De nombreuses études anatomopathologiques ont démontré l’accumulation de glycogène au niveau de la cellule de Schwann du nerf périphérique, particulièrement au niveau des petites fibres, myélinisées et amyéliniques, au niveau de la jonction neuro-musculaire (12), et aussi au niveau de la corne antérieur de la moelle (13), cliniquement il s’agit respectivement de neuropathies à petites fibres (NPF), d’expression clinique sensitive, à type de douleurs et de paresthésies douloureuses distales des membres inférieurs, associées souvent à des troubles dysautonomiques (14). En cas d’atteinte de la corne antérieure, le déficit moteur est de topographie distale des 4 membres, avec des pieds tombants et une aréflexie tendineuse (16).
Atteinte du SNC et cérébro-vasculaire
L’accumulation de glycogène au niveau des muscles lisses des artères et artérioles cérébrales altère leurs propriétés anti-thrombotiques, suggérant ainsi un mécanisme pathogène hypoxique-ischémique, responsable d’accidents vasculaires cérébraux ischémiques ou hémorragiques (4). Les malformations vasculaires à type d’anévrysme ont été rapportées (4). L’atteinte vasculaire extra cérébrale est possible. Les manifestations vasculaires et parenchymateuses du SNC ne sont pas à rechercher systématiquement, sauf si le patient présente des symptômes ou signes évocateurs d’une complication vasculaire cérébrale.
Perte auditive
La perte auditive est fréquente, et est associée à des troubles cochléaires, elle est secondaire à une accumulation de glycogène au niveau du muscle stapédien ou muscle de l’étrier (16).
Manifestations ophtalmologiques
Les plus fréquemment observées sont le strabisme, le ptosis et l’ophtalmoplégie.
L’accumulation de glycogène dans le cristallin et la rétine serait responsable de cataracte et d’anomalies rétiniennes (16).
Manifestations dentaires et gastro-intestinales
Les anomalies dentaires sont rares, il s’agit surtout d’anomalies du développement dentaire et de prolifération gingivale (16).
L’accumulation de glycogène dans les muscles lisses des intestins s’exprime par une gêne abdominale, diarrhée chronique, crampes, constipation, ballonnements postprandiaux, satiété précoce, difficultés d’alimentation et de déglutition, faible gain de poids, diminution du réflexe nauséeux et hépatomégalie (16).
Manifestations génito-urinaires
Souvent négligées, les manifestations génito-urinaires altèrent la qualité de vie des patients. La miction impérieuse est le symptôme le plus fréquent. L’incontinence urinaire et fécale ont été rapportées (17).
Atteinte de sang périphérique
Dans le sang, les anomalies lymphocytaires sont au premier plan, il s’agit de lymphocytes vacuolés. Cet aspect est présent dans les autres maladies de surcharge lysosomale (18).
Atteinte endocrinienne
L’atteinte endocrinienne la plus fréquente est l’hypothyroïdie, suggérant un lien entre le déficit enzymatique et la fonction thyroïdienne (19).
Conclusion
Le spectre phénotypique de la forme à révélation tardive de la maladie de Pompe s’est élargi. Il ne s’agit plus de la myopathie progressive des ceintures, associée à des troubles respiratoires précoces, mais d’une affection multisystémique, faisant intervenir l’atteinte de plusieurs tissus de différents organes. Le dosage de l’activité de la maltase acide permet le diagnostic définitif. L’intérêt du diagnostic précoce de cette maladie réside dans la mise en route d’un traitement substitutif précoce par enzyme recombinante, disponible depuis 2007 en Algérie.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
H. Hoefsloot, M. Hoogeveen-Westerveld, A.J. Reuser, B.A. Oostra, Characterization of the human lysosomal alpha-glucosidase gene, Biochem. J. 272(1990) 493–497.
S. Kishnani, R.D. Steiner, D. Bali, K. Berger, B.J. Byrne, L.E. Case, et al., Pompe disease diagnosis and management guideline, Genet. Med. 8 (2006) 267–288.
Antonio Toscano, Carmelo Rodolico, Olimpia Musumeci, Multisystem late onset Pompe disease (LOPD): an update on clinical aspects, Ann Transl Med 2019;7(13):284.
Justin Chan, Zoheb B. Kazi, Ankit K. Desai, Kaitlyn Corey, Stephanie Austin, Lisa D. Hobson Webb, et al. The emerging phenotype of late-onset pompe disease: A systematic literature review de la forme tardive de la MP. Molecular Genetics and Metabolism, Volume 120, Issue 3, March 2017, Pages 163-172.
I. Soliman, N.A. van der Beek, P.A. van Doorn, W.B. Vletter, A. Nemes, B.M. Van Dalen, and al. Cardiac involvement in adults with Pompe disease, J. Intern. Med. 264 (2008) 333–339.
Sifi, M. Medjroubi, R. Froissart, N. Taghane, K. Sifi, A. Benhabiles, et al. Clinical Analysis of Algerian Patients with Pompe Disease, Hindawi Publishing Corporation Journal of Neurodegenerative Diseases Volume 2017, Article ID 9427269, https://doi.org/10.1155/2017/9427269.
Jones, H.N., et al., Quantitative assessment of lingual strength in late-onset Pompe disease. Muscle Nerve, 2015. 51(5): p. 731-5.
Hossain MA, Miyajima T, Akiyama K, Eto Y. A Case of Adult-onset Pompe Disease with Cerebral Stroke and Left Ventricular Hypertrophy. J Stroke Cerebrovasc Dis. 2018 Nov;27(11):3046-3052.
Muller-Felber W, Horvath R, Gempel K, Podskarbi T, Shin Y, Pongratz D, et al. Late onset Pompe disease: clinical and neurophysiological spectrum of 38 patients including long-term follow-up in 18 patients. Neuromuscul Disord. 2007;17:698–706.
Hagemans, M.L., et al., Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain, 2005. 128(Pt 3): p. 671-7.
Falk DJ, Todd AG, Lee S, Soustek MS, El Mallah MK, Fuller DD, et al. Peripheral nerve and neuromuscular junction pathology in Pompe disease. Hum Mol Genet. 2015;24:625-36.
Lee NC, Hwu WL, Muramatsu SI, Falk DJ, Byrne BJ, Cheng CH, et al. A Neuron-specific gene therapy relieves motor deficits in Pompe disease mice. Mol Neurobiol 2018;55:5299-309.
Li-Kai Tsai, Wuh-Liang Hwu, Ni-Chung Lee, Pei-Hsin Huang, Yin-Hsiu Chien, Clinical Features of Pompe Disease with Motor Neuronopathy, Neuromuscular Disorders (2019), doi:https://doi.org/10.1016/j.nmd.2019.09.011.
Chan J, Desai AK, Kazi ZB, Corey K, Austin S, Hobson-Webb LD, and al. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol Genet Metab. 2017 Mar;120(3):163-172.
Pardo, J., T. Garcia-Sobrino, and A. Lopez-Ferreiro, Gastrointestinal symptoms in late-onset Pompe disease: Early response to enzyme replacement therapy. J Neurol Sci, 2015. 353(1-2): p.181-2.
Anderson G, Smith VV, Malone M, et al. Blood film examination for vacuolated lymphocytes in the diagnosis of metabolic disorders; retrospective experience of more than 2500 cases from a single centre. J Clin Pathol 2005;58:1305-10
Schneider J, Burmeister LA, Rudser K, et al. Hypothyroidism in late-onset Pompe disease. Mol Genet Metab Rep 2016;8:24-7.
[1] HyperCKémie = Augmentation du taux sérique de créatine kinase (NDLR).
Introduction. Le syndrome de RETT (MIM #312 750) est une maladie génétique neuro- développementale dominante liée à l’X, touchant essentiellement les filles, le plus souvent due à des mutations du gène MECP2. Matériels et méthodes : Étude clinique et génétique de 16 patientes répondant aux critères diagnostiques Rett révisés
A. Saadi¹, W. Amer Elkhoudoud², S. Lougani², Hallal S³, L. Moussa², B. Imessaoudene4, N. Kassouri², L. Ali Pacha¹,
(1) Service de Neurologie CHU Mustapha Bacha, Alger
(2) Service de Neurologie, EHS Ben Aknoun, Alger,
(3) Laboratoire Centrale de Biochimie, CHU Mustapha Bacha, Alger
(4) Laboratoire de Biochimie, EHS Ben Aknoun, Alger.
Date de soumission : 11 Février 2020.
Abstract: Introduction: RETT syndrome (MIM # 312 750) is a dominant X-linked neurodevelopmental genetic disorder, primarily affecting girls, most commonly caused by mutations in the MECP2 gene. Materials and methods: Clinical and genetic study of 16 patients with the revised Rett diagnostic criteria, followed at the neurology department of Ben-Aknoun hospital. A sequencing analysis of the exons coding for the MECP2 gene was performed in all patients and supplemented by an MLPA study in 2 patients. Results :Analysis of the MECP2 gene made it possible to identify in the 16 patients 5 nonsense mutations (R255X, R168X, R270X, Tyr141X, R294X), 2 missense mutations (c.301C> T, c.455 C> G), 5 intragenic deletions and 2 large deletions. All patients have the classic form and one has an atypical form with early epilepsy. The age of diagnosis at 10 is slightly late. Epilepsy is present in 75% with an age of onset between 1-8 years. Acquired microcephaly is present in 75%. Sleep disturbances in 50%, bruxism in 80%, hyperpnea in 75%, scoliosis is found in 4 patients, two of whom lost walking between the ages of 7 and 12. One patient had a prolonged QT interval. Conclusion: It is difficult to retain significant clinical-genetic correlations from this study, due to the small number of cases. Genotype-phenotype correlations are inconsistent, in part due to the pattern of X chromosome inactivation. However, it can serve as a starting point for a national Rett syndrome database. The quantitative MLPA study of patients with a negative MECP2 mutation allowed the identification of the association of Rett syndrome and periventricular nodular heterotopia not reported in the literature to date.
Keywords: Rett syndrome, epilepsy, MECP2, MLPA.
Résumé :Introduction. Le syndrome de RETT (MIM #312 750) est une maladie génétique neuro- développementale dominante liée à l’X, touchant essentiellement les filles, le plus souvent due à des mutations du gène MECP2. Matériels et méthodes : Étude clinique et génétique de 16 patientes répondant aux critères diagnostiques Rett révisés, suivies au service de neurologie de l’établissement hospitalier spécialisé de Ben-Aknoun. Une analyse par séquençage des exons codants du gène MECP2, a été réalisée chez toutes les patientes et complétée par une étude MLPA chez 2 patientes. Résultats : L’analyse du gène MECP2 a permis d’identifier chez les 16 patientes ; 5 mutations stop (R255X, R168X, R270X, Tyr141X, R294X), 2 faux sens (c.301C>T, c.455 C>G), 5 délétions intragéniques et 2 grandes délétions par méthode semi quantitative. Toutes les patientes présentent la forme classique et une la forme atypique avec épilepsie précoce. L’âge de diagnostic à 10 ans est un peu tardif. L’épilepsie est présente dans 75% avec un âge de début entre 1-8 ans. La microcéphalie acquise est présente dans 75%. Les troubles du sommeil dans 50%, le bruxisme dans 80%, l’hyperpnée dans 75%, la scoliose est retrouvée chez 4 patientes dont deux ont perdu la marche entre l’âge de 7 et 12 ans. Une seule patiente a présenté un allongement de l’intervalle QT. Conclusion : Il est difficile de retenir des corrélations clinico-génétiques significatives à partir de cette étude, en raison du faible nombre de cas. Les corrélations génotype-phénotype sont incohérentes, en partie à cause du schéma d’inactivation des chromosomes X. Néanmoins, elle peut servir comme point de départ pour une base de données nationale du syndrome de Rett. L’étude quantitative MLPA des patientes mutation MECP2 négative a permis l’identification d’une association Rett-hétérotopie nodulaire périventriculaire non rapportée dans la littérature à ce jour.
Mots clés : Syndrome de Rett, épilepsie, MECP2, MLPA.
Introduction
Le syndrome de Rett est une maladie neuro-développementale rare, touchant essentiellement les filles, en se transmettant selon le mode dominant lié à l’X. Elle est caractérisée dans sa forme typique, par une phase de développement normal ou subnormal, suivie d’une décélération globale du développement psychomoteur, puis une perte des acquisitions cognitives et motrices ; puis une longue période de stabilité. La quatrième phase de détérioration motrice tardive commence à partir de l’âge de 10 ans [1,2]. Les signes typiques du syndrome de Rett sont : la perte de l’utilisation volontaire des mains, l’absence d’apparition ou la perte du langage, des troubles de la coordination et de l’équilibre avec une perte possible de la marche, des stéréotypies manuelles, des accès d’hyperventilation intermittente, une déficience intellectuelle sévère, des troubles du comportement et de la communication, et d’autres signes tels que l’épilepsie, le refroidissement des extrémités, le retard de croissance, l’ostéogénie et la scoliose. Les formes atypiques ou variantes sont : la forme avec préservation du langage, la forme congénitale sans phase de développement normal, la forme avec une épilepsie sévère et précoce.
La prévalence du syndrome de Rett dans le monde serait d’environ 1/10.000 à 1/23.000 filles [3]. Le syndrome de Rett est causé dans plus de 95% des formes typiques par des mutations dans le gène MECP2. Les gènes CDKL5, pour la forme avec épilepsie précoce, le gène FOXG1 pour la forme congénitale [4,5].
Matériels et méthodes
C’est une étude clinique et génétique de 16 patientes répondant aux critères diagnostiques du syndrome de Rett, suivies au service de neurologie de l’établissement hospitalier spécialisé de Ben Aknoun. Une analyse par séquençage des exons codants du gène MECP2, a été réalisée chez toutes les patientes et complétée par une étude MLPA chez 2 patientes.
Résultats
La présente étude comprend 16 filles, âgées de 3 à 34 ans. Dix d’entre elles sont originaires d’Alger, les autres de Bordj Bou Areridj, Msila Tizi Ouzou, Ain Defla, Miliana et Djelfa. Il s’agit de cas sporadiques de syndrome de RETT, selon les critères diagnostiques Rett révisés par Neuil en 2010 [6] (Tableau A). Toutes les patientes sont nées au terme d’une grossesse bien suivie sans incident sauf pour la patiente P4 qui a présenté une hypotonie néonatale. Le développement psychomoteur s’est fait avec retard de gravité variable chez l’ensemble des patientes. Les patientes (P3, P4, P6, P8, P9), soit 33% n’ont jamais acquis la marche, alors que 1/3 des patientes ont acquis la marche dans les limites de la normale. Cinq patientes (P2, P4, P6, P8, P9) n’ont jamais acquis le langage, les autres pouvaient prononcer 2 à 3 mots à partir de l’âge de 12 à 24 mois. Le profil évolutif de la maladie chez nos patientes est compatible avec les critères diagnostiques révisés du syndrome de Rett.
La période de régression débute entre l’âge de 11 à 30 mois, et dans la moitié des cas, elle survient entre 18 et 20 mois. La durée de cette période varie entre 6 mois à 5 ans. Cette phase évolutive est caractérisée par la perte de l’utilisation des mains, l’apparition des stéréotypies, des troubles de la communication, du comportement et une instabilité à la marche.
La période de stabilisation qui correspond à une amélioration notamment des capacités de communication, du contrôle visuel et de l’utilisation partielle des mains, a débuté chez nos patientes entre l’âge de 2 ans et 7 ans. Les deux patientes P1 (17 ans) et P10 (7 ans) ont présenté respectivement à partir de l’âge de 12 ans et 7 ans, une aggravation nette du syndrome pyramidal avec perte de la marche, une scoliose et une atrophie musculaire. La préservation du contact visuel et quelques aspects de la communication non verbale, nous permet de les classer au stade 4 de détérioration motrice. Alors que la plus âgée (34 ans) conserve toujours une marche instable avec cyphose dorsale importante et n’a pas présenté de fractures osseuses.
L’épilepsie est fréquente chez nos patientes, elle est retrouvée dans 75% des cas soit 12/16 patientes (tableau B). Le début varie entre l’âge de 4 mois (forme épileptique précoce) et 8 ans, soit un âge de début moyen de 4 ans sans corrélations entre l’âge d’apparition des crises et le profil évolutif. Il s’agit souvent de crises généralisées convulsives et non convulsives nocturnes et diurnes. Trois patientes (P2, P4, P15) ont présenté des crises myocloniques diffuses. La fréquence des crises est variable, les patientes (P2, P7, P11) présentent une épilepsie sévère à début précoce survenue entre l’âge de 4 mois et 12 mois, résistante au traitement et nécessitant la prise de 2 médicaments antiépileptiques (acide valproïque et/ou leviracetam, lamotrigine). Les patientes (P1, P4, P6, P15, P16) n’ont pas fait de crises depuis plus de 3 à 4 années. Toutes les patientes épileptiques ont présenté des anomalies non spécifiques à l’EEG à type de P, PO généralisées en bouffées diffuses parfois à prédominance hémisphérique. Des anomalies EEG diffuses ont été retrouvées chez 3 patientes (P9, 10, 12) en l’absence de crises d’épilepsie. Une seule patiente (P2) a présenté une épilepsie précoce dès l’âge de 4 mois avec des myoclonies et crises tonico-cloniques généralisées réfractaires, persistant encore jusqu’à ce jour, soit à l’âge de 10 ans. Aussi, elle n’a acquis ni le langage ni la marche.
Parmi les critères accessoires (Tableau C), la microcéphalie acquise est présente chez 3/4 des patientes (12/16) après l’âge de 2 ans. La microcéphalie est sévère avec un PC à -4DS chez les patientes 1 et 9 et absente chez 4 patientes. Les troubles du sommeil modérés surviennent chez 50% des patientes au cours de l’évolution de la maladie. Le bruxisme est observé chez 80% des patientes. Alors que les troubles respiratoires à type d’accès d’hyperpnée d’intensité variable survenant à des périodes différentes du développement, sont absents chez 8 patientes. La scoliose est retrouvée chez 4 patientes mais ce sont les filles les plus âgées (17 ans et 34 ans), qui présentent la forme sévère. La moitié des patientes présente des troubles vasomoteurs à type de refroidissement des extrémités (pieds), par contre la baisse de la sensibilité à la douleur est signalée par les parents de 3 patientes.
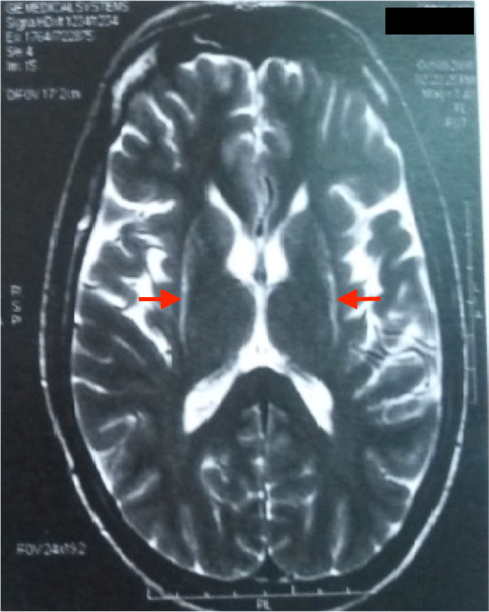
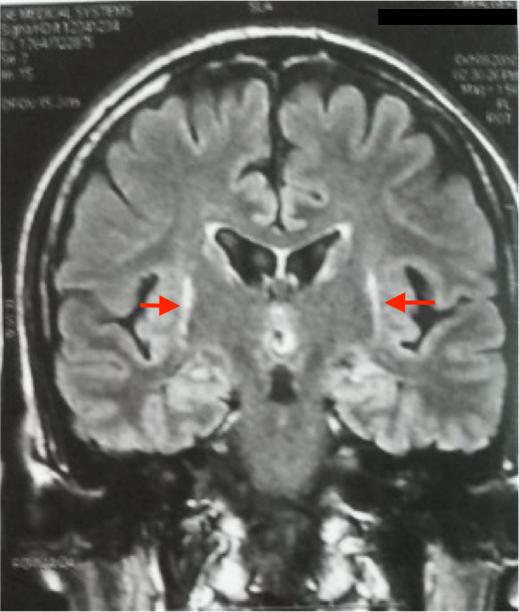
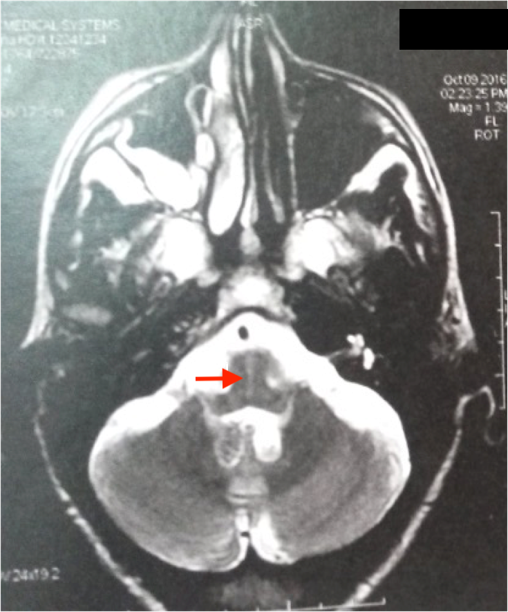
Le bilan métabolique standard est normal chez toutes les patientes. L’IRM cérébrale pratiquée chez toutes les patientes ne révèle aucune anomalie sauf une atrophie corticale fronto-pariétale chez la patiente P3 ; et une hétérotopie nodulaire périventriculaire chez la patiente P15. Il s’agit d’une fillette âgée de 8 ans, née de parents non apparentés, qui a présenté un retard du langage avec régression cognitive, perte de l’usage des mains, stéréotypies manuelles dès l’âge de 2 ans, suivies de troubles du sommeil, de crises myocloniques, de pseudo-absences et microcéphalie acquise à l’âge de 4 ans. L’EEG a montré des anomalies paroxystiques généralisées et focalisées, diffuses sur un rythme de fond structuré. L’électrocardiogramme pratiqué chez 7 patientes a mis en évidence chez la patiente P9 des troubles de la repolarisation diffuse avec une onde T négative, espace QT : 0,43s (0,32s).
L’analyse du gène MECP2 par séquençage direct des exons 2, 3 et 4 a permis l’identification de 12 mutations différentes (tableau D). Cinq mutations non-sens : c.763c>T(p.R255X) x2 fois , c.502C>T (p.R168X) x2 fois, c.808C>T (p.R270X) , c.423 C>G.(p.Tyr141X ), c.880 C>T. (p.R294X ) chez les patientes P1, 3, 6, 8, 10, 11, 13. Deux mutations faux sens : c.301C>T (p.101P>S) et c.455 C>G. (p.P152A). Cinq délétions intra géniques : c.753delC(p.G252A fs). c.1157-1197 del 41, c.1119-1189 del 71, c.1450-1453 del 4 (AGAG), c.del 32(1157-1188) toutes situées au niveau de l’exon 4 chez les patientes P2, P5, P7, P9, P16.
Cependant, l’étude cytogénétique (MLPA) pratiquée chez les 2 patientes sans mutation MECP2 (P14 et P15) a retrouvé respectivement une délétion de 1,7-61,7Kb et une délétion hétérozygote de la région Xq28 d’une taille de 450 kb à 600 kb emportant :
La totalité du gène MECP2 ;
Tout ou partie des gènes FLNA et IRAK1.
Discussion
Cette étude moléculaire nous a permis de confirmer le diagnostic clinique de syndrome de Rett chez 16 filles, âgées entre 3 et 34 ans avec une moyenne d’âge de 20 ans. L’âge du diagnostic varie entre 2 et 18 ans, soit une moyenne d’âge de 10 ans. Toutes les patientes présentent une forme classique sauf la patiente P2, qui présente une forme atypique avec épilepsie précoce due à une délétion intragénique c.753delC (p.G252A fs) avec décalage du cadre de lecture (Tableau D).
Dans notre petite série, nous constatons que la forme atypique représente 6% des cas, ce qui est faible en comparaison avec les résultats de grandes études portant sur 1.928 cas, 315 cas, 638 cas et 1.085 cas, où le rapport des formes typiques/atypiques varie entre 85-92%/13-15% [7,8,9]. L’âge moyen du diagnostic à 10 ans est un peu plus tardif comparativement à une étude récente de 1.928 cas où l’âge moyen varie entre 2,8 ans pour le Rett classique et de 3,8 ans pour la forme atypique [10]. Le retard de croissance peut s’aggraver avec l’âge et s’observer dans 85% à 90% des filles avec un syndrome de Rett classique [11].
L’épilepsie est aussi fréquente dans notre série (75%), que dans les études rapportées à ce jour (50-90%). Dans une étude de 602 cas de Rett, Claze et al trouvent une épilepsie dans 60% survenant vers l’âge de 4,5 ans, souvent associée à un tableau clinique sévère [12]. Les crises généralisées tonico-cloniques et partielles complexes sont les plus fréquentes, d’autres crises peuvent se voir comme les myoclonies et les spasmes [13]. La prévalence de l’épilepsie pharmaco-résistante est de 23% dans notre série et varie entre 16 et 45% des cas selon les séries. Des anomalies EEG précédant la survenue de crises d’épilepsie sont observées chez 2 patientes, ainsi que des phénomènes paroxystiques non épileptiques, fréquemment rapportés dans la littérature qui rendent difficile le diagnostic d’épilepsie.
La scoliose observée dans plus de 80% des personnes âgées de 25 ans [14], est retrouvée chez les 2 patientes les plus âgées (17 et 34 ans) et la forme atypique. L’ostéopénie survient chez 74% vers l’âge de 20 ans, la diminution de la densité minérale osseuse augmente le risque de fractures surtout chez les patientes qui n’ont pas encore perdu la marche [15]. Les femmes ayant un syndrome de Rett survivent généralement à l’âge adulte, mais la mort subite peut être en partie causée par l’incidence plus élevée des anomalies de l’onde T et l’allongement de l’intervalle QT. Dans notre série, une seule patiente présente un allongement de l’intervalle QT.
Dans notre série, les mutations ponctuelles (5 non sens, 2 faux sens, 5 délétions intragéniques) représentent 87% des cas (14/16), suivies de 2 grandes délétions 13% des cas (2/16). Dans la littérature, la fréquence moyenne des mutations ponctuelles est à 80%, beaucoup plus élevée que celle des micro-délétions, 8%. Parmi les 8 mutations ponctuelles (R106W, R133C, T158M, R168X, R255X, R270X, R294X, R306C) les plus fréquentes à l’origine de 67,4% des cas de Rett [344], nous avons retrouvé chez nos patientes 4 mutations stop : c.763c>T (p.R255X) , c.502C>T (p.R168X) , c.808C>T (p.R270X), c.880 C>T. (p.R294X). Dans une étude égyptienne portant sur une dizaine de cas, les mutations les plus fréquentes sont : R255X (3cas), R270X (3cas), R168X (4cas) [17]. Le séquençage du gène MECP2 chez 7 patientes tunisiennes a permis d’identifier la mutation T528M dans 50% des cas et la mutation R168X une seule fois [18]. Les mutations faux sens et non-sens sont retrouvées à une fréquence presque identique entre 35%-40% [7,8]. Dans notre série, les patientes P4 et P12 présentent une mutation faux sens c.301C>T et c.455 C>G. Cheadle a trouvé que la maladie est significativement atténuée chez les personnes présentant des mutations faux-sens que chez celles avec des mutations tronquantes [19].
Les différentes études concernant la sévérité du syndrome et les mutations spécifiques ne sont pas concluantes à cause de la variabilité génotype/phénotype, mais certains auteurs rapportent quelques conclusions. Neul, en 2008, en étudiant la sévérité du syndrome de Rett dans 3 domaines, la marche, l’utilisation des mains et le langage, trouve que les mutations (R133C, R294X, R306C) sont moins sévères que les autres mutations R168X et larges délétions [9,16]. Amir et al, ont trouvé une corrélation positive entre les mutations tronquantes et des anomalies respiratoires, alors que la scoliose était plus fréquente chez les personnes présentant des mutations faux-sens [20], associe les mutations p.R255X, p.T158M et p.C306C à une augmentation du risque de survenue de crises d’épilepsie sévères [21].
Il est difficile de tirer des conclusions à partir de cette étude, vu le faible nombre de cas. Néanmoins, nous avons remarqué que la même mutation R168X est à l’origine d’une forme classique plutôt sévère avec arrêt de la marche à l’âge de 7 ans chez la patiente P10. Ce résultat rejoint les données de la littérature qui rapportent l’association de cette mutation R168X à des formes sévères [16]. Mais la même mutation est à l’origine d’une forme modérée chez la patiente P3. Parmi les 3 mutations (p.R255X, p.T158M et p.C306C), pouvant jouer un rôle dans l’augmentation du risque de survenue de l’épilepsie, une seule, la p.R255X a été retrouvée chez la patiente P1et P13. Cependant, d’autres études de phénotype-génotype ont montré que les mutations spécifiques ne peuvent pas être le seul déterminant de la gravité, en raison de l’existence d’autres facteurs tels que l’inactivation du chromosome X et d’autres variations génétiques.
Concernant les cas particuliers des filles MECP2 négatives, il existe une proportion de (5-10%) de filles MECP2 mutation négative [22], dont 25% présentent de grandes délétions intragéniques MECP2 multi-éxoniques [23,24,25]. Les quelques cas de micro-délétion Xq28 décrits à ce jour concernent seulement les gènes FLNA-EDMD ou MECP2-IRAK1 [26]. Les rares cas de grande délétion MECP2 étendue au gène IRAK1 rapportés présentent souvent un syndrome de Rett classique avec parfois un trouble de l’ossification ou des infections, mais sans anomalies congénitales majeures [27]. C’est le premier cas de micro-délétion impliquant les gènes MECP2 et FLNA et IRAK1 non rapporté dans la littérature, à l’origine d’un syndrome de Rett typique et d’une hétérotopie périventriculaire chez une fille non porteuse de mutation MECP2. L’épilepsie et les anomalies EEG observées chez notre patiente ne sont pas spécifiques, et font partie du phénotype Rett, car les crises d’épilepsie dues à la mutation FLNA surviennent vers l’âge de 15 ans en moyenne. La recherche de malformations cardiaques congénitales (persistance du canal artériel, dilatation de l’aorte thoracique) décrites dans le phénotype FLNA, s’est révélée négative.
Conclusion
Il est difficile de tirer des conclusions et retenir des corrélations clinico-génétiques significatives à partir de cette étude, vu le faible nombre de cas. Néanmoins, elle peut servir comme point de départ pour une base de données nationale du syndrome de Rett. L’étude quantitative MLPA des patientes mutation MECP2 négative a permis l’identification d’une association Rett, hétérotopie nodulaire périventriculaire non rapportée dans la littérature à ce jour et de réduire le risque de faux négatifs. La recherche de troubles de la repolarisation cardiaque pouvant être la cause de mort subite doit être systématique chez toutes les filles Rett.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Tableau A : Caractéristiques cliniques et évolutives des patientes Rett
Tableau B : caractéristiques des crises épileptiques
P
Age
Age de début
Type
EEG
Traitement
Dernière crise (âge)
1
17ans
8 ans
Crises généralisées convulsives 2C/j au début
Tracé mal organisé, activité lente Type thêta (5-6c/s)
Ac. valproïque
14ans
2
10ans
4mois
Myoclonies diffuses et oculaires Crises généralisées convulsives
Tracé mal organisé, P, POL en bouffées généralisées à prédominance hémisphérique gauche
Ac. valproïque Clonazepam
1crise /3mois
3
8ans
30mois
Crises généralisées convulsives
Tracé désorganisé P, PO continues diffuses. Tracé organisé, bouffées OL d’aspect crocheté fronto-polaire bilatérale, P centro-temporale droite
Ac. valproïque Lamictal
1crise /4mois
4
7ans
2ans
Myoclonies diffuses, crises généralisées convulsives et non convulsives
Stade sommeil P, PP, PO multifocales DT ou gauche, parfois en bouffées. Tracé veille mal organisé activité épileptique quasi continue prédominance hémisphérique gauche
Ac. valproïque
3ans
5
7ans
6ans
Crises généralisées
Bouffées O, PO centrales droite parfois bilatérales
Ac. valproïque
6ans
6
7ans
1an
Crises généralisées pendant le sommeil
Tracé sans anomalies épileptiques
DPK puis Lamictal
2ans 1/2
7
7ans
1an
Crises généralisées nocturnes très fréquentes au début
Tracé ralenti, anomalies paroxystiques diffuses
DPK et Keppra Lamictal
1crise/ 30j
8
3ans
–
Pas de crises
Tracé normal
9
5ans
–
Pas de crises
Anomalies paroxystiques temporales postérieures droites qui diffusent dans les régions homologues controlatérales
–
10
3ans
–
Pas de crises
Rythme de fond organisé, pointes et pointes ondes localisées
–
11
8ans
2ans
Crises généralisées non convulsives
Tracé mal organisé, lent. PO lentes, bouffées de longue durée, parfois continues, de grandes amplitudes, régions antérieures bilatérales.
Leviracetam Clobazam Puis Lamotrigine
1 crise/2j
12
14ans
5ans
Agitation pendant le sommeil
Tracé de fond organisé. polypointes ondes diffuses
–
13
34ans
4ans
quelques crises à type d’absence
Bouffée de P et PO généralisées
Ac. valproïque
4ans1/2
14
7ans
5ans
Crise partielles motrices
– Activités paroxystiques bifrontorolandiques – Anomalies paroxystiques prédominant à gauche
Ac. valproïque
1crise/sem
15
11ans
8ans
Type de frayeurs nocturnes, – myoclonies avec lâchage d’objet survenant au réveil, – fixité du regard, contraction tonique main droite quelques secondes
– Anomalies paroxystiques généralisées. – Anomalies irritatives focalisées et diffuses sur un rythme de fond structuré
Ac. valproïque
6ans
16
8ans
3ans
Perte de connaissance, chute non convulsive
Sommeil mal organisé Nombreuses figures paroxystiques diffuses et généralisées.
Tableau D: Formes cliniques avec mutation MECP2 et anomalies cytogénétiques correspondantes
Patiente
Age diagnostique
Forme clinique
Mutation gène MECP2
1
11 ans
Classique
c.763c>T (p.R255X)
Non sens
2
4 ans
Épilepsie précoce
c.753delC (p.G252A fs)
Délétion 1 pb
3
8 ans
Classique
c.502C>T (pR168X)
Non sens
4
4 ans
Classique
c.301C>T (p.101P>S)
Faux sens
5
3 ans
Classique
c.1157-1197 Del41
Délétion 41pb
6
3 ans
Classique
c.808C>T (p.R270X)
Non sens
7
5 ans
Classique
c.del (1119-1189)
Délétion 68pb
8
2 ans
Classique
c.423 C>G. (p.Tyr141X )
Non sens
9
5 ans
Classique
c.1450-1453 del4 (AGAG)
Délétion 4pb
10
3 ans
Classique
c.502 C>T. (p.R168X )
Non sens
11
8 ans
Classique
c.880 C>T. (p.R294X )
Non sens
12
14 ans
Classique
c.455 C>G. (p.P152A)
Faux sens
13
18 ans
Classique
c.763 C>T. (p.R255X )
Non sens
14
6 ans
Classique
Pas de mutation
MLPA : Del 1,7-61,7Kb
15
8 ans
Classique
Pas de mutation
MLPA : Délétion hétérozygote de la région Xq28 d’une taille de450 kb à 600 kb emportant : -la totalité du gène MECP2 -tout ou partie des gènes FLNA et IRAK1
16
8 ans
Classique
c.del 32 (1157-1188)
Délétion 38pb
Références :
Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, Leonard H, Bailey ME, Schanen NC, Zappella M, Renieri A, Huppke P, Percy AK. Rett Search Consortium. Rett syndrome: revised, diagnostic criteria and nomenclature. Ann Neurol. 2010 Dec;68(6):944-50.
Einspieler C, Marschik PB. Regression in Rett syndrome: Developmental pathways to its onset. Neurosci Biobehav Rev. 2019 Mar;98:320-332.
Armstrong AH, Hangauer J, Agazzi H, Nunez A, Gieron-Korthals M. Individuals with intellectual and developmental disabilities. In: David AS, ed. Handbook of Pediatric Neuropsychology. New York: Springer. 2010;537-50
Hagberg B, Gillberg C. Rett variants-rettoid phenotypes. In: Hagberg B, Anvret M, Wahlstrom J, eds. Rett Syndrome: Clinical and Biological Aspects. London, UK: MacKeith Press; 1993:40-60
Gold WA, Krishnaraj R, Ellaway C, Christodoulou J. Rett syndrome: a genetic update and clinical review focusing on comorbidities. ACS Chem Neurosci. 2018;9:167–76
Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, Leonard H, Bailey ME, Schanen NC, Zappella M, Renieri A, Huppke P, Percy AK. Rett Search Consortium. Rett syndrome: revised, diagnostic criteria and nomenclature. Ann Neurol. 2010 Dec;68(6):944-50.
Percy AK, Lane JB, Childers J, Skinner S, Annese F, Barrish J, Caeg E, Glaze DG, Mac Leod P. Rett syndrome: North American database. J Child Neurol.Dec,22 (12):1338-41.
Zhang X,Bao X, Zhang J, Zhao Y, Cao G, Pan H, Zhang J, Wei L, Wu X. Molecular characteristics of Chinese patients with Rett syndrome. Eur J Med Genet. 2012 Dec; 55(12): 677-81.
Neul JL, Lane JB, Lee HS, et al. Developmental delay in Rett syndrome: data from the natural history study. Journal of neurodevelopmental disorders. 2014;6(1):20
Tarquinio DC, Hou W, Neul JL, Lane JB, Barnes KV, O’Leary HM, et al. Age of diagnosis in Rett syndrome: patterns of recognition among diagnosticians and risk factors for late diagnosis. Pediatr Neurol. 2015 Jun;52(6):585-91
Motil KJ, Schultz RJ, Wong WW, Glaze DG. Increased energy expenditure associated with repetitive involuntary movement does not contribute to growth failure in girls with Rett syndrome. J Pediatr. 1998;132:228–33.
Glaze DG, Percy AK, Skinner S, Motil KJ, Neul JL, Barrish JO, Lane JB, Geerts SP, Annese F, Graham J, McNair L, Lee HS. Epilepsy and the natural history of Rett syndrome. Neurology. 2010 Mar 16;74(11):909-12.
Steffenburg U, Hagberg G, Hagberg B. Epilepsy in a representative series of Rett syndrome. Acta Paediatr. 2001;90:34–9.
Kerr AM, Webb P, Prescott RJ, Milne Y. Results of surgery for scoliosis in Rett syndrome. J Child Neurol.2003;18:703–8.
Leonard H, Thomson MR, Glasson EJ, Fyfe S, Leonard S, Bower C, Christodoulou J, Ellaway C. A population-based approach to the investigation of osteopenia in Rett syndrome. Dev Med Child Neurol. 1999;41:323–8.
Neul JL, Fang P, Barrish J, Lane J, Caeg EB, Smith EO, Zoghbi H, Percy A, Glaze DG. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology. 2008 Apr 15;70(16):1313-21.
Mansour L1, El Sobky E, Mohamed SM, Marzouk H, Tarek LA. Genotype-phenotype relationship among Egyptian children with Rett syndrome. J Egypt Public Health Assoc. 2015 Sep;90(3):133-7
Fendri-Kriaa N, Mkaouar-Rebai E, Moalla D, Belguith N, Louhichi N, Zemni R, Slama F, Triki C, Fakhfakh F. Tunisian Network on Mental Retardation. Mutational analysis of the MECP2 gene in Tunisian Patients with Rett Syndrome: a novel double mutation. J Child Neurol 2010;25(8):1042–6.
Cheadle JP, Gill H, Fleming N, Maynard J, et al. Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: correlation of disease severity with mutation type and location. Hum Mol Genet. 2000 Apr 12 ;9(7):1119–29.
Amir RE, Van den Veyver IB, Schultz R, Malicki DM, Tran CQ, Dahle EJ, Philippi A, Timar L, Percy AK, Motil KJ, Lichtarge O, Smith EO, Glaze DG, Zoghbi HY. Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann Neurol. 2000 May;47:670–9.
Nissenkorn A, Levy-Drummer RS, Bondi O, Renieri A, Villard L, Mari F,et al. Epilepsy in Rett syndrome–lessons from the Rett networked database. Epilepsia 2005 Apr;56(4)569-7.
Matsuishi T, Yamashita Y, Takahashi T, Nagamitsu S Rett syndrome: the state of clinical and basic research, and future perspectives. Brain Dev 2011 Sep;33(8):627-31
Bourdon V, Philippe C, Grandemenge A, Reichwald K, Jonveaux P, Deletion screening by fluorescencein situ hybridization in Rett syndrome patients. Ann Genet 2001. Oct-Dec;44(4) :191-4
Schollen E, Smeets E, Deflem E, Fryns JP, Matthijs G. Gross rearrangements in the MECP2 gene in three patients with Rett syndrome: implications for routine diagnosis of Rett syndrome. Hum Mutat2003 Aug;22(2):116-20.
IOUROV I Y, Vorsanova SG, Voinova VY, Kurinnaia OS, Zelenova MA, Demidova IA, Yurov YB microdeletions are common in mutation-negative females with Rett syndrome and cause mild subtypes of the disease. Mol Cytogenet Nov 2013. 27;6(1):53
Clapham K R, Yu TW, Ganesh VS, Barry B, Chan Y, Mei D, Parrini E, Funalot B, Dupuis L, Nezarati MM, du Souich C, van Karnebeek C, Guerrini R, Walsh CA..FLNA genomic rearrangements cause periventricular nodular heterotopia. Neurology 2012 Jan 24;78(4):269-78
Scala IE, Longo I, Ottimo F, Speciale C, Sampieri K, Katzaki E, Artuso R, Mencarelli MA, D’Ambrogio T, Vonella G, Zappella M, Hayek G, Battaglia A, Mari F, Renieri A, Ariani F. MECP2 Deletions and Genotype–Phenotype Correlation in Rett Syndrome AJMG part A 2007. Dec 1;143A(23):2775-84
Les leucodystrophies métaboliques, affections rares et génétiques, sont caractérisées par une atteinte prédominante de la myéline. Elles constituent un grand nombre de maladies très hétérogènes dans leur expression clinique et leur physiopathologie. La majorité sont diagnostiquées biochimiquement et génétiquement et bénéficient d’un diagnostic prénatal. Nous rapportons l’expérience de l’unité neuropédiatrique du service de neurologie de l’établissement Ali Ait-Idir sur une durée de quinze ans concernant les leucodystrophies métaboliques pédiatriques.
S.Makri-Mokrane, S. Meziche, I. Talaboulma. Service de Neurologie. EHS Ali Ait-Idir, Bab El Oued, Alger.
Date de soumission : 11 Février 2020.
Abstract: Metabolic leukodystrophies are rare and genetic conditions, characterized by predominantly myelin damage. It constitutes a large number of very heterogeneous diseases in their clinical expression and their pathophysiology. The majority are biochemically and genetically diagnosed and benefit from prenatal diagnosis. We report the experience of the neuropediatric unit of the neurology service of the Ali Ait-Idir hospital over a period of fifteen years concerning paediatric metabolic leukodystrophies. Of 24 children with a picture of metabolic leukodystrophies, 50% had X-linked adrenoleukodystrophy and 4 cases had metabolic leukodystrophy. In the absence of enzymatic and/or molecular confirmation accurate phenotyping and careful interpretation of the imaging make it possible to identify leukodystrophy.
Résumé : Les leucodystrophies métaboliques, affections rares et génétiques, sont caractérisées par une atteinte prédominante de la myéline. Elles constituent un grand nombre de maladies très hétérogènes dans leur expression clinique et leur physiopathologie. La majorité sont diagnostiquées biochimiquement et génétiquement et bénéficient d’un diagnostic prénatal. Nous rapportons l’expérience de l’unité neuropédiatrique du service de neurologie de l’établissement Ali Ait-Idir sur une durée de quinze ans concernant les leucodystrophies métaboliques pédiatriques. Sur 24 enfants présentant un tableau de leucodystrophies métaboliques, 50% avaient une adrénoleucodystrophie liée à l’X, et 4 cas une leucodystrophie métabolique. En l’absence de confirmation enzymatique et/ou moléculaire, un phénotypage précis et une interprétation minutieuse de l’imagerie permettent d’identifier une leucodystrophie.
Mots clés : leucodystrophie, IRM, adrénoleucodystrophie liée à l’X.
Introduction
Les leucodystrophies métaboliques (LM) sont des affections neuro-dégénératives rares. Les LM sont des maladies caractérisées par une atteinte prédominante et primitive de la myéline du système nerveux central (SNC), avec une préservation relative des axones, parfois associée à une atteinte du système nerveux périphérique (SNP). Les LM sont des affections génétiques causées par des déficits enzymatiques, elles sont appelées également leucoencéphalopathies héréditaires. La majorité sont diagnostiquées biochimiquement et génétiquement et bénéficient d’un diagnostic prénatal. Le terme de LM rassemble un grand nombre de maladies très hétérogènes dans leur expression et leur physiopathologie. Elles débutent à des âges variables, de la naissance à l’âge adulte mais elles restent plus fréquentes chez l’enfant. En pratique, le diagnostic d’une LM est difficile et il est nécessaire de connaitre les étapes de myélinisation du SNC de l’enfant. Cliniquement, devant la découverte d’une anomalie de la substance blanche (SB) à l’IRM cérébrale, éliminer initialement les pathologies acquises et plus particulièrement la leucomalacie chez le jeune enfant. Si la symptomatologie des LM est dominée par des troubles moteurs (syndrome cérébello-spastique) associés à une atteinte mentale variable, l’âge de début, l’aspect évolutif et la présence de certains signes cliniques tels une neuropathie, une mégalencephalie, une atteinte sensorielle et extra-neurologique vont orienter le diagnostic étiologique. Les potentiels évoqués cérébraux (PEC) sont utiles pour objectiver l’atteinte myélinique du SNC qui se traduit par un allongement des temps de conduction centraux. L’électroneuromyographie (EMG-VC), l’électrorétinogramme (ERG) et les potentiels évoqués auditifs (PEA) permettent de rechercher respectivement une atteinte du SNP, une rétinopathie ou une surdité associée. L’IRM cérébrale est sensible pour détecter les anomalies de la SB qui apparaissent en hypersignal sur les séquences pondérées en T2. L’IRM cérébrale est un examen essentiel car il contribue au diagnostic, au suivi et à l’évolution progressive des lésions. La topographie des lésions de la SB, leur signal sur les différentes séquences de l’IRM (T1 et FLAIR en particulier), ainsi que la présence de lésions associées permettent également d’identifier des aspects suggestifs de certaines étiologies.
Classification actuelle
Leucodystrophies peroxysomales
Adrénoleucodystrophie/ Adrénomyéloneuropathie,
Maladie de Refsum adulte,
Maladies du spectre Zellweger, maladies avec défaut de formation des peroxysomes, c’est-à-dire : syndrome de Zellweger, adrénoleucodystrophie néonatale et maladie de Refsum infantile.
Leucodystrophies lysosomales
Leucodystrophie métachromatique,
Maladie de Krabbe.
Leucodystrophies cavitaires
Maladie d’Alexander,
Maladie de Canavan,
Leucodystrophie mégalencéphalique avec kystes sous-corticaux,
Syndrome CACH/VWM.
Leucodystrophies hypomyélinisantes
Maladie de Pelizaeus-Merzbacher,
Maladie de Pelizaeus-Merzbacher like,
Paraplégie spastique II,
Les leucodystrophies à polymérases III.
Leucodystrophies atypiques
Syndrome d’Aicardi-Goutières,
Syndrome Ravine.
Leucodystrophies indéterminées.
Matériel et méthodes
Nous rapportons l’expérience de l’unité neuropédiatrique du service de Neurologie de l’établissement Ali Ait-Idir sur une durée de quinze ans concernant les LM. Tous les enfants avec suspicion de LM ont eu le protocole suivant : anamnèse détaillée précisant les différentes étapes du développement psychomoteur, l’histoire de la maladie et l’arbre généalogique ; un examen neurologique et somatique minutieux ; un examen neurophysiologique et plus précisément des PEC, un EMG-VC, un électrorétinogramme et un EEG ; une étude cytochimique du LCR ; un examen ophtalmologique ; des tests psychométriques et une IRM cérébrale. Seuls les cas avec un tableau d’adrénoleucodystrophie liée à l’X ont eu en plus le dosage du cortisol sérique. La confirmation biochimique a été réalisée dans 5 cas.
Résultats
24 enfants présentaient une LM, par ordre de fréquence :
Adrénoleucodystrophie liée à l’X : 12 cas, âge entre 4-10 ans. La maladie s’est manifestée par des difficultés scolaires ou des modifications comportementales hormis un cas âgé de 4 ans, qui avait présenté une mélanodermie sur insuffisance surrénalienne comme symptomatologie initiale. L’examen clinique a mis en évidence un syndrome pyramidal et cérébelleux dans 02 cas. Une détérioration mentale, une agitation, une diminution de l’acuité auditive et visuelle chez tous les enfants et présence d’un œdème papillaire dans un cas. La mélanodermie était constante avec une sévérité de degrés variables. Une hyperprotéinorrachie isolée était présente ainsi qu’un allongement des latences centrales au potentiel évoqué somesthésique (PES).
Absence d’atteinte neurogène périphérique à l’examen électromyographique (EMG). Le bilan endocrinien a révélé une diminution du cortisol sérique. Le dosage des acides gras à très longues chaines n’a été pratiqué que chez deux enfants et a mis en évidence une élévation significative.
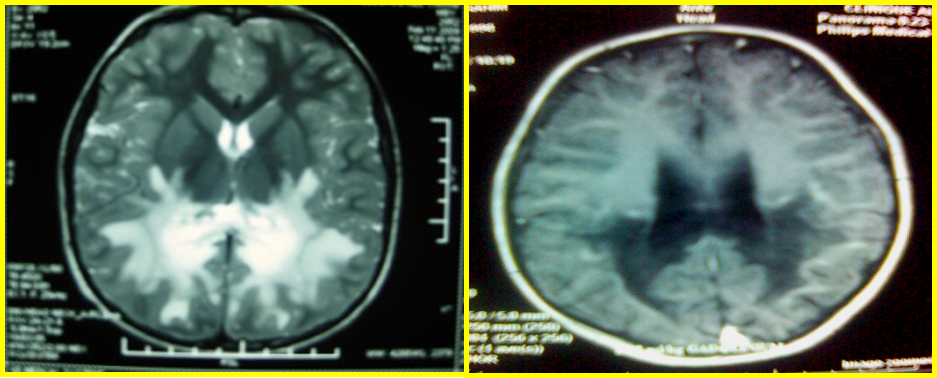
L’IRM cérébrale a objectivé des anomalies bilatérales et symétriques de la SB le plus souvent pariéto-occipitale avec prise de contraste en périphérie qui traduit toujours des lésions évolutives, une atteinte spécifique de splénium du corps calleux, une atteinte des fibres en U avec progression des lésions d’arrière en avant.
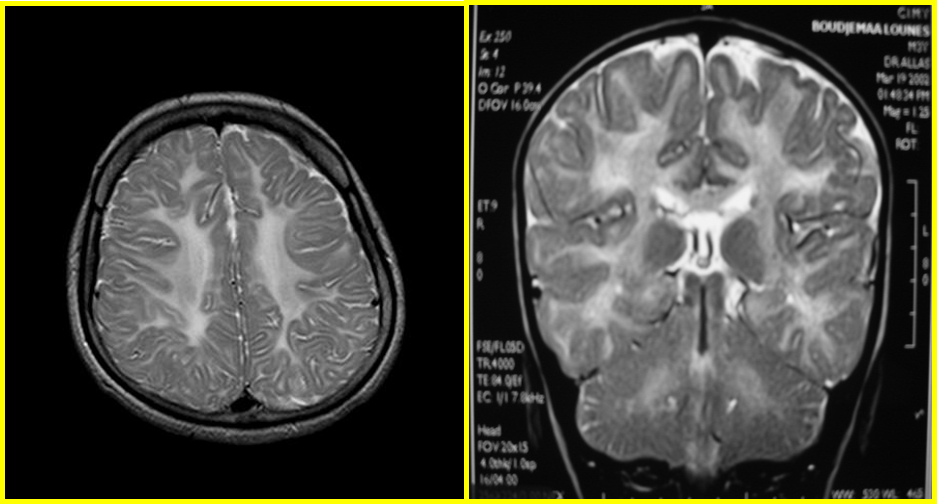
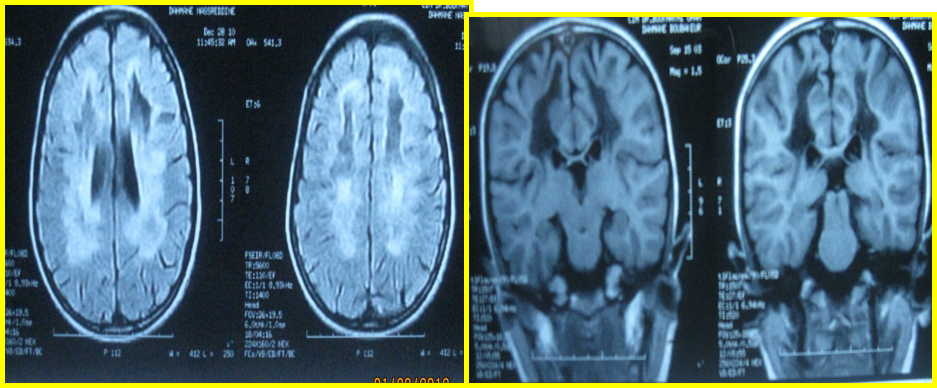
Fig. 1 : Coupes axiales T2 FLAIR et T1 avec prise de contraste : hypersignal pariéto-occipitale avec prise de contraste en périphérie et atteinte des fibres en U. Coupe sagittale T2 FLAIR : atteinte du splénium du corps calleux.
Leucodystrophie métachromatique : 4 cas, dont l’âge est compris entre 2 et 3 ans, qui consultent pour une perte progressive des acquisitions psychomotrices.
Le tableau clinique se résumait à une tétraparésie spastique, une aréflexie aux 4 membres sur un retard psychomoteur sévère.
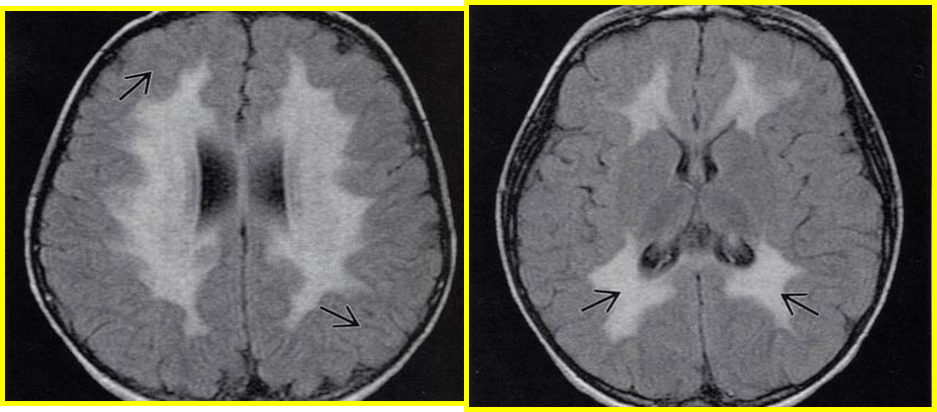
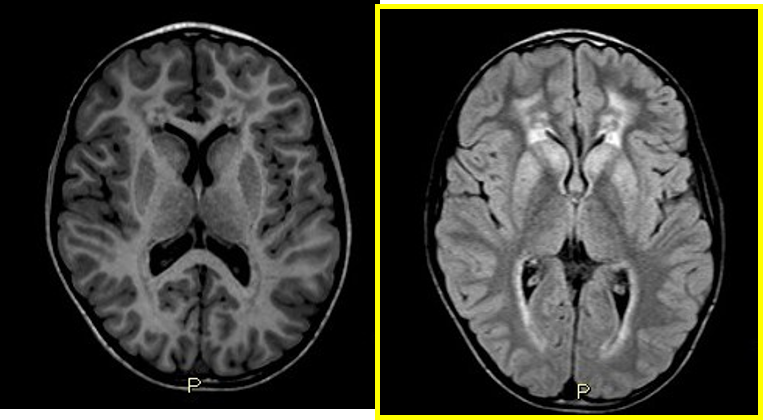
Le bilan a été comme suit : une hyperprotéinorrachie à la ponction lombaire (PL), un allongement des latences centrales et périphériques aux PES et une atteinte neurogène périphérique de type myélinique aux 4 membres à l’EMG. Un déficit en arylsulfatase A détecté dans les leucocytes a permis un diagnostic biochimique. Un hypersignal diffus de la SB périventiculaire, en forme de papillon, à prédominance postérieure, épargnant les fibres en U sous corticales ; avec un aspect tigré des centres semi ovales (témoignant de la préservation de la myélinisation périvasculaire) est observé à l’imagerie cérébrale.
Fig. 2 : Séquence axiale T2. Anomalie diffuse de la SB à prédominance postérieure, les fibres en U sous corticales sont épargnées.
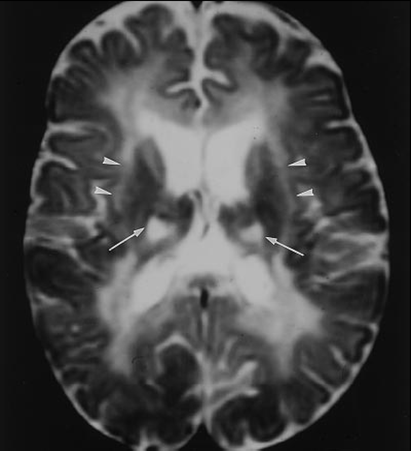
Maladie de Krabbe : une fillette de 8 mois consulte pour une régression psychomotrice et vomissements incessants. Cliniquement, elle présentait une tétraparésie spastique, des spasmes toniques et une aréflexie aux membres inférieurs. Les explorations suivantes ont été réalisées avec mise en évidence d’une hyperprotéinorrachie isolée à la PL, un allongement des latences centrales et périphérique aux PES et un effondrement des vitesses motrices à l’EMG. Confirmation biochimique et déficit de la cérébroside-β-galactosidase sérique. À l’imagerie cérébrale, démyélinisation diffuse et atteinte spécifique des capsules internes et externes et des thalami sont retrouvées.
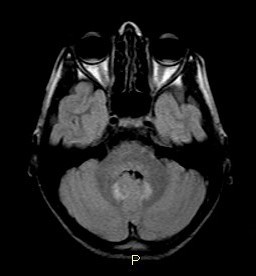
Fig. 3 : Coupe axiale T2. Démyélinisation diffuse et atteinte spécifique des capsules internes et externes et des thalami.
Maladie de Pelizaeus-Merzbacher : 3 cas : 2 frères âgés respectivement de 9 et 16 ans, issus de parents cousins germains, avaient présenté un retard psychomoteur modéré, une marche instable et un retard scolaire, ainsi qu’un garçon âgé de 04 ans issu également d’un mariage consanguin avec notion de cas similaire dans la famille présentait depuis une année une maladresse gestuelle, un tremblement du chef et un trouble de l’équilibre. Le tableau clinique de la fratrie était comme suit : un syndrome cérébelleux, une paraparésie spastique et nystagmus pendulaire présent uniquement chez le cadet ainsi qu’un retard mental modéré et un retard staturo-pondéral. L’examen du cas sporadique, un nystagmus pendulaire unilatéral gauche, un syndrome cérébelleux et un syndrome pyramidal sont notés. L’exploration pratiquée a révélé une cytochimie du LCR et un EMG normaux, un allongement des latences centrales au PES et une démyélinisation discrète intéressant la SB cérébrale épargnant les fibres en U ainsi qu’une démyélinisation cérébelleuse à l’IRM cérébrale.
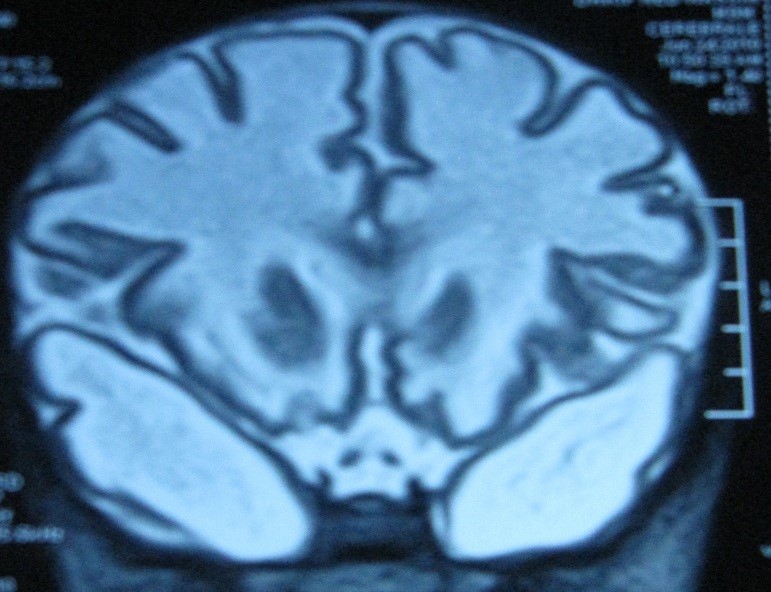
Fig. 4 : Coupe axiale T2 et coronale T2 : démyélinisation discrète, intéressant la SB cérébrale épargnant les fibres en U et démyélinisation cérébelleuse.
Maladie d’Alexander : un garçon de 3 ans présentant un développement psychomoteur quasi absent et une épilepsie pour laquelle un traitement à base de Depakine[1], (DPK) a été prescrit. Une macrocranie, une tétraparésie spastique ont été objectivées ainsi que des crises d’épilepsie fréquentes. Présence d’une hyperprotéinorrachie et normalité des PEC. Présence de pointes ondes, PO, lentes diffuses sur un tracé déstructuré et ralenti pour l’âge à l’EEG de veille ; absence des fuseaux du sommeil physiologique à l’EEG de sommeil ; présence d’anomalies de la SB à prédominance frontale et des hypersignaux des noyaux gris centraux, des thalami et du tronc cérébral à l’IRM cérébrale.
Fig. 5 : Coupes axiales T2 Flair. Anomalies de la SB à prédominance frontale et des hypersignaux des noyaux gris centraux, des thalami et du tronc cérébral.
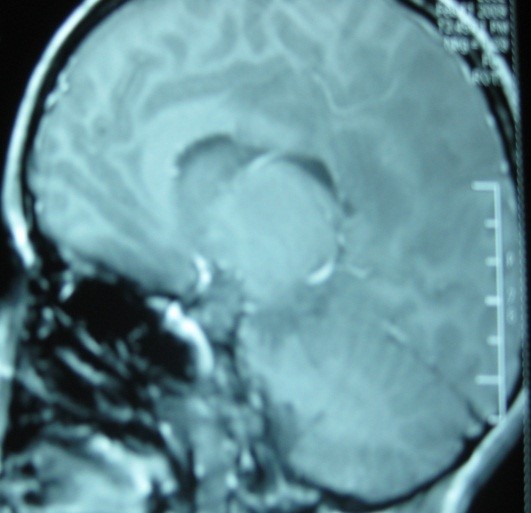
Leucodystrophie mégalencéphalique avec kystes subcorticaux : enfant âgé de 26 mois, de parents cousins germains, consulte pour une régression psychomotrice, une augmentation progressive du périmètre crânien (PC), et une épilepsie équilibrée sous DPK. L’examen clinique initial a mis en évidence une macrocranie majeure (6DS), une hyperréflexie ainsi que des troubles comportementaux à type d’hyperactivité avec néanmoins un bon contact. Atteinte démyélinisante centrale aux PES et PEA. Sur le tracé EEG de veille un ralentissement à 4-6 c/s postérieur est noté et sur le tracé EEG de sommeil, présence des figures du sommeil et des paroxysmes de PO généralisées lors du sommeil profond. Normalité de l’examen EMG et atteinte sévère et précoce de la SB (bien que les manifestations neurologiques durant les 1ers stades de la maladie soient relativement modérées) avec effacement des espaces sous arachnoïdiens épargnant le corps calleux, le tronc cérébral et des kystes dans les lobes temporaux subcorticaux à l’IRM cérébrale.
Fig. 6 : Coupes coronales T2 et T1. Atteinte sévère et précoce de la SB avec effacement des espaces sous arachnoïdiens et des kystes dans les lobes temporaux subcorticaux.
Syndrome CACH (Childhood Ataxia with Central Hypomyelination) / VWM (Vanishing White Matter) : 2 frères, âgés respectivement de 11 ans et 16 ans, de parents consanguins présentent un retard du langage et de la marche. À l’âge de 8 ans, l’ainé a présenté brutalement après une chute de sa hauteur une faiblesse de la jambe droite puis une année plus tard une lourdeur des jambes et troubles de l’équilibre s’aggravant transitoirement lors des syndromes fébriles. Un syndrome cérébello-pyramidal, des déformations spinale et orthopédique, un retard mental modéré et un retard staturo-pondéral discret sont objectivés à l’examen neurologique, la déambulation était conservée. Aux PES : atteinte somesthésique centrale, normalité des PEV et des PEA ainsi que l’EMG et les bilans endocrinien et ophtalmologique. À l’EEG de veille, le tracé de fond est fait d’un rythme alpha avec présence de bouffées paroxystiques d’ondes et de pointes en centro-temporales bilatérales prédominant à droite. À l’IRM cérébrale : anomalie de la SB diffuse, bilatérale et symétrique sus- et tentorielle avec un aspect cavitaire (en T2 FLAIR) à prédominance périventriculaire.
Fig. 7 : Coupes axiales T2 FLAIR et coupes coronales T1. Anomalie de la SB diffuse, bilatérale et symétrique sus- et tentorielle avec un aspect cavitaire.
Discussion et conclusion
L’adrénoleucodystrophie liée à l’X est la plus fréquente des LM. Un phénotypage précis et une interprétation minutieuse de l’imagerie permettent d’identifier une leucodystrophie même en l’absence de confirmation enzymatique et/ou moléculaire.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Photos : Les photographies sont celles des patients. Celles des figures 2 et 3 qui m’ont été remises gracieusement par un confrère radiologue.
Références
Vanderver A, Tonduti D, Schiffmann R, and al. Editors In: Adam MPand al, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors2014 Feb 6. Leukodystrophy Overview
Bonkowsky JL, Nelson C, Kingston JL, Filloux FM, Mundorff MB, Srivastava R. The burden of inherited leukodystrophies in children. 2010;75:718–25.
Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. 2009;72:750–9.
La maladie de Charcot Marie Tooth (CMT), neuropathie héréditaire la plus fréquente partout dans le monde, est caractérisée par une hétérogénéité clinique et génétique avec plus de 80 gènes pouvant être impliqués dans sa pathogénie.
M. Tazir, S. Nouioua, M. Bellatache, L. Ali Pacha, Laboratoire de Neurosciences, Université Benyoucef Benkhedda, Service de Neurologie CHU Mustapha Bacha, Alger.
Date de soumission : 10 Février 2020.
Abstract: Charcot Marie Tooth disease (CMT), the most common hereditary neuropathy worldwide, is characterized by clinical and genetic heterogeneity with more than 80 genes that may be involved in its pathogenesis. The classic form beginning between the first and the third decade, marked by a slowly progressive deficit and an amyotrophy of the legs, a steppage gait, pescavus and scoliosis, is compatible with almost a normal life expectancy, and in more than 50% of cases itis due to the duplication of the PMP22 gene, characterizing the autosomal dominant demyelinating form. The X-linked form of CMT due to a Connexin 32 mutation is in 2nd place in the world frequency. In Algeria and the Maghreb countries, the autosomal recessive forms (AR) of CMT constitute more than half of the diagnosed cases. The axonal AR form caused by a founding mutation of the LMNA gene is characterized by a more severe phenotype with a rapid progression of the motor deficit of the distal muscles of the lower limbs to the proximal muscles, aggravating ambulation and leading to early cessation of walking. The GDAP1 gene in its axonal and demyelinating AR forms is characterized by an early onset, significant skeletal deformities (pescavus varus and scoliosis) quickly confining the patient to the wheelchair. The advent of large-scale gene sequencing techniques (exome sequencing) has made it possible to identify many new genes involved in CMTs and corresponding deficient proteins and thus broaden the understanding of the pathophysiological mechanisms, making it possible to open wide avenues for therapeutic trials. The multitude of genes involved has led experts in these pathologies to consider another way of classifying the different forms of CMT, replacing the alphanumeric classification system with a patient identification system based on the mode of inheritance, the type of neuropathy and the involved gene.
Résumé : La maladie de Charcot Marie Tooth (CMT), neuropathie héréditaire la plus fréquente partout dans le monde, est caractérisée par une hétérogénéité clinique et génétique avec plus de 80 gènes pouvant être impliqués dans sa pathogénie. La forme classique débutant entre la première et la 3ème décade, marquée par un déficit et une amyotrophie des jambes lentement progressifs, un steppage à la marche, des pieds creux et parfois une scoliose, est compatible avec une espérance de vie quasi normale, et dans plus de 50% des cas, elle est due à la duplication du gène PMP22, caractérisant la forme démyélinisante autosomique dominante. La forme de CMT liée à l’X due à une mutation de la connexine 32 vient en 2ème position dans la fréquence mondiale. En Algérie et dans les pays du Maghreb, les formes autosomiques récessives (AR) de CMT constituent plus de la moitié des cas diagnostiqués. La forme axonale AR causée par une mutation fondatrice du gène LMNA est caractérisée par un phénotype plus sévère avec une progression rapide du déficit moteur des muscles distaux des membres inférieurs aux muscles proximaux aggravant la déambulation et menant à un arrêt précoce de la marche. Le gène GDAP1 dans ses formes AR axonale et démyélinisante est caractérisé par un début précoce, des déformations squelettiques importantes (pieds creux varus et scoliose), confinant le patient rapidement à la chaise roulante. L’avènement des techniques de séquençage des gènes à large échelle (exome sequencing) a permis d’identifier de nombreux nouveaux gènes impliqués dans les CMT et les protéines correspondantes déficientes et ainsi élargir la compréhension des mécanismes physiopathologiques responsables, permettant d’ouvrir de larges avenues aux essais thérapeutiques. La multitude des gènes impliqués a amené les experts de ces pathologies à réfléchir sur une autre manière de classer les différentes formes de CMT, remplaçant le système de classification alphanumérique par un système d’identification du patient basé sur le mode de transmission héréditaire, le type de neuropathie et le gène impliqué.
Mots clés : Maladie de Charcot-Marie-Tooth, ADCMT, ARCMT, XCMT, LMNA, GDAP1, NGS, WES.
Introduction
La maladie de Charcot Marie Tooth (CMT) fait partie du vaste chapitre des neuropathies héréditaires qui comprend, en plus des neuropathies héréditaires motrices et sensitives (HMNS) ou CMT, les neuropathies héréditaires motrices (HMN) et les neuropathies héréditaires sensitives et dysautonomiques (HSAN). Ces deux dernières entités, beaucoup plus rares, ne sont pas abordées dans cet article, mais on peut dire que malgré la diversité génétique, il est possible d’établir une distinction entre les HMSN qui comportent des troubles moteurs et sensitifs, les HSAN où prédominent les manifestations sensitives et dysautonomiques et les HMN dans lesquelles seules les fibres motrices sont atteintes (1, 2).
Les progrès récents dans les domaines de la génétique moléculaire et la biologie cellulaire ont révolutionné nos connaissances sur les neuropathies héréditaires, sachant que plus de 80 gènes associés aux CMT ont été identifiés et plus d’une vingtaine d’anomalies génétiques sont en relation avec les HMN et HSAN.
Les HMSN ou CMT sont les maladies neurodégénératives du système nerveux périphérique les plus courantes, avec une fréquence variable dans les différentes populations, et dont la caractéristique est une hétérogénéité clinique, neuropathologie et génétique.
Cliniquement, la maladie de Charcot Marie Tooth est caractérisée par une perte progressive de la motricité distale des membres inférieurs avec amyotrophie, et de la sensibilité distale associée à des déformations des pieds : pieds creux et orteils déformés en marteau.
L’étude électrophysiologique demeure le premier examen à réaliser pour le diagnostic et la classification des HMSN.
CMT1 et CMT2 sont classiquement transmis selon le mode autosomique dominant, cependant des formes autosomiques récessives ou liées à l’X sont de plus en plus décrites. Les formes dominantes sont plus fréquentes aux États Unis, en Europe de l’ouest et au Japon, alors que dans d’autres pays comme ceux du bassin méditerranéen et notamment d’Afrique du Nord, où la prévalence des mariages consanguins est élevée, les formes autosomiques récessives peuvent représenter plus de 50% des cas (3-4).
L’étude génétique des CMT a débuté en 1991 avec l’identification d’une duplication de 1,4Mb dans le chromosome 17 contenant le gène PMP22 (protéine myélinique périphérique 22) qui est à l’origine, pour une grande part, des formes CMT1 (5, 6).
Depuis ce temps, il y a eu des progrès supplémentaires importants ayant permis la compréhension des bases moléculaires de nombreuses formes de CMT. En effet, de très nombreux gènes sont actuellement connus comme étant impliqués dans les CMT, ce qui a conduit à une meilleure connaissance de la physiopathologie de ces affections et de la biologie cellulaire du système nerveux périphérique.
L’étude neuropathologique qui s’effectue sur des biopsies nerveuses d’un nerf sensitif comme le sural ou le péronier superficiel, plus récemment sur des biopsies de peau, et qui peut montrer les lésions de neurodégénération des fibres nerveuses, a contribué grandement à la compréhension des mécanismes pathophysiologiques des mutations génétiques à l’origine des CMT. Cependant, le développement des techniques génétiques modernes permettant l’identification de mutations de gènes connus ou de gènes candidats, a modifié l’approche diagnostique des CMT. Actuellement, la biopsie nerveuse est rarement indiquée pour le diagnostic des neuropathies héréditaires. Néanmoins, la biopsie nerveuse et l’analyse des fibres nerveuses des biopsies cutanées sont utiles dans une perspective de recherche et d’études de fonctionnalité couplée à une corrélation avec les altérations de gènes découverts dans l’exome ou le génome de patients, grâce aux nouvelles méthodes de séquençage de l’ADN à large échelle.
Phénotype classique des CMT et classification
Comme indiqué précédemment, les CMT sont un groupe hétérogène cliniquement et génétiquement, l’âge de début, l’évolution clinique et les résultats électrophysiologiques étant variables selon les différentes formes clinico-génétiques. Dans la majorité des cas, l’évolution des troubles est lente avec un âge de début habituellement dans la première ou deuxième décade.
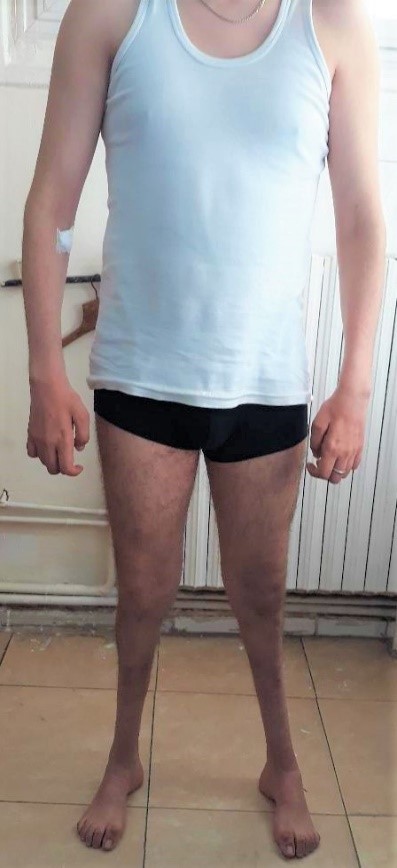
Le phénotype commun comprend un steppage à la marche, des pieds creux, une hypoesthésie distale et une amyotrophie distale des membres inférieurs donnant l’aspect de « jambes de coq ». Les membres supérieurs sont atteints plus tardivement avec une amyotrophie progressive pouvant aboutir à un aspect de « mains en griffe ». (Photo 1 et 2)
Photo1 : Amyotrophie des jambes avec aspect en « jambes de coq ». Amyotrophie des mains et rétraction réductible des doigts.
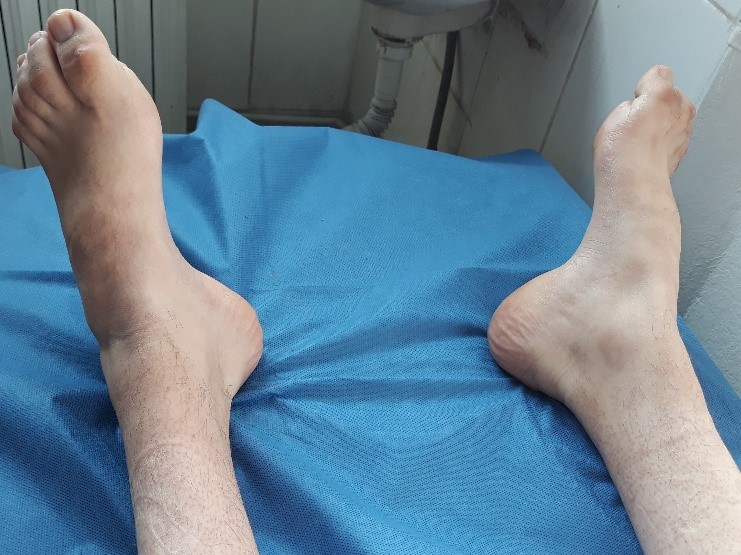
Photo 2 : Pieds creux avec aspect des orteils en marteau.
L’examen neurologique révèle une diminution ou une abolition des réflexes avec une atteinte motrice et sensitive distale symétrique. La marche et la stabilité sont habituellement affectées en raison des troubles proprioceptifs et des déformations squelettiques (pieds creux et orteils en marteau, voir photo 2).
L’étude des conductions nerveuses (VCN) permet de classer les CMT en formes démyélinisantes, intermédiaires ou axonales. Les VCN des membres supérieurs et les amplitudes des potentiels d’action musculaire (CMAP) ainsi que les potentiels sensitifs sont nécessaires alors que les nerfs des membres inférieurs sont souvent inexcitables.
Quand la vitesse de conduction motrice (VCM) du médian est <38m/s, la forme démyélinisante ou CMT1 est diagnostiquée, alors qu’une VCM du nerf médian >38m/s associée à des CMAP réduites avec absence des potentiels d’action sensitifs conduisent au diagnostic de la forme axonale ou CMT2. La forme intermédiaire ou CMT I est diagnostiquée quand la VCM du médian se situe entre 38 et 45m/s (en pratique, ces chiffres pouvant aller au-delà de ces limites). L’électromyographie de détection met en évidence des signes de dénervation chronique diffus aux quatre membres.
Certains enfants atteints de CMT ont un tableau clinique sévère avec un retard de la marche et des déformations orthopédiques des pieds et souvent du rachis et une VCM du médian très réduite (15m/s). Ces formes précoces ont été classées CMT3 et décrites auparavant comme syndrome de Déjerine-Sottas (DSS) ou neuropathie congénitale avec hypomyélination (CHN).
Actuellement, la sévérité de la neuropathie peut être évaluée par le score CMT comportant 9 paramètres cliniques et électrophysiologiques (7).
La classification des CMT est également basée sur le mode de transmission génétique. CMT1 et CMT2 sont de transmission autosomique dominante, CMTX est liée à l’X. Les formes autosomiques récessives sont classées CMT4 ou ARCMT1 pour les formes démyélinisantes et ARCMT2 pour les formes axonales ; sachant qu’une transmission autosomique récessive est fortement suspectée lorsqu’il y a au moins deux enfants atteints d’une même fratrie issue de parents consanguins indemnes.
Ces différentes formes de CMT sont subdivisées selon l’anomalie génétique constatée. Actuellement plus de 80 gènes, dont la mutation peut entrainer un phénotype CMT, sont identifiés. Cependant, des études récentes ont montré qu’environ 90% des diagnostics moléculaires dans les populations occidentales concernaient des mutations ou réarrangements dans les gènes PMP22 (CMT1A), GJB1 (CMTX1), MPZ (CMT1B) et MFN2 (CMT2A) dans l’ordre décroissant de fréquence, alors que les mutations des autres gènes sont relativement beaucoup plus rares (8-9). Dans les formes axonales, les mutations des gènes LMNA, GDAP1et SH3TC1 se sont révélées relativement fréquentes dans les populations du bassin méditerranéen et Maghrébines (10).
Certains patients avec des tableaux de neuropathies congénitales (DSS ou CHN) peuvent avoir des mutations dans les gènes dominants ou récessifs tels que MPZ, PMP22, EGR2, GDAP1, MTPR2 et PRX (11).
Le diagnostic de neuropathie héréditaire est plus difficile à établir en l’absence d’histoire familiale ; néanmoins ce diagnostic est évoqué devant des troubles symétriques à début infantile ou juvénile d’une neuropathie distale lentement progressive associée à des pieds creux. L’électrodiagnostic contribue également au diagnostic en montrant une réduction uniforme de toutes les vitesses de conduction nerveuse, par opposition aux neuropathies acquises où les VCM sont réduites de façon asymétrique avec des signes de dispersion temporelle ou des blocs de conduction.
La biopsie nerveuse n’est pas recommandée dans les formes ADCMT1 et les CMT liés à l’X mais peut être considérée comme un moyen nécessaire pour mieux caractériser la neuropathie dans certaines formes non communes (CMT2 et ARCMT) afin d’orienter les tests moléculaires. En effet, les anomalies histologiques peuvent être suffisamment caractéristiques de l’anomalie génétique sous-jacente, comme celles rencontrées dans les mutations MPZ, GJB1, PRX, FGD4 et LMNA.
En définitive, l’orientation des tests génétiques est avant tout basée sur le phénotype clinique, l’électrodiagnostic et le mode de transmission héréditaire. Un algorithme de diagnostic CMT devrait être basé en premier lieu sur l’étude électrophysiologique avec les vitesses de conduction nerveuses et l’électromyographie pour établir si la neuropathie est démyélinisante ou axonale, en second lieu sur l’histoire familiale et l’arbre généalogique de façon à déterminer si le mode de transmission héréditaire est AD, AR ou lié à l’X. Ces données permettent de planifier ensuite l’étude génétique par la recherche de mutations des gènes correspondants les plus fréquents, à savoir PMP22, GJB1, MPZ, MFN2 et SH3TC2, et pour notre région, LMNA et GDAP1, et de rechercher les plus rares, éventuellement, dans un deuxième temps. En fait, nous rencontrons dans nos régions toutes les formes de CMT, que ce soit les dominantes, liées à l’X ou intermédiaires. Cependant, les formes AR y sont aussi fréquentes, sinon plus, que les autres formes, alors qu’elles sont très rares voire inexistantes dans les pays où la consanguinité est rarissime. Dans cet article nous détaillerons les formes prévalentes au bassin méditerranéen et notamment en Afrique du Nord.
Actuellement, les équipes de diagnostic génétique les plus performantes utilisent les méthodes de séquençage à grande échelle comme le séquençage haut débit appelé next generation sequencing (NGS), et le séquençage à haut débit de l’exome. En effet, l’exome (qui désigne tous les exons ou séquences codantes) de plusieurs patients peut être séquencé simultanément. Plusieurs gènes ciblés ou panel de gènes pour de nombreux patients peuvent être séquencés en une fois. Alors qu’auparavant le séquençage était fastidieux et « time consuming », se faisant gène par gène à la recherche de mutation. Ces nouvelles techniques, y compris celle de bio-informatique, considérées maintenant comme étant les meilleures méthodes d’exploration génétique des CMT et des autres affections neurogénétiques, ont permis la découverte de nombreux nouveaux gènes ces dernières années, permettant d’accroitre la connaissance de nouveaux mécanismes physiopathologiques de ces affections qui conduiront certainement à de nouvelles approches thérapeutiques.
Des mécanismes et voies moléculaires très variés ont été mis en évidence dans les neuropathies héréditaires grâce au nombre élevé des gènes et protéines altérés identifiés. Les structures myéliniques, la dynamique mitochondriale, la régulation transcriptionnelle, le turnover protéique ainsi que le transport axonal sont ainsi impliqués dans les différentes formes de CMT, permettant d’explorer de nouvelles stratégies thérapeutiques, malgré leur très grande complexité physiopathologique.
Caractéristiques cliniques et pathologiques des différentes formes de CMT :
Les CMT autosomiques dominants
Les CMT dominants sont les formes les plus fréquentes en général, notamment dans les pays occidentaux.
Les formes AD-CMT1 (démyélinisantes) sont les plus communes (80% des cas). La plupart des patients ont un phénotype CMT classique avec une VCM sur le nerf médian inférieure à 38m/s, des signes neuropathologiques comprenant une réduction importante des axones myélinisés et un aspect en « bulbes d’oignon » des cellules de Schwann (neuropathie hypertrophique) recouvrant des axones entourés d’une myéline fine (Figure). Cliniquement, cette neuropathie hypertrophique peut être diagnostiquée en palpant un gros nerf comme le cubital au coude. À l’heure actuelle, les sous-types CMT1 sont classés de CMT1A à CMT1G. De nouveaux gènes CMT1 sont apparus récemment : FBLN5 et c1orf194 . (Voir le lien https://neuromuscular.wustl.edu/time/hmsn.html pour les détails cliniques correspondant aux CMT1).
Une nouvelle classification proposant de remplacer les lettres par le nom du gène impliqué et prenant en compte la forme démyélinisante (De), axonale (Ax) ou intermédiaire (In), ainsi que l’hérédité (AD, liée à l’X ou AR) a été publiée récemment par Mathis et al (12), et reprise par d’autres auteurs experts dans le domaine (13, 14). L’argument principal pour changer la classification étant que les lettres de l’alphabet ne suffisent plus pour le nombre de gènes impliqués sans cesse croissant. Ainsi, CMT1A devient pour ce système de classification : AD-CMTDe-PMP22. L’avantage d’une telle classification c’est la suppression des lettres et des chiffres remplacés par les noms des gènes et la pathologie (démyélinisante ou axonale).
La plupart des patients AD-CMT1 (ou AD-CMTDe) ont un handicap bénin à modéré interférant peu avec une vie active, peu d’entre eux évoluant vers la chaise roulante. Rarement le phénotype est sévère avec un tableau de neuropathie congénitale type DSS.
Figure : Microscopie électronique du nerf sural, section transverse. Les axones myélinisés sont entourés de proliférations de cellules de Schwann en bulbes d’oignons. (Etoile : un axone entouré de myéline anormalement fine). Photo JM Vallat, ref. 18.
Les formes AD-CMT2 (ou AD-CMTAx) sont relativement fréquentes pouvant représenter le tiers des formes dominantes. La plus fréquente est CMT2A (AD-CMTAx-MFN2), liée au gène de la mitofusine (MFN2), protéine mitochondriale, dans laquelle on peut observer un phénotype CMT classique avec des VCM du nerf médian supérieures à 45m/s, des potentiels sensitifs souvent absents, mais avec parfois des signes d’atteinte du système nerveux central comme des réflexes vifs, un signe de Babinski ou une atrophie optique. La biopsie nerveuse montre une raréfaction neuronale avec des signes de régénération (axonal sprouting), sans signe de démyélinisation. La plupart des CMT2A sont sévèrement atteints et perdent la marche à l’âge adulte.
En dehors des AD-CMTAx-MFN2 ou CMT2A, les autres sous-types sont très rares, décrits dans une ou deux familles. Elles figurent néanmoins dans la classification actuelle jusqu’à CMT2Z lié au gène MORC2, prenant la dernière lettre de l’alphabet, montant ainsi les limites de cette classification. (Voir le lien https://neuromuscular.wustl.edu/time/hmsn.htmlpour les détails cliniques correspondant à chaque forme de CMT2)
Certaines formes de CMT2 comme CMT2C (TRPV4), CMT2D (GARS), CMT2F (HSPB1) et CMT2L (HSPB8) sont alléliques à certaines formes de neuropathies motrices héréditaires distales, confortant l’hypothèse d’un continuum entre les CMT axonales et les HMN distales (15).
Les CMT liés à l’X
Un CMT lié à l’X est suspecté lorsque la transmission père-fils est absente sur les arbres généalogiques et quand les patients males sont plus sévèrement atteints (16). Quatre formes dominantes et récessives de CMT liés à l’X ont été décrites. CMTX1 lié à une mutation du gène GJB1 encodant la connexine 32 (Cx32), est la forme la plus fréquente après CMT1A dans les cohortes américaines et européennes.
Les CMT dominants intermédiaires (DI-CMT) sont caractérisés par un phénotype bénin à modérément sévère et des VCM du médian allant de 25 à 50m/s (17). Les formes intermédiaires affectent la myéline et l’axone, ce qui met en exergue la relation étroite entre ces deux structures du nerf périphérique. Six formes cliniques ont été identifiées (Voir classification des CMT intermédiaires dans le lien : https://neuromuscular.wustl.edu/time/hmsn.html)
Les CMT autosomiques récessifs.
Les CMT autosomiques récessifs (AR-CMT) sont décrits dans les pays où les mariages consanguins sont fréquents (pays du pourtour méditerranéen, du Moyen-Orient et les populations Roms d’Europe).
La plupart des cas AR-CMT sont caractérisés par un début précoce de la maladie et une progression clinique rapide conduisant à des déformations orthopédiques (pieds varus équin, mains déformées Aran-Duchenne et déformations vertébrales majeures) et un arrêt précoce de la marche.
Les CMT autosomiques récessifs comprennent des formes démyélinisantes (AR-CMT1 ou CMT4) avec des VCM <38m/s, et les formes axonales (AR-CMT2) avec des VCM >45m/s aux membres supérieurs, des amplitudes réduites des potentiels moteurs ainsi que des potentiels sensitifs réduits ou absents aux quatre membres. La biopsie nerveuse dans les AR-CMT1 montre une démyélinisation importante avec parfois des anomalies spécifiques de la myéline et une perte axonale secondaire, alors que dans les AR-CMT2 les fibres myélinisées sont très réduites en nombre sans signes évidents de démyélinisation et remyélinisation ou de régénération axonale active.
Au plan génétique, plusieurs types de ARCMT ont été identifiés, avec des caractéristiques cliniques, pathologiques et ethniques propres (18). On peut consulter des formes rares nouvellement identifiées dans le site : https://neuromuscular.wustl.edu/time/hmsn.html.
Le diagnostic moléculaire des AR-CMT est plus complexe du fait de la faible fréquence des gènes identifiés et de leur grande hétérogénéité. Cependant, ce diagnostic génétique peut être orienté par les caractéristiques épidémiologiques, cliniques et pathologiques. Certaines formes de AR-CMT comme celles associées aux gènes récessifs NGRD-1 et LMNA sont dues à des mutations fondatrices dans des populations spécifiques, Roms (Gitans) en Europe et du Maghreb, respectivement ; alors que d’autres formes liées aux gènes GDAP1 et SH3TC2 sont décrites dans tous les pays méditerranéens (19-20), y compris en Algérie : voir la thèse de DESM de Ameur El Khoudoud, Université d’Alger.
AR-CMT1A (CMT4A) correspondant au premier locus AR décrit, lié au gène GDAP1 codant pour une protéine de la membrane mitochondriale, est caractérisé par un début précoce et une neuropathie grave et des déformations distales des membres conduisant à un arrêt de la marche à l’adolescence. Cette forme de CMT liée au gène GDAP1 peut avoir aussi une transmission AD avec un phénotype moins sévère (21).
D’autres formes de AR-CMT telles que celles associées à des anomalies de la myotubularine (MTMR2 et MTMR13) donnent des tableaux de CMT sévères, caractérisées par une myéline redondante avec des outfoldings et infoldings à la biopsie nerveuse, pouvant orienter le diagnostic moléculaire, lorsqu’elle est possible, ce qui n’est pas évident. Autrement, dans certaines formes, des particularités cliniques comme une cyphoscoliose au premier plan et une hypoacousie, (AR-CMT1C, SH3TC2), une parésie des cordes vocales avec une respiration stertoreuse (AR-CMT1A, GDAP1), une ataxie sensitive avec des troubles moteurs discrets et des VCM très réduites (AR-CMT1F, PRX), permettent d’orienter le diagnostic génétique.
Parmi les formes axonales de ARCMT, la forme ARCMT2A, ou AR-CMTAx-LMNA (selon la classification récemment proposée mettant en évidence le gène impliqué à la place des lettres de l’alphabet), identifiée dans une famille marocaine de neuf membres et dont la mutation fondatrice c.892C>T (p. Arg298Cys) du gène LMNA a été par la suite caractérisée chez des familles algériennes, est une forme de CMT axonale qui débute entre 11 et 14 ans et qui s’aggrave assez rapidement avec une atteinte précoce des muscles proximaux des membres inférieurs rendant la marche de plus en plus difficile avec steppage et dandinement (22-23). Certains patients atteignent le stade de la chaise roulante après 15 à 20 ans d’évolution mais d’autres peuvent rester ambulants plus longtemps (23). Cette mutation fondatrice prédomine dans une région du Maghreb située entre le Centre et l’Ouest de l’Algérie et le Maroc. L’ancêtre commun le plus récent aurait vécu il y a 800 à 900 ans (24).
Mesures thérapeutiques
Actuellement, aucun traitement spécifique n’est disponible pour guérir ou arrêter la progression des neuropathies héréditaires, en sachant que des essais thérapeutiques divers notamment de thérapies géniques, sont en cours d’étude ; l’un des plus récents essais médicamenteux étant l’essai au PXT3003, une combinaison de trois médications déjà connues (baclofene, naltrexone et D-sorbitol) (25, 26).
Cependant, il est bien reconnu que les traitements symptomatiques sont nécessaires, entre autres pour corriger des déformations squelettiques et ainsi améliorer la qualité de vie du patient. La physiothérapie et les mesures de réadaptation demeurent les meilleures approches thérapeutiques pour les patients qui présentent un phénotype classique de CMT. La kinésithérapie est indispensable pour lutter contre l’atrophie musculaire et surtout les rétractions capsulo-tendineuses, permettant ainsi d’éviter, ou du moins retarder et limiter, les déformations articulaires. La prescription de chaussures orthopédiques et/ou des orthèses spécifiques (releveurs du pied le plus souvent) est fortement recommandée, ainsi que des équipements disponibles adaptés aux déformations des mains.
La prise en charge des déformations orthopédiques des pieds et de la colonne vertébrale nécessite parfois un recours à la chirurgie.
La paralysie des cordes vocales que l’on peut observer dans certaines formes de ARCMT (GDAP1, MTMR2) peut bénéficier d’un traitement au laser efficace, réduisant de façon satisfaisante les troubles de la phonation et de la respiration (27).
La douleur et la dépression peuvent être présentes dans certaines formes de CMT, pouvant nuire à la qualité de vie du patient. Les douleurs chroniques sont souvent associées à ces neuropathies et peuvent être liées à une origine musculo-squelettique ou neuropathique. Dans ces cas des traitements symptomatiques comme la thérapie physique, les interventions orthopédiques, le soutien psychologique et les médicaments s’avèrent souvent très utiles.
Conclusion
Le diagnostic positif des CMT est relativement aisé, reposant sur le tableau clinique et l’électrophysiologie ainsi que l’évolution lentement progressive. Le diagnostic des différentes formes cliniques repose sur l’électrodiagnostic mais aussi sur le type de transmission héréditaire et le testing moléculaire. Celui-ci est nettement facilité par les méthodes modernes de diagnostic génétique comme le NGS, le séquençage d’exome et l’analyse bio-informatique.
Dans la littérature européenne et nord-américaine les formes dominantes et liées à l’X représentent plus de 80% des cas, alors que dans les pays comme l’Algérie et les autres pays du Maghreb où le taux de consanguinité est encore important, les formes autosomiques récessives représentent plus de 50% des cas.
La prise en charge des patients et des familles atteintes de neuropathies héréditaires consiste à effectuer un conseil génétique, et surtout à programmer une physiothérapie au long cours avec des spécialistes en médecine physique pour prévenir et traiter les complications orthopédiques de ces affections, en attendant les thérapies curatrices spécifiques.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Photos : Originales. Collection personnelle de l’auteure.
Références
Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular Neurologic, genetic, and electrophysiological findings in hereditary polyneuropathies. Arch Neurol 1968; 18:603–8.
Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980; 103:259–80.
Harding AE, Thomas PK. Autosomal recessive forms of hereditary motor and sensory neuropathy. J NeurolNeurosurgPsychiatry1980; 43:669–78.
Dubourg O, Azzedine H, Verny C, Durosier G, Birouk N, Gouider R, et al. Autosomal recessive forms of demyelinating Charcot–Marie–Tooth disease. Neuro molecular Med 2006; 8:75–86.
Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, et al. DNA duplication associated with Charcot–Marie–Tooth disease type 1A. Cell 1991;66:219–32.
Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, Hoogendijk JE, Baas F, et al. Duplication in chromosome 17p11.2 in Charcot–Marie–Tooth neuropathy type 1a (CMT 1a). Neuromuscul Disord 1991; 1:93–7.
Murphy SM, Herrmann DN, McDermott MP, Scherer SS, Shy ME, Reilly MM, et al. Reliability of the CMT neuropathy score (second version) in Charcot–Marie–Tooth disease. J Peripher Nerv Syst 2011; 16:191–8.
Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot–Marie–Tooth disease subtypes and genetic testing strategies. Ann Neurol 2011; 69:22–33.
Gess B, Schirmacher A, Boentert M, Young P. Charcot–Marie–Tooth disease: frequency of genetic subtypes in a German neuromuscular center population. Neuromuscular Disorder 2013; 23:647–51.
Tazir M, Bellatache M, Nouioua S, Vallat JM. Autosomal recessive Charcot Marie Tooth disease: from genes to phenotypes. J Peripher NervSyst. 2013;18(2):113–129.
El-Abassi R, England JD, Carter GT. Charcot–Marie–Tooth disease: an overview of genotypes, phenotypes, and clinical management strategies. PMR 2014; 6:342–55.
Mathis S, Goizet C, Tazir M, Magdelaine C, Lia AS, Magy L, et al. Charcot Marie Tooth diseases: an update and some new proposals for the classification. J Med Genet 2015;52(10):681–90
Magy L, Mathis S, Le Masson G, Goizet C, Tazir M, Vallat JM. Updating the classification of inherited neuropathies: results of an international survey. Neurology. 2018;90:e870–6
Bird TD. Charcot Marie Tooth (CMT) Hereditary Neuropathy 1998 Sep 28 [updated 2020 Jan 2. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. Gene Reviews. Seattle (WA): University of Washington, Seattle; 1993-2020
Solla P, Vannelli A, Bolino A, Marrosu G, Coviello S, Murru MR, et al. Heat shock protein 27 R127W mutation: evidence of a continuum between axonal Charcot–Marie–Tooth and distal hereditary motor neuropathy. J Neurol Neurosurg Psychiatry 2010; 81:958–62.
Boerkoel CF, Takashima H, Garcia CA, Olney RK, Johnson J, Berry K, et al. Charcot–Marie–Tooth disease and related neuropathies: mutation distribution and genotype–phenotype correlation. Ann Neurol 2002; 51:190–201.
Nicholson G, Myers S. Intermediate forms of Charcot–Marie–Tooth neuropathy: a review. Neuromolecular Med 2006; 8:123–30.
Tazir M, Hamadouche T, Nouioua S, Mathis S, Vallat JM. Hereditary motor and sensory neuropathies or Charcot Marie Tooth diseases: an update. J Neurol Sci. 2014;347(1-2):14–22
Kalaydjieva L, Gresham D, Gooding R, Heather L, Baas F, de Jonge R, et al. N-myc downstream-regulated gene 1 is mutated in hereditary motor and sensory neuropathy-Lom. Am J Hum Genet 2000; 67:47–58.
Gosselin I, Thiffault I, Tétreault M, Chau V, Dicaire MJ, Loisel L, et al. Founder SH3TC mutations are responsible for a CMT4C French-Canadians cluster. Neuromuscul Disord 2008; 18:483–92.
Sivera R, Frasquet M, Lupo V, et al. Distribution and genotype-phenotype correlation of GDAP1 mutations in Spain. Sci Rep. 2017;7(1):6677.)
Bouhouche A, Benomar A, Birouk N, et al. A locus for an axonal form of autosomal recessive Charcot Marie Tooth disease maps to chromosome 1q21.2-q21.3. Am J Hum Genet 1999; 65: 722-727.
Tazir M, Azzedine H, Assami S, et al. Phenotypic variability in autosomal recessive axonal Charcot Marie Tooth disease due to the R298C mutation in lamin A/C. Brain. 2004;127(Pt 1):154–163.
Hamadouche T, Poitelon Y, Genin E, et al. Founder effect and estimation of the age of the c.892C>T (p.Arg298Cys) mutation in LMNA associated to Charcot Marie Tooth subtype CMT2B1 in families from North Western Africa. Ann Hum Genet 2008; 72: 590-597.
Attarian S, Dubourg O, Funalot B, Gonnaud PM, Lacour A, Magy L, et al. A phase II randomized, placebo-controlled multicenter clinical trial of three doses of PXT3003 in 80adult patients with CMT1A treated for 1 year. J Peripher Nerv Syst 2013; 18(Suppl.): S7-8.
Morena J, Gupta A, Hoyle JC. Charcot-Marie-Tooth: From Molecules to Therapy. Int J Mol Sci. 2019;20(14):3419.
Nouioua S, Hamadouche T, Funalot B, et al. Novel mutations in the PRX and the MTMR2 genes are responsible for unusual Charcot Marie Tooth disease phenotypes. Neuromuscul Disord. 2011;21(8):543–550.
L’atrophie multisystématisée (AMS) est une pathologie neurodégénérative rare, sporadique, survenant le plus souvent dans la sixième décennie avec une prédominance masculine.Atrophie multisystématisée
W.Amer El Khedoud, A. Saadi, A. Benhaddadi, B. Mammeri, S. Lougani, N. Kassouri, F. Ferrat, Service de Neurologie, EHS Ben Aknoun, Alger
Date de soumission : 10 Février 2020.
Abstract: Multiple system atrophy (MSA) is a rare and sporadic, progressive neurodegenerative disorder which occurs most frequently in the sixth decade with a male predominance, characterized by progressive autonomic failure, parkinsonian features, and cerebellar and pyramidal features in various combinations. Patients are classified as MSA-C or MSA-P depending on the predominance of cerebellar ataxia or parkinsonism. It is considered to be one of the two main etiologies of atypical parkinsonian syndrome with progressive supranuclear palsy (PSP). It is classified among “synucleinopathies” such as idiopathic Parkinson’s disease (MPI) and dementia with Lewy bodies (DCL). AMS is the most common atypical neurodegenerative parkinsonian syndrome. Their prognosis is worse than that of idiopathic Parkinson’s disease (DPI). Half of the patients are in a wheelchair after 5 years, with an average survival of less than 10 years. In a consensus conference on diagnosis held in 1998 and reviewed in 2008, three levels of certainty were established, possible, probable, and definite MSA, with the diagnosis of definite MSA requiring autopsy confirmation. We report the clinical and radiological study of three cases of MSA.
Résumé : L’atrophie multisystématisée (AMS) est une pathologie neurodégénérative rare, sporadique, survenant le plus souvent dans la sixième décennie avec une prédominance masculine. Sur le plan clinique, l’AMS se caractérise par l’association variable d’un syndrome Parkinsonien peu dopa sensible, d’un syndrome cérébelleux, d’un syndrome pyramidal et de troubles dysautonomiques. La prédominance d’un des deux syndromes, Parkinsonien ou cérébelleux, permet de les classer en deux formes cliniques AMS-P et AMS-C. Elle est considérée comme l’une des deux principales étiologies de syndrome Parkinsonien atypique avec la paralysie supranucléaire progressive (PSP). Elle est classée parmi les “synucléinopathies”, comme la maladie de Parkinson idiopathique (MPI) et la démence à corps de Lewy (DCL). L’AMS est le syndrome Parkinsonien atypique neurodégénératif le plus fréquent. Son pronostic est plus sombre que celui de la maladie de Parkinson idiopathique (MPI). La moitié des patients est en fauteuil roulant après 5 ans, avec une survie médiane inférieure à 10 ans. L’AMS se diagnostique à partir de critères cliniques définissant un niveau « probable » ou « possible » établis par Gilman et al, en 1998 et révisés en 2008. Le diagnostic de certitude repose sur une confirmation anatomopathologique. Nous rapportons l’étude clinique et radiologique de trois cas d’AMS.
L’atrophie multisystématisée (AMS) est une pathologie neurodégénérative rare, sporadique, survenant le plus souvent dans la sixième décennie avec une prédominance masculine [1,2]. Sur le plan clinique, l’AMS se caractérise par l’association variable d’un syndrome Parkinsonien peu dopasensible, d’un syndrome cérébelleux, d’un syndrome pyramidal et de troubles dysautonomiques. La prédominance d’un des deux syndromes, Parkinsonien ou cérébelleux, permet de les classer en deux formes cliniques AMS-P et AMS-C [3,4]. Elle est considérée comme l’une des deux principales étiologies de syndrome Parkinsonien atypique avec la paralysie supranucléaire progressive (PSP). Elle est classée parmi les “synucléinopathies”, comme la maladie de Parkinson idiopathique (MPI) et la démence à corps de Lewy (DCL). L’AMS est le syndrome Parkinsonien atypique neurodégénératif le plus fréquent. Elle peut être parfois confondue avec la maladie de Parkinson idiopathique (MPI) notamment parce que certaines formes d’AMS sont dopasensibles. La prévalence de l’AMS est faible, 10 fois moindre que la MPI.
Son pronostic est plus sombre que celui de la maladie de Parkinson idiopathique (MPI), la moitié des patients est en fauteuil roulant après 5 ans, avec une survie médiane inférieure à 10 ans [2].
Les facteurs de mauvais pronostic seraient l’apparition précoce de troubles urinaires, la présence de signes dysautonomiques sévères ou d’un stridor [5]. Les causes de décès les plus fréquentes sont les infections broncho-pulmonaires et les morts subites [6].
L’AMS se diagnostique à partir de critères cliniques définissant un niveau « probable » ou « possible » établis par Gilman et al, en 1998 et révisés en 2008 [3,4]. Le diagnostic de certitude repose sur une confirmation anatomopathologique retrouvant notamment des inclusions oligodendrogliales intracytoplasmiques (ou glial cytoplasmic inclusion [GCI]) majoritairement constituées d’alpha-synucléine [7] présentes dans l’ensemble du système nerveux central avec une forte densité dans les régions appartenant au système olivo-ponto-cérébelleux et aux boucles motrices cortico-striato-corticales [8].
Cas Cliniques
Figure 01
Figure 02
Figure 03
Cas 1 : Mme G.H.F. âgée de 50 ans, sans antécédents particuliers, a présenté une lourdeur des membres inférieurs, suivie un an après par l’installation d’un déséquilibre à la marche avec chutes fréquentes, incontinence urinaire et lenteur à l’élocution. L’évolution s’est faite vers l’aggravation progressive avec arrêt de la marche au bout de deux ans confinant la patiente au fauteuil roulant.
L’examen neurologique retrouve un syndrome Parkinsonien akineto-rigide bilatéral et symétrique, (amimie, rareté du clignement, hypokinésie, hypertonie plastique), une dysarthrie mixte cérébelleuse et extrapyramidale, des réflexes ostéo-tendineux exagérés aux 4 membres et un syndrome dysautonomique fait d’incontinence urinaire et d’une hypotension orthostatique.
L’IRM cérébrale a montré des anomalies typiques d’une MSA. Sur les séquences T2 et FLAIR, on note un hypersignal de la bordure postérolatérale du putamen (pointe des flèches rouges) (figure 1,2) et un hypersignal au niveau du pont : début du « signe de la croix » dû à une atrophie des fibres pontiques, qui débute habituellement par l’hypersignal vertical (figure 3).
Cas 2 : Mr D.S. âgé de 62 ans sans antécédents particuliers qui a présenté un déséquilibre à la marche entrainant des chutes, suivi un an plus tard d’une lourdeur aux membres inferieurs associée à des troubles de l’élocution, troubles sexuels et incontinence urinaire.
L’examen neurologique a objectivé un syndrome cérébelleux stato-cinétique très important, un syndrome dysautonomique (troubles mictionnels et hypotension orthostatique : chute de la pression artérielle systolique de 30 mmHg lors du passage de la position couchée à debout), un discret syndrome Parkinsonien akinéto-rigide et des réflexes ostéo-tendineux exagérés aux 4 membres. L’IRM cérébrale est sans anomalie.
Cas 3 : Mr A. A. âgé de 52 ans, a présenté à l’âge de 48 ans des mictions impérieuses associées à une impuissance sexuelle suivie en quelques mois de pertes de connaissance brèves lors du passage rapide de la position assise à la position debout (le bilan cardiaque est sans anomalies), quelques mois après son hospitalisation ; apparition d’une lourdeur aux MI avec déséquilibre à la marche.
Examen neurologique : Syndrome dysautonomique sévère fait d’une hypotension orthostatique symptomatique, une impériosité mictionnelle avec un résidu post mictionnel à l’échographie vésicale, un discret syndrome Parkinsonien akinéto-rigide et des réflexes exagérés aux 4 membres. L’IRM cérébrale est sans anomalies.
Le bilan thyroïdien, carentiel (Vitamine B12, E), sérologies infectieuses, le bilan auto-immun et paranéoplasique étaient négatifs chez les trois patients.
Le bilan neuropsychologique : MMSE et BREF n’ont pas trouvé de déclin cognitif.
Le traitement par la L-Dopa n’a entrainé aucune amélioration sauf pour le cas 3 où la réponse était partielle au début.
Discussion
Le diagnostic de MSA probable selon les critères de Gilman a été retenu chez nos 3 patients devant la présence constante de signes dysautonomiques associés à un syndrome Parkinsonien bilatéral et symétrique ainsi que des réflexes vifs avec des anomalies à l’IRM cérébrale en faveur d’une (MSA-P) chez le 1er cas, et à un syndrome cérébelleux stato-cinétique dominant le tableau clinique avec un discret syndrome Parkinsonien et reflexes vifs chez le 2ème cas, (MSA-C).
Le 3ème cas a présenté des signes dysautonomiques, une hypotension orthostatique symptomatique parfois syncopale, isolée pendant cinq ans entrainant un retard important au diagnostic.
Les critères diagnostiques de l’AMS établis en 1998 (4, 5) ont été récemment revus lors d’une nouvelle conférence de consensus (3, 7). Ces nouveaux critères sont destinés à affiner le diagnostic et à inclure des signes additionnels ainsi que le résultat d’explorations paracliniques.
Le diagnostic d’AMS repose avant tout sur des critères cliniques consensuels [3] au sein desquels des anomalies spécifiques ont été identifiées à l’IRM.
Les critères diagnostiques définissent deux types d’AMS : l’AMS-P où le syndrome Parkinsonien prédomine comme dans notre cas 1 et l’AMS-C où le syndrome cérébelleux est au premier plan comme chez notre patient cas 2.
Par ailleurs, on classe la maladie selon trois niveaux de certitude : AMS « possible » (Tableaux II et III), « probable » (Tableau I) et « certaine » [3,4]. La certitude diagnostique est obtenue à l’examen neuropathologique post-mortem, montrant une dégénérescence des structures olivo-ponto-cérébelleuses et de la voie nigrostriée associée à d’abondantes inclusions gliales intracytoplasmiques d’alpha-synucléine (8).
Nos 3 patients répondent aux critères d’AMS probable. (Tableau I).
Tableau I. Critères pour le diagnostic d’AMS probable.
Maladie de début sporadique, progressive, chez un adulte (> 30 ans), caractérisée par : – une dysautonomie avec incontinence urinaire (associée à une dysfonction érectile chez l’homme) ou hypotension orthostatique (HO) survenant dans les 3 minutes du lever avec chute de pression artérielle (PA) d’au moins 30 mmHg pour la systolique (PAS) ou de 15 mmHg pour la diastolique (PAD) • et un syndrome parkinsonien (bradykinésie avec rigidité, tremblement ou instabilité posturale) peu dopasensible • ou un syndrome cérébelleux (ataxie à la marche avec dysarthrie cérébelleuse, ataxie des membres ou dysfonction oculomotrice cérébelleuse).
Tableau II. Critères pour le diagnostic d’AMS possible.
Maladie de début sporadique, progressive, chez un adulte (> 30 ans), caractérisée par : – un signe suggérant une dysautonomie : mictions impérieuses sans autres explications, vidange vésicale incomplète, dysfonction érectile chez les hommes, ou HO n’ayant pas les critères exigés dans l’AMS probable • et un syndrome parkinsonien (bradykinésie avec rigidité, tremblement ou instabilité posturale) • ou un syndrome cérébelleux (ataxie à la marche avec dysarthrie cérébelleuse, ataxie des membres ou dysfonction oculomotrice cérébelleuse) • et au moins un des critères additionnels du tableau III
Les signes cliniques principaux
Le syndrome Parkinsonien, plus fréquemment de forme akinéto-rigide, est dit « atypique » car il ne répond pas ou peu au traitement dopaminergique, et est associé à d’autres signes qui sont inhabituels dans la maladie de Parkinson idiopathique (MPI) comme l’instabilité posturale et les chutes précoces chez nos 3 patients ; la progression rapide vers le fauteuil roulant pour le cas 1 et le confinement au lit pour le cas 3 ainsi que la dysarthrie pour les cas 1 et 2 (signes considérés comme des drapeaux rouges dans la MPI et en faveur d’une AMS-P) (tableau 3). Néanmoins, une dopasensibilité est retrouvée chez environ un tiers des patients avec une excellente dopasensibilité en début de maladie pour 10 % d’entre eux comme chez notre patient, cas 3, qui a répondu partiellement à la L-dopa.
Le syndrome cérébelleux est au premier plan dans l’AMS-C, plus tardif et inconstant dans l’AMS-P où il est parfois difficile à distinguer des troubles posturaux du syndrome Parkinsonien. Notre patient cas 2 a présenté un syndrome cérébelleux au 1er plan associé à un discret syndrome Parkinsonien en faveur d’une AMS-C.
La dysautonomie est quasi constante, précoce et sévère contrairement à la MPI ou à la paralysie supra nucléaire progressive (PSP). Elle est un pilier central pour le diagnostic, son dépistage est donc primordial.
La dysautonomie, définie dans les critères consensuels, comprend une hypotension orthostatique (HO) et/ou des troubles vésico-sphinctériens.
L’Hypotension orthostatique est définie par une chute de la pression artérielle systolique d’au moins 20 mm d’Hg ou une baisse de la pression artérielle diastolique d’au moins 10 mm d’Hg, enregistrée dans les 3 minutes suivant le passage à l’orthostatisme comme ce fut le cas chez nos patients 1 et 2 alors qu’elle était symptomatique pour le cas 3.
Les troubles vésicosphinctériens sont souvent inauguraux dans l’AMS et précèdent généralement l’HO (10, 11). Les manifestations cliniques d’hyperactivité vésicale sont fréquentes au début de la maladie (urgences mictionnelles, impériosités, pollakiurie, fuites par impériosités) [12-15]. L’incontinence urinaire est le symptôme le plus fréquemment rapporté (13, 16, 18, 19) comme ce fut le cas chez nos trois patients.
L’impuissance est souvent précoce, et elle est quasi constante chez l’homme (16, 18).
Les autres signes de dysautonomie sont la dysfonction érectile, retrouvée chez nos 2 patients cas 2 et 3 et les troubles gastro-intestinaux dominés par la constipation.
Il existe aussi des troubles de la thermorégulation avec une intolérance à la chaleur ou la présence de mains froides et violacées.
La présence d’un syndrome pyramidal est retrouvée chez environ 50 % des patients mais il n’est jamais au premier plan du tableau clinique. Chez nos 3 patients on a retrouvé des réflexes ostéo- tendineux vifs aux 4 membres.
La dysarthrie est précoce, mixte (parkinsonienne et cérébelleuse), retrouvée chez nos patients 1 et 2 ; elle s’associe souvent à des troubles de la déglutition et une dysphonie. Ces troubles sont pourvoyeurs de pneumopathie de déglutition qui sont une des principales causes de décès [6].
Les troubles cognitifs sont le plus souvent modérés, sous la forme d’anomalies de nature sous-cortico-frontale touchant les tâches attentionnelles, l’exécution, la planification et la mémoire de travail. Ils se rapprochent plus de ceux rencontrés dans la MPI et semblent plus sévères dans les AMS-P.
Pour nos 3 patients ; il n’a pas été retrouvé de troubles cognitifs.
L’imagerie est actuellement incluse dans le diagnostic d’AMS possible (3-20).
L’IRM cérébrale et l’imagerie fonctionnelle peuvent apporter des arguments supplémentaires pour le diagnostic d’AMS. L’IRM cérébrale peut montrer une atrophie putaminale, pontique et des pédoncules cérébelleux moyens. Sur les séquences pondérées en T2, on retrouve souvent un hyposignal de la partie postérieure du putamen, parfois associé à un hypersignal de la bordure postérolatérale du putamen, un hypersignal en forme de « croix » pontique et parfois des hypersignaux floconneux des pédoncules cérébelleux moyens [21] comme retrouvé chez notre patient cas 1.
Elle peut cependant être normale au début de la maladie.
Les études en tomographie par émission de positons (TEP) avec le 18 F-fluorodésoxyglucose (FDG) ont montré un hypométabolisme au niveau du putamen et du cervelet [22].
Chez un patient présentant un syndrome Parkinsonien atypique sans syndrome cérébelleux, la démonstration en TEP-FDG d’un hypométabolisme cérébelleux permet le diagnostic d’une AMS-P « possible » (Tableau II).
De même, chez un patient présentant un syndrome cérébelleux sans syndrome Parkinsonien, l’observation d’une dénervation dopaminergique nigrostriée par TEP ou tomographie d’émission monophotonique (TEMP) (DATSCAN)), est en faveur d’un diagnostic d’AMS-C « possible » [23]. Ces examens ne sont cependant pas des examens de routine.
Un tiers des patients AMS répond au traitement par lévodopa mais souvent de manière transitoire. Une réponse excellente à la lévodopa est néanmoins retrouvée chez 10 % des patients en début de maladie [24]. Les effets secondaires à type d’hypotension orthostatique sont des facteurs limitant à l’augmentation du traitement.
Un traitement à base de L-dopa a été institué chez nos 3 patients, il n’y a pas eu de réponse pour les cas 1 et 2, et une réponse partielle pour le cas 3.
Il n’existe pas de traitements validés pour agir sur le syndrome cérébelleux.
Une prise en charge rééducative est indispensable au décours de l’AMS afin de faciliter les déplacements et de prévenir les chutes.
Une prise en charge multidisciplinaire (cardiovasculaire et réadaptative) de l’hypotension orthostatique et des troubles vésicosphinctériens (24, 25) permet d’améliorer le quotidien des patients. Pour l’hypotension orthostatique, nos patients ont bénéficié de conseils hygiéno-diététiques : une bonne hydratation, un régime riche en sel, le port de bas de contention dès le lever et dormir avec une surélévation de la tête de 30°. Ils ont été adressés en cardiologie pour prise en charge de leur hypotension orthostatique et en rééducation fonctionnelle pour exploration et prise en charge de leurs troubles vésicosphinctériens.
Conclusion
L’AMS est une affection neurodégénérative dont le diagnostic reste essentiellement clinique. Les examens d’imagerie morphologique et fonctionnelle peuvent toutefois permettre un diagnostic un peu plus précoce.
Les ressources thérapeutiques de cette affection restent limitées sur le plan médicamenteux notamment pour la prise en charge des troubles moteurs. Le traitement symptomatique de la dysautonomie à la fois cardiovasculaire et vésicosphinctérienne ainsi que le traitement dopaminergique permettent d’améliorer les conditions de vie du patient.
Une prise en charge multidisciplinaire semble indispensable à l’accompagnement de ces patients et de leur famille.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Photos : Service de Neurologie de l’EHS Ben Aknoun.
Références
Bjornsdottir A, Gudmundsson G, Blondal H, Olafsson E. Incidence and prevalence of multiple system atrophy: a nation-wide study in Iceland. J Neurol Neurosurg Psychiatry 2013;84 (2):136–40.
Wenning GK, Geser F, Krismer F, Seppi K, Duerr S, Boesch S, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013; 12(3):264–74.
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008; 71(9): 670–6.
Gilman S, Low PA, Quinn N, Albanese A, Ben-Shlomo Y, Fowler CJ, et al. Consensus statement on the diagnosis of multiple system atrophy. J Auton Nerv Syst 1998; 74(2–3):189–92.
Kollensperger M, Geser F, Ndayisaba JP, Boesch S, Seppi K, Ostergaard K, et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: final analysis of the European multiple system atrophy registry. Mov Disord 2010; 25(15): 2604–12.
Papapetropoulos S, Tuchman A, Laufer D, Papatsoris AG, Papapetropoulos N, Mash DC. Causes of death in multiple system atrophy. J Neurol Neurosurg Psychiatry 2007; 78(3):327–9.
rojanowski JQ, Revesz T. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol 2007;33(6):615–20.
Jellinger KA. Neuropathological spectrum of synucleinopathies. Mov Disord 2003; 18(Suppl. 6):S2–12.
Kollensperger M, Geser F, Seppi K, Stampfer-Kountchev M, Sawires M, Scherfler C, et al. Red flags for multiple system atrophy. Mov Disord 2008;23(8):1093–9.
Sakakibara R, Hattori T, Uchiyama T et al. Urinary dysfunction and orthostatic hypotension in multiple system atrophy: which is the more common and earlier manifestation? J Neurol Neurosurg Psychiatry 2000; 68(1):65-9.
Mabuchi N, Hirayama M, Koike Y et al. Progression and prognosis in pure autonomic failure (PAF): comparison with multiple system atrophy. J Neurol Neurosurg Psychiatry 2005;76(7):947-52.
Wenning GK, Tison F, Ben Shlomo Y et al. Multiple system atrophy: a review of 203 pathologically proven cases. Mov Disord 1997;12(2):133-47.
Wenning GK, Scherfler C, Granata R et al. Time course of symptomatic orthostatic hypotension and urinary incontinence in patients with postmortem confirmed parkinsonian syndromes: a clinicopathological study. J Neurol Neurosurg Psychiatry 1999; 67(5):620-3.
Joseph PA, Arné P, Barat M et al. Troubles vésicosphinctériens et explorations du sytème urinaire. In: Les syndromes parkinsoniens atypiques et secondaires. Paris : Masson (ed) 2006:106-12.
Bonnet AM, Pichon J, Vidailhet M et al. Urinary disturbances in striatonigral degeneration and Parkinson’s disease: clinical and urodynamic aspects. Mov Disord 1997; 12(4):509-13.
Wenning GK, Ben Shlomo Y, Magalhaes M et al. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain 1994;117(Pt 4):835-45.
Wenning GK, Ben Shlomo Y, Magalhaes M et al. Clinicopathological study of 35 cases of multiple system atrophy. J Neurol Neurosurg Psychiatry 1995; 58(2):160-6.
Kirchhof K, Apostolidis AN, Mathias CJ et al. Erectile and urinary dysfunction may be the presenting features in patients with multiple system atrophy: a retrospective study. Int J Impot Res 2003; 15(4):293-8.
Kollensperger M, Stampfer-Kountchev M, Seppi K et al. Progression of dysautonomia in multiple system atrophy: a prospective study of self-perceived impairment. Eur J Neurol 2007; 14(1):66-72.
Brooks DJ, Seppi K. Proposed neuro-imaging criteria for the diagnosis of multiple system atrophy. Mov Disord 2009; 24(7):949-64.
Schrag A, Good CD, Miszkiel K, Morris HR, Mathias CJ, Lees AJ, et al. Differentiation of atypical parkinsonian syndromes with routine MRI. Neurology 2000; 54(3):697–702.
Eckert T, Barnes A, Dhawan V, Frucht S, Gordon MF, Feigin AS, et al. FDG PET in the differential diagnosis of parkinsonian disorders. Neuroimage 2005; 26(3):912–21.
Munoz E, Iranzo A, Rauek S, Lomeña F, Gallego J, Ros D, et al. Subclinical nigrostriatal dopaminergic denervation in the cerebellar subtype of multiple
Wenning GK, Geser F, Poewe W. Therapeutic strategies in multiple system atrophy. Mov Disord 2005; 20(Suppl. 12):S67–76.
Freeman R. Clinical practice. Neurogenic orthostatic hypotension. N Engl J Med 2008; 358(6):615–24.
La stimulation cérébrale profonde (SCP) dans la Maladie de Parkinson (MP) n’est envisagée qu’en cas de symptômes invalidants, malgré une pharmacothérapie optimisée. Elle est contre-indiquée en cas de démence, de pathologie psychiatrique non contrôlée et/ou d’affection concomitante à potentiel évolutif à moyen terme.
H.Leklou, F. Ysmail Dahlouk, Service de Neurologie, CHU Mohamed Lamine Debaghine, Bab El Oued, Alger.
Date de soumission : 09 Février 2020.
Abstract: Deep Brain Stimulation (DBS) in Parkinson’s disease (PD) is only considered in the case of invalid symptoms despite optimized pharmacotherapy. It is contra-indicated in the case of dementia, non-controlled psychiatric pathology and/or associated affection with risk of progression in the medium term. The main target is the sub-thalamic nucleus. DBS is remarkably effective on symptoms. The correct positioning of the electrodes limits the risk of occurrence of undesirable effects which can usually be avoided by adjusting the stimulation parameters.
Keywords: Deep brain stimulation, Parkinson disease, surgical treatment.
Résumé : La stimulation cérébrale profonde (SCP) dans la Maladie de Parkinson (MP) n’est envisagée qu’en cas de symptômes invalidants, malgré une pharmacothérapie optimisée. Elle est contre-indiquée en cas de démence, de pathologie psychiatrique non contrôlée et/ou d’affection concomitante à potentiel évolutif à moyen terme. La principale cible est le noyau sous-thalamique. La SCP est remarquablement efficace sur les symptômes. Le bon positionnement des électrodes limite le risque de survenue d’effets indésirables qui peuvent être généralement évités par l’ajustement des paramètres de stimulation.
Mots clés : stimulation cérébrale profonde, maladie de Parkinson, traitement chirurgical.
Introduction
Dans les années cinquante, des lésions des noyaux gris centraux furent les premiers traitements de la maladie de parkinson (MP). Ces lésions consistaient en thermo coagulations réalisées en condition stéréotaxique au sein du thalamus (thalamotomies) pour le contrôle du tremblement.
Par la suite, la coagulation d’une partie limitée du pallidum interne fut proposée pour contrôler les dyskinésies dopa-induites invalidantes et plus partiellement le syndrome akinéto-rigide et trémulant. Après l’avènement de la dopathérapie, le nombre de ces interventions a fortement diminué.
Au cours de ces interventions lésionnelles, la stimulation électrique était un moyen permettant d’identifier la cible stéréotaxique ; elle était utilisée chez un patient sous anesthésie locale afin de rechercher un effet indésirable et la thermolésion n’étant alors effectuée qu’en son absence. En effet, cette procédure thérapeutique, surtout quand elle est bilatérale, pouvait être à l’origine de troubles cognitifs, de l’équilibre et de dystonie.
Progressivement, cette stimulation électrique fut utilisée pendant des jours ou des semaines, afin de sélectionner les électrodes les plus efficaces parmi plusieurs implantées, avant de procéder à la chirurgie lésionnelle. Cette stimulation électrique était effectuée à diverses fréquences mais le plus souvent à 50 Hz, elle pouvait modifier le tremblement d’un patient éveillé, soit l’aggraver, soit l’améliorer. Ce n’est qu’en 1987 que Benabid a pu montrer que, seule une fréquence de stimulation du thalamus supérieure à 100 Hz, dite à haute fréquence, permettait de supprimer le tremblement, mimant ainsi l’effet d’une lésion dans le même site, mais avec l’avantage crucial de la réversibilité et de l’adaptabilité des paramètres électriques.
Une meilleure connaissance de l’organisation fonctionnelle des noyaux gris centraux a permis ces dernières années un développement spectaculaire de la neurochirurgie fonctionnelle avec tout particulièrement l’avènement de la stimulation au sein d’une structure cible : initialement le thalamus et plus récemment dans deux autres structures profondes que sont le pallidum interne et le noyau subthalamique.
Bases physiopathologiques de la stimulation cérébrale profonde dans la MP
Les symptômes moteurs de la maladie résultent principalement d’une dégénérescence de la voie directe inhibitrice entre le striatum et le pallidum interne ; la diminution de l’inhibition sur le pallidum est à l’origine de son hyperactivité.
Parallèlement, l’effet inhibiteur exercé par la dopamine sur la voie indirecte, passant par le pallidum externe, le noyau subthalamique, puis le pallidum interne, est aussi diminué.
La conséquence principale est une augmentation de l’activité des neurones glutaminergiques du noyau subthalamique qui stimulent de façon excessive les deux structures cibles : la substance noire réticulée et le pallidum interne.
Ainsi, cette hyperactivité subthalamique à l’origine de l’hyperactivité pallidale interne entraine une inhibition tonique majeure du thalamus moteur et, par la même, la perte de l’activation normale des aires motrices corticales qui rend compte de la symptomatologie. L’activité excessive du noyau subthalamique joue donc un rôle clé dans la physiopathologie de la maladie de Parkinson.
La méthode de stimulation du noyau subthalamique a été introduite en 1993 par l’équipe Grenobloise. La stimulation du noyau subthalamique inhibe l’hyperactivité de cette structure et par conséquent réduit l’action inhibitrice du pallidum interne sur le thalamus, facilitant la volée efférente excitatrice du thalamus vers le cortex.
La stimulation cérébrale profonde à haute fréquence a connu beaucoup de progrès et suscite un immense espoir ; même en l’absence d’une compréhension claire et universellement admise de ses principes et ses mécanismes d’action, qui font même encore l’objet de controverse.
Intérêt de la stimulation cérébrale profonde
La stimulation cérébrale profonde de ces structures reproduit l’effet thérapeutique souhaité sans induire de lésion anatomique. La stimulation cérébrale et le traitement médicamenteux ont une action antiparkinsonienne similaire, la différence est que la chirurgie permet de diminuer de façon drastique le traitement L-Dopa, ce qui permet par conséquent de réduire la fréquence des dyskinésies.
Elle présente par rapport à la technique lésionnelle de nombreux avantages :
Elle est hyper sélective et modulable ;
Elle peut être réalisée de façon uni ou bilatérale ;
Les effets secondaires, qui sont parfois rencontrés, sont réversibles après arrêt de la stimulation ou diminution de son intensité.
En revanche, son inconvénient majeur est son coût élevé.
Les multiples avantages de la stimulation cérébrale profonde font que cette technique, moins invasive, est actuellement largement préférée à la thermo lésion.
Indications
Cette chirurgie reste réservée à des patients :
Ayant une forme évoluée et sévère de maladie de Parkinson idiopathique (au moins 5 ans) ;
Ne répondant pas au traitement médical classique optimisé (les différentes associations médicamenteuses ne permettent qu’un contrôle limité). Un traitement est considéré comme optimisé lorsque les options thérapeutiques ont été correctement tentées (utilisation d’un agoniste dopaminergique, L-dopa fractionnée à la dose de 800 mg/j, pour au moins trois mois, essai d’un inhibiteur de la COMT (catéchol-O-méthyltransférase), etc.). Un essai d’ajustement sur une période de l’ordre de six mois par un neurologue expert en MP est légitime. Vu l’amélioration de la qualité de vie par la SCP comparée aux ajustements médicamenteux, et la stabilité à long terme de ses effets (> 10 ans), l’épuisement de toutes les possibilités médicamenteuses avant d’envisager une opération est à proscrire ;
Présentant des effets indésirables sévères secondaires aux médicaments antiparkinsoniens. Il s’agit généralement de fluctuations motrices (c’est-à-dire de fluctuations d’effet des médicaments tels que les blocages de fin de dose ou imprévisibles), et de dyskinésies (mouvements anormaux secondaires au traitement dopaminergique pulsatile), compromettant les activités de la vie quotidienne,
Conservant une excellente sensibilité à la L- dopa (très bonne amélioration de la symptomatologie parkinsonienne au maximum d’effets du traitement dopaminergique).
Contre-indications
Pour être candidats à la SCP, les patients ne doivent pas présenter :
· De démence. En cas de doute sur une démence débutante, il est important de réévaluer le patient au minimum six mois plus tard pour voir s’il existe une aggravation progressive.
· D’anomalies à l’IRM encéphalique pouvant augmenter le risque hémorragique lors de l’implantation.
· D’affection concomitante à potentiel évolutif à moyen terme (cancer non contrôlé par exemple) ou augmentant le risque opératoire (pathologie cardiaque instable, encéphalopathie vasculaire sévère, nécessité d’un traitement anticoagulant permanent pour une valve cardiaque mécanique, etc.).
· De troubles psychiatriques florides et non contrôlés. Un syndrome dépressif majeur doit être corrigé avant l’indication opératoire. Des hallucinations ou une psychose, secondaires à de fortes doses de traitement dopaminergique ne contre-indiquent pas formellement une SCP.
Les contre-indications relatives sont
· Un âge avancé. Entre 75 et 80 ans, chaque patient doit être évalué en fonction de son statut cognitif et de son état général car il existe un risque plus grand d’aggravation cognitive et de complications chirurgicales ainsi qu’un moins bon rapport bénéfice-risque. L’intervention est contre-indiquée chez les patients de plus de 80 ans sauf pour la cible thalamique.
· Les signes axiaux (dysarthrie, trouble de l’équilibre, troubles de la marche, tels que le freezing et les chutes) dopa-résistants. Ils résultent généralement de lésions non dopaminergiques, et ne sont pas améliorés par le traitement dopaminergique ni par la SCP. S’ils répondent à la L-dopa, ils ne sont pas une contre-indication.
Une fois l’ensemble de ces critères mesurés, l’indication opératoire est retenue après une appréciation du rapport bénéfice-risque pour chaque patient. La décision chirurgicale est prise en concertation multidisciplinaire (neurologue, neurochirurgien, psychiatre et neuropsychologue).
Quel bilan pré-chirurgical ?
L’évaluation préopératoire minutieuse en milieu spécialisé se fait en collaboration étroite entre les neurologues et les neurochirurgiens. Elle a pour but :
D’éliminer un autre syndrome parkinsonien, notamment d’origine dégénérative ;
De vérifier si la maladie est sévère et handicapante ;
De vérifier si le traitement médical a été bien conduit, et bien toléré ;
De vérifier s’il existe encore une bonne sensibilité à la L-dopa appréciée par le score UPDRS III (une amélioration de plus de 30% est considérée comme cliniquement significative) ;
D’écarter les éventuelles contre-indications à la chirurgie (voir plus haut).
Cibles de stimulation
Nous avons vu qu’elles étaient classiquement au nombre de trois :
Le noyau ventral intermédiaire du thalamus, le pallidum interne et le noyau subthalamique.
Le noyau ventral intermédiaire du thalamus :
La stimulation du noyau ventral intermédiaire du thalamus a été très longtemps proposée dans les formes tremblantes sévères, invalidantes dans la vie quotidienne et résistantes à un traitement médical bien conduit, ou en cas d’effets secondaires limitant l’augmentation posologique.
L’efficacité reste très limitée sur la rigidité et sur les dyskinésies, alors que cette stimulation n’agit pas sur l’akinésie, justifiant de poursuivre après l’intervention un traitement antiparkinsonien classique.
Les effets secondaires se traduisent par des dyskinésies et plus rarement des manifestations dystoniques mineures et réversibles après réduction de l’intensité de la stimulation.
Le pallidum interne :
La stimulation de la partie ventro-postéro-latérale du pallidum interne fut proposée dans un premier temps pour le traitement des dyskinésies sévères induites par la L-dopa ; le caractère le plus souvent bilatéral des symptômes justifiait une stimulation bilatérale d’emblée. En cas de dyskinésies unilatérales, la stimulation est unilatérale mais les patients doivent être opérés du côté controlatéral dans quelques années.
L’efficacité de la stimulation pallidale sur la triade parkinsonienne reste très variable avec un bénéfice chiffré entre 30 et 80% selon les cas. Les effets secondaires peuvent se traduire notamment par une dysarthrie ou des troubles cognitifs.
La stimulation du noyau subthalamique :
La stimulation du noyau subthalamique permet un contrôle de la plupart des symptômes de la maladie de Parkinson, que ce soit le syndrome akinéto-rigide et trémulant, mais aussi les mouvements involontaires dont le contrôle est obtenu secondairement après la diminution, voire la suppression totale dans certains cas, des traitements antiparkinsoniens.
Un recul de quelques années confirme que, si l’effet thérapeutique de la stimulation de cette structure se maintient ; il ne semble pas empêcher la survenue de certains symptômes tels que les troubles posturaux et intellectuels.
L’apparition secondaire de troubles axiaux dopa-résistants (instabilité posturale, enrayages cinétiques de la marche, dysarthrie) des troubles cognitifs et d’une apathie, observés chez certains patients, peut être la conséquence de l’évolution naturelle de la maladie mais semble parfois précipitée par la stimulation subthalamique, sans qu’il soit possible à l’heure actuelle de prévoir une telle évolution.
Implantation des électrodes et du neurostimulateur
La thermo lésion et la mise en place des électrodes dans les cibles choisies sont fondées sur la fusion de l’imagerie entre la tomodensitométrie et l’imagerie par résonance magnétique cérébrale mais aussi sur l’enregistrement électrophysiologique per-opératoire dans la stimulation cérébrale profonde. La cible, le trou trépan et la trajectoire sont définis par l’image de l’unité de fusion afin d’éviter les ventricules et les structures vasculaires. Les séquences IRM T1 et T2 aident à visualiser le STN (noyau subthalamique). La précision de ces emplacements est obtenue avec la macro stimulation en cas de thalamotomie ou pallidotomie et des micro-enregistrements per-opératoires en cas de stimulation cérébrale profonde (image 1).
Image 1 : image de la série personnelle du Dr Y. Smail Dahlouk
Le patient subira aussi une implantation d’un boîtier de stimulation (neurostimulateur), qu’il utilisera pour déclencher l’impulsion électrique (> 80 Hz) (image 2).
Image 2 : Boîtier de stimulation (neurostimulateur)
Comment ça se passe au bloc opératoire en pratique ?
La fusion des images entre scanner et IRM se fait en utilisant la station de travail neuronavigation.
Les coordonnées de la cible STN ainsi que la trajectoire sont choisies selon les coordonnées théoriques du STN selon le plan AC-PC (STN).
La même procédure est utilisée pour l’autre côté.
Les micro-enregistrements effectués par le neurologue (Figure 1).
(Figure 1) : enregistrement réalisé par Dr Ysmail Dahlouk
La programmation est le temps du neurologue et consiste à programmer plusieurs paramètres électriques : (fréquence, voltage, durée d’impulsion, configuration des quatre contacts de l’électrode). Ces paramètres peuvent être adaptés tout au long de la surveillance post-opératoire des patients afin d’optimiser la réponse thérapeutique.
Suivi post opératoire
Une bonne coopération du patient est indispensable ; ce dernier devra notamment accepter un suivi régulier pour le réglage des différents paramètres de stimulation ; en effet, il est important de réajuster les paramètres de stimulation en fonction du devenir post-opératoire des patients.
Résultats
La stimulation du NST[1] reproduit les effets de la dopathérapie et élimine les fluctuations propres à ce traitement quand il est administré au long cours. Le patient pourra donc se retrouver de façon permanente en période ON sans dyskinésie. Après un an de SCP du NST, les activités de la vie quotidienne et les symptômes moteurs sont améliorés de 60% comparés à l’état pré-chirurgical sans médicament. La stimulation permet d’améliorer en moyenne de 80% le tremblement, 67% la rigidité, 56% l’akinésie, 55% la marche et 73% la durée des blocages journaliers. La dopathérapie est diminuée d’environ 50% avec amélioration, voire disparition des dyskinésies et des phénomènes dystoniques. La SCP améliore aussi la douleur, les fluctuations psychiques, les symptômes dysautonomiques, la qualité du sommeil ainsi que les troubles du contrôle des impulsions grâce à la diminution du traitement dopaminergique. L’amélioration des scores de qualité de vie est de 13 à 24% lorsque des échelles incluent les aspects psychologiques, sociaux et moteurs de la vie quotidienne. Des mauvais scores de qualité de vie sont associés à une humeur dépressive et à une apathie. La réduction des traitements, l’efficacité motrice de la SCP et les altérations de la fluence verbale secondaire à la SCP n’ont pas d’impact sur la qualité de vie.
L’amélioration motrice globale se maintient à 54% à cinq ans (75% pour le tremblement, 71% pour la rigidité, 48% pour l’akinésie, 52% pour la marche) et 36% à onze ans (69% pour le tremblement, 44% pour la rigidité, 28% pour l’akinésie, 30% pour la marche). En revanche, la SCP ne permet pas de stopper l’évolution naturelle de la maladie et l’apparition des signes dopa- et SCP-résistants tels que les signes axiaux (dysarthrie, dysphagie, instabilité posturale, troubles de la marche) et les troubles cognitifs. Ainsi, les signes cardinaux de la maladie et les fluctuations motrices restent bien contrôlés, mais la qualité de vie des patients se dégrade progressivement par l’apparition de chutes, de dysphagie, d’incontinence urinaire et de démence.
Complications non chirurgicales
Les risques de l’intervention chirurgicale
Confusion mentale ;
Hémorragie cérébrale ;
Infections.
Les risques liés au matériel implanté
Mauvais fonctionnement de l’électrode ;
Sa fracture ;
Sa migration ;
Érosion cutanée.
Complications non chirurgicales liées à la stimulation
Le bon positionnement des électrodes limite le risque de survenue d’effets indésirables. Ceux provoqués par la SCP sont réversibles à son arrêt et peuvent être généralement évités par l’ajustement des paramètres de stimulation.
Problèmes moteurs :
Dyskinésies
La survenue de dyskinésies lors de l’augmentation de l’intensité de stimulation du NST est le signe d’un positionnement optimal des électrodes. Les dyskinésies s’estompent au fil des semaines, et avec la réduction du traitement médicamenteux. La survenue de dyskinésies implique une augmentation prudente des paramètres de stimulation et un ajustement rapide du traitement dopaminergique.
Troubles de la marche
Contrairement aux troubles de la marche et de l’équilibre dopa-sensibles qui sont améliorés par la SCP, les troubles de la marche sont parfois directement induits par la SCP ; ils seraient dus à la diffusion du courant aux fibres pallidothalamiques, à l’effet négatif de la stimulation à haute fréquence ou à l’effet suboptimal de la SCP augmentant l’asymétrie de l’akinésie aux jambes.
Troubles de la parole
L’aggravation de l’hypophonie et la dysarthrie est multifactorielle et elle serait liée à la diffusion du courant aux faisceaux corticobulbaire ou cérébellothalamique, et/ou à l’évolution de la maladie. Lorsqu’elle est due à la SCP, elle s’améliore par la réduction de l’amplitude de stimulation ou par le recours à une stimulation plus focale.
Problèmes généraux :
Syndrome des jambes sans repos (SJSR)
Ce phénomène nécessite une réintroduction de la médication dopaminergique vespérale quand celle-ci a été arrêtée. Une légère augmentation de la SCP peut être tentée, car ce syndrome réagit parfois à la stimulation du NST.
Prise de poids
La prise de poids serait due à une amélioration de la capacité du patient à s’alimenter et l’effet de la SCP sur l’hypothalamus. En pratique, il faut prévenir les patients et donner des conseils diététiques afin d’éviter des prises de poids excessives.
Problèmes neuropsychologiques
La SCP du NST ne module pas uniquement les circuits cortico-sous-corticaux moteurs mais aussi les circuits limbiques et ainsi provoque des troubles neuropsychiatriques.
Dépression ou euphorie
Une euphorie, voire un état maniaque, peuvent être observés après SCP du NST. Une dépression, avec risque suicidaire, est aussi observée et elle est favorisée par les antécédents dépressifs, la baisse du traitement dopaminergique postopératoire et les difficultés sociales et psychologiques à s’adapter aux modifications rapides induites par la chirurgie. Ces effets opposés s’expliquent par les différences de localisation des électrodes, de dénervation mésolimbique et de prise en charge médicamenteuse.
Apathie
L’apathie ou manque de motivation et d’initiative est fréquemment rencontrée dans la MP. Après SCP du NST, elle peut apparaître ou s’aggraver dans 12 à 24% des cas. Elle résulterait non seulement de la réduction postopératoire des traitements dopaminergiques mais serait aussi en lien avec un profil dégénératif dopaminergique prédominant à l’aire tegmentale ventrale, induisant une plus grande déplétion dopaminergique mésocorticolimbique chez certains patients. La réintroduction ou l’augmentation d’un traitement par agoniste dopaminergique permet d’améliorer l’apathie. Ainsi, l’attitude générale actuelle est de ne pas complètement supprimer les traitements dopaminergiques après la chirurgie.
Troubles cognitifs
La SCP du NST peut entraîner une réduction de la fluence verbale et une augmentation de l’impulsivité. Un déclin de la mémoire de travail et du fonctionnement cognitif global peut être observé. Il n’est cependant pas lié à la SCP du NST mais à la lésion de la tête du noyau caudé lors de la chirurgie. Une trajectoire via le noyau caudé doit donc être évitée.
Conclusion
Le recours à un traitement neurochirurgical au cours de la maladie de Parkinson ne concerne qu’un nombre relativement restreint de patients répondant à des critères de sélection bien définis.
C’est une chirurgie lourde mais qui donne d’excellents résultats si les indications sont bien respectées.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références :
Pollak Deep brain stimulation for Parkinson’s disease – patient selection. Handb Clin Neurol 2013 (116)
WMM Schuepbach J Rau K Knudsen Neurostimulation for Parkinson’s disease with early motor complications. N Engl J Med 2013 (368)
P Limousin P Krack P Pollak Electrical stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med 1998 (339)
KA Follett FM Weaver M Stern Pallidal versus subthalamic deep-brain stimulation for Parkinson’s disease. N Engl J Med 2010 (362)
VJJ Odekerken T van Laar MJ Staal Subthalamic nucleus versus globus pallidus bilateral deep brain stimulation for advanced Parkinson’s disease (NSTAPS study): A randomised controlled trial. Lancet Neurol 2013 (12)
AL Benabid P Pollak E Seigneuret Chronic VIM thalamic stimulation in Parkinson’s disease, essential tremor and extra-pyramidal dyskinesias. Acta Neurochir Suppl (Wien) 1993 (58)
T Witjas E Kaphan J Régis Effects of chronic subthalamic stimulation on nonmotor fluctuations in Parkinson’s disease. Mov Disord 2007 (22)
D Floden SE Cooper SD Griffith AG Machado Predicting quality of life outcomes after subthalamic nucleus deep brain stimulation. Neurology 2014 (83)
MG Rizzone, A Fasano, A Daniele, Long-term outcome of subthalamic nucleus DBS in Parkinson’s disease: From the advanced phase towards the late stage of the disease? Parkinsonism Relat Disord 2014 (20)
S Thobois C Ardouin E Lhommée Non-motor dopamine withdrawal syndrome after surgery for Parkinson’s disease: Predictors and underlying mesolimbic denervation. Brain 2010 (133)
K Witt O Granert C Daniels Relation of lead trajectory and electrode position to neuropsychological outcomes of subthalamic neurostimulation in Parkinson’s disease: Results from a randomized trial. Brain 2013 (136)
La maladie de Parkinson est une maladie dégénérative chronique caractérisée par des symptômes moteurs et non moteurs. La prévalence de la maladie de Parkinson dans le monde est variable, suggérant fortement l’implication de facteurs génétiques et environnementaux dans sa pathogénèse.
M.Bensaadi, N. Slimani, Z. Amamra, Y. Koubci, H. Bouzenada, Service de Neurologie, Hôpital Central de l’Armée, Aïn Naâdja, Alger.
Date de soumission : 09 Février 2020
Abstract:Parkinson disease (PD) is a chronic, progressive neurodegeneration disease characterized by both motor and non motor features. The variable prevalence of PD throught the world suggests that environmental and genetic factors may play an important role in disease pathogenisis. Helicobacter pylori (H.pylori) infection is one of the most common chronic infection, with high prevalence in the Mediterranean area. H.pylori mostly causes gastritis and peptic ulcer but is also linked to other gastrointestinal disorders including neurological diseases such as dementia, multiple sclerosis and PD. The study aimed to investigate the effect of H. pylori eradication on the motor symptoms of Parkinson’s disease.
Keywords: Parkinson disease, Helicobacter pylori.
Résumé : La maladie de Parkinson est une maladie dégénérative chronique caractérisée par des symptômes moteurs et non moteurs. La prévalence de la maladie de Parkinson dans le monde est variable, suggérant fortement l’implication de facteurs génétiques et environnementaux dans sa pathogénèse. Helicobacter pylori (H. pylori) est une infection commune, avec une haute prévalence dans le pourtour méditerranéen, principalement impliquée dans la survenue de gastrite et d’ulcères chroniques, elle a été récemment impliquée dans la physiopathologie de pathologies neurologiques diverses, tel que les démences, la sclérose en plaque et la maladie de Parkinson. Cette étude a pour but d’évaluer les effets de l’éradication de l’H. pylori sur les symptômes moteurs de la maladie de Parkinson.
Mots clés : Maladie de Parkinson, Helicobacter pylori.
Introduction
La maladie de Parkinson (PD) est une maladie neurodégénérative chronique. La variabilité de sa prévalence à travers le monde suggère le rôle prépondérant de facteurs génétiques et environnementaux dans sa pathogénèse.
L’infection par Helicobacter pylori (H. pylori) a une haute prévalence dans notre pays.
pylori est mis en cause dans la survenue de gastrites chroniques et d’ulcères, mais également dans la survenue de pathologies extradigestives, telles que les démences, la sclérose en plaques et la maladie de Parkinson.
Plusieurs hypothèses ont été formulées pour expliquer la physiopathologie et le rôle de l’H. pylori dans la genèse de la maladie de Parkinson. La principale hypothèse décrite dans la littérature fait référence à la sécrétion de neurotoxines induisant une dégénérescence des neurones dopaminergiques.
D’autres hypothèses sont liées aux différents phénomènes pro-inflammatoires consécutifs à l’infection chronique. Le duodénum étant le site d’absorption préférentiel de la L-Dopa, plusieurs articles de la littérature supposent que H. pylori pourrait influencer la pharmacocinétique de l’absorption de la L-Dopa et même l’inactiver, en altérant la muqueuse duodénale.
Ci-contre un schéma des différentes hypothèses actuelles :
Méthodologie
Il s’agit d’une étude prospective expérimentale type évaluation avant et après traitement. Elle a concerné 20 patients atteints de la maladie de Parkinson, chez lesquels nous avons réalisé une FOGD (fibroscopie oeso-gastroduodénale) avec biopsie, confirmant leur atteinte par H. pylori. Une évaluation de leur pathologie a été réalisée grâce au score MDS-UPDS[1] moteur, avant et après traitement de l’infection par H. pylori.
Critères d’inclusion
Critères de non inclusion
1- Age ≥ 60 2- Patients répondant aux critères de maladie de Parkinson
1. Antécédents de chirurgie gastrique 2. Antécédents de traitement de l’H. pylori 3. Récent changement dans la thérapeutique (réajustement du traitement dopaminergique) 4. Antécédents de chirurgie de la maladie de Parkinson
Test statistique réalisé : Test de Wilcoxon à échantillons appariés, avec un niveau de signification à 0,05.
Résultats
Sur les 20 patients de l’étude (13 hommes / 7 femmes) :
Figure 1 : Répartition selon le sexe
70% des patients ont amélioré leur score UPDRS moteur d’une moyenne de 5,4 points lors du contrôle à 1 mois après éradication de l’H. pylori .
Aucune amélioration n’a été notée après éradication chez 30% des patients.
Figure 2 : Score UPDRS moteur après traitement de H. pylori.
On note une nette amélioration du score après éradication de l’Helicobacter pylori et cette amélioration est significative sur le plan statistique (p=0,001).
Discussion
Plusieurs études ont démontré l’implication de l’H. pylori dans la maladie de Parkinson. Cette relation est en premier lieu née lorsque plusieurs études cliniques ont montré la haute prévalence des sérologies H. pylori positives chez les patients parkinsoniens. Puis la relation est devenue encore plus significative lorsqu’une étude réalisée en 2017 a montré une corrélation entre l’infection par l’H. pylori et le risque de développer une maladie de Parkinson [1].
Nilsen et al, ont démontré que la prescription du traitement d’éradication de l’H. pylori cinq ans avant le diagnostic de la maladie de parkinson est associée à une diminution de 45% du risque de développer la maladie. Ils ont conclu que l’infection chronique par H. pylori contribue à la pathogénèse de la maladie de Parkinson, bien avant l’apparition des signes moteurs de la maladie.
Bien que la plupart des études indiquent que la prévalence de l’infection par H. pylori est bien supérieure dans la maladie de Parkinson que chez les sujets témoins, plusieurs controverses subsistent concernant la physiopathologie précise de l’implication de l’H. pylori dans la maladie de Parkinson.
Cinq études [2,3,4,5,6] suggèrent que les patients infectés avaient une augmentation des fluctuations motrices, ainsi qu’un UPDRS score plus élevé. L’éradication de l’H. pylori a montré une amélioration des symptômes de la maladie de Parkinson [6] ; cette amélioration a été notable principalement chez les patients traités par levodopa, avec augmentation de la biodisponibilité et une diminution des fluctuations motrices.
Auparavant, cela a été notifié dans trois études [2,7,8] dont l’une réalisée en 2018, prouvant l’augmentation de l’absorption de levodopa après éradication de l’infection H. pylori versus les sujets témoins recevant un placebo.
Conclusion
Cette étude a été réalisée dans le but d’observer les effets de l’éradication de l’H. pylori sur les symptômes moteurs de la maladie de Parkinson. Un échantillon plus grand est souhaitable afin de mieux préciser l’impact et l’implication de cette bactérie sur la pathologie extra digestive, en particulier sur la maladie de Parkinson.
.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
Shen, H. Yang, Y. Wu, D. Zhang, H. Jiang, Association of Helicobacter pylori infection with Parkinson’s diseases: a meta-analysis, Helicobacter (2017).
Y. Lee, W.T. Yoon, H.Y. Shin, S.H. Jeon, P.L. Rhee, Helicobacter pylori infection and motor fluctuations in patients with Parkinson’s disease, Mov. Disord. 23 (12) (2008) 1696–1700.
Hashim, S. Azmin, H. Razlan, N.W. Yahya, H.J. Tan, M.R. Manaf, N.M. Ibrahim, Eradication of Helicobacter pylori infection improves levodopa action, clinical symptoms and quality of life in patients with Parkinson’s disease, PLoS One 9 (11) (2014) e112330.
R. Mridula, R. Borgohain, V. Chandrasekhar Reddy, V. Bandaru, T. Suryaprabha, Association of Helicobacter pylori with Parkinson’s disease, J. Clin. Neurol. 13 (2) (2017) 181–186.
Liu, W. Su, S. Li, W. Du, X. Ma, Y. Jin, K. Li, H. Chen, Eradication of Helicobacter pylori infection might improve clinical status of patients with Parkinson’s disease, especially on bradykinesia, Clin. Neurol. Neurosurg. 160 (2017) 101–104.
Pierantozzi, A. Pietroiusti, A. Galante, G. Sancesario, G. Lunardi, E. Fedele, P. Giacomini, P. Stanzione, Helicobacter pylori-induced reduction of acute levodopa absorption in Parkinson’s disease patients, Ann. Neurol. 50 (5) (2001) 686–687.
Pierantozzi, A. Pietroiusti, L. Brusa, S. Galati, A. Stefani, G. Lunardi, E. Fedele, G. Sancesario, G. Bernardi, A. Bergamaschi, A. Magrini, P. Stanzione, A. Galante, Helicobacter pylori eradication and l-dopa absorption in patients with PD and motor fluctuations, Neurology 66 (12) (2006) 1824–1829.
D.J. McGee, X.H. Lu, E.A. Disbrow, Stomaching the possibility of a pathogenic role for Helicobacter pylori in Parkinson’s disease, J. Parkinsons D
[1] MDS-UPDRS = Echelle MDS-UPDRS, Movement Disorder Society – Unified Parkinson’s Disease Rating Scale (NDLR).
La sclérose latérale amyotrophique (SLA) est la plus fréquente des maladies du motoneurone de l’adulte. Différentes formes cliniques sont classiquement individualisées. Elle est de pronostic habituellement fatal. Sa prise en charge est actuellement orientée vers la pluridisciplinarité. Elle repose sur des objectifs de soins médicaux et rééducatifs permettant de prévenir au mieux les complications. Un diagnostic précoce avec l’identification des différentes formes de début serait nécessaire pour une meilleure prise en charge.
Bourezg, A. Boulefkhad, Y. Sifi, A. Hamri, A. Mzahem, Service de Neurologie, CHU Dr Benbadis, Constantine. Faculté de médecine, Université Salah Boubnider Constantine 3. Laboratoire de génétique et de biologie moléculaire, Université Salah Boubnider Constantine 3.
Date de soumission : 21 Février 2020.
Abstract: Amyotrophic lateral sclerosis (ALS) is the most common motor neuron disease in adults. Different clinical forms are classically described. It is known to be of poor prognosis. The management of ALS involves a wide range of specialized practitioners and approaches including medical treatmentand physical therapy in order to prevent its complications. Early diagnosis and identification of the onset clinical variant are necessary for a better management. The objective of this work was to report the epidemiological, clinical characteristics and the state of management of patients with ALS followed in the neurology department of Constantine University Hospital.This is a retrospective, descriptive study covering the period of 7 years from January 2013 to December 2019. Included are patients diagnosed with ascertained ALS according to the revised Awaji-Shima and El Escorial criteria. Clinical and para-clinical data were collected from medical records.Forty-nine eastern algerian patients were enrolled with an average age of 56 and a sex ratio of 2.26. A family history of ALS was found in 3 cases. The diagnostic delay was 12 months. The onset of the disease was spinal in 61% of patients. Treatment with Riluzole was prescribed in 28.6% of patients. Our results show similarity to the major series in the literature.
Résumé : La sclérose latérale amyotrophique (SLA) est la plus fréquente des maladies du motoneurone de l’adulte. Différentes formes cliniques sont classiquement individualisées. Elle est de pronostic habituellement fatal. Sa prise en charge est actuellement orientée vers la pluridisciplinarité. Elle repose sur des objectifs de soins médicaux et rééducatifs permettant de prévenir au mieux les complications. Un diagnostic précoce avec l’identification des différentes formes de début serait nécessaire pour une meilleure prise en charge. L’objectif de ce travail était de rapporter les caractéristiques épidémiologiques, cliniques et l’état de la prise en charge des patients atteints de SLA suivis au service de neurologie du CHU Benbadis de Constantine. Il s’agit d’une étude rétrospective, descriptive réalisée sur une période de 7 ans allant de Janvier 2013 à Décembre 2019. Ont été inclus tous les patients diagnostiqués SLA certaine, selon les critères d’Awaji-Shima et El Escorial révisés. Les données cliniques et paracliniques ont été recueillies à partir des dossiers médicaux. Quarante-neuf patients ont été colligés, tous de l’est algérien. Un antécédent familial de SLA était retrouvé chez 3 patients. La moyenne d’âge était de 56 ans avec un sex-ratio de 2,26. Le délai de diagnostic était de 12 mois. Le début de la maladie était spinal chez 61% des patients. Le traitement par Riluzole était prescrit chez 28,6% des patients. Nos résultats se rapprochent des grandes séries de la littérature.
Mots clés : sclérose latérale amyotrophique, SLA, neurodégénérative, maladie du motoneurone, Riluzole.
Introduction
La sclérose latérale amyotrophique (SLA), dénommée aussi « maladie de Charcot » en référence au neurologue français, Jean-Martin Charcot qui l’a décrite pour la première fois en 1865 (1). Aux États-Unis, elle est connue sous le nom de maladie de Lou Gehrig, célèbre joueur de base-ball décédé de cette maladie en 1941 (2). La SLAest une affection neurodégénérative du système nerveux touchant aussi bien le motoneurone périphérique que central, responsable d’une sclérose siégeant au niveau de la partie latérale de la moelle épinière, s’exprimant cliniquement par une amyotrophie (2). Elle est souvent précédée de crampes et de fasciculations exprimées à l’examen électroneuromyographique (ENMG) par des décharges asynchrones d’une fibre ou d’un groupe de fibres. La destruction irréversible et progressive de ces motoneurones (2) est responsable de paralysies progressives des membres, labio-glosso-pharyngo-laryngés et des muscles respiratoires à l’origine d’une insuffisance respiratoire restrictive (3).
L’âge moyen de début de la SLA se situe autour de 60 ans, sa prévalence est de l’ordre de 4 à 6 pour 100.000 personnes et son incidence est de 1,7 pour 100.000 personnes (4).
Sa présentation clinique est très hétérogène, elle dépend surtout de la prédominance de l’atteinte du motoneurone, de la topographie du déficit moteur, du mode évolutif et des signes associés (5).
Elle apparait dans la plupart des cas de manière sporadique, cependant 5 à 10% des patients ont une histoire familiale antérieure de SLA (2). Le diagnostic repose essentiellement sur les données cliniques et électroneuromyographiques. La neuro-imagerie n’a d’intérêt que pour le diagnostic différentiel (6). On décrit actuellement 8 phénotypes distinctifs : la SLA classique à début spinal, la SLA à début bulbaire, les formes focalisées aux membres flail arm et flail leg, les formes spastiques pyramidales ou à début respiratoire, les formes pures d’atteinte du motoneurone périphérique (PMNP) ou pures d’atteinte du neurone moteur central (NMC) (5). Son pronostic est habituellement sévère, avec une médiane de survie de 40 mois et des extrêmes allant de 6 mois à 15 ans (7). Le mécanisme exact de la dégénérescence sélective des deux neurones moteurs est encore mal élucidé, cependant la théorie multifactorielle reste probable (2).
La prescription du Riluzole a permis de retarder le recours à la ventilation mécanique et de prolonger de façon modeste la durée de vie. Dans les formes avancées, le Riluzole n’apporte aucun bénéfice pour le malade (2). En réalité, la prise en charge du patient atteint de SLA se limite à un traitement symptomatique et palliatif dans un cadre d’interventions multidisciplinaires (kinésithérapie, orthophonie, mesures diététiques, prise en charge de l’insuffisance respiratoire et support psychologique) visant à améliorer la qualité de vie des patients.
Dans notre pays, les données épidémiologiques de cette affection sont très pauvres. Notre travail a pour objectif de rapporter les caractéristiques épidémiologiques, cliniques et l’état de la prise en charge des patients atteints de SLA suivis au service de neurologie du CHU Benbadis de Constantine.
Matériel et méthodes
Notre étude est descriptive, rétrospective incluant 49 patients atteints de SLA colligés au sein du service de Neurologie du CHU Constantine entre janvier 2013 et décembre 2019.
Le recueil des informations a été réalisé à partir des dossiers médicaux. La population cible est définie par l’ensemble des patients diagnostiqués SLA certaine, selon les critères d’Awaji Shima et El Escorial modifiés (8). Toutes les SLA probables, possibles, ou toutes autres affections simulant une atteinte du motoneurone central et périphérique ont été exclues.
Les paramètres étudiés étaient : le sexe, l’âge, les antécédents personnels et familiaux, la consanguinité, l’origine géographique, la profession, le mode de début de la maladie, le délai de diagnostic, les résultats de l’ENMG et de la ponction lombaire ainsi que les modalités de la prise en charge.
La collecte des données a été codée et saisie sur le logiciel SPSS version 21.0 et exprimée en moyennes et écarts-types pour les variables quantitatives et en pourcentages pour les variables qualitatives.
Résultats
Notre étude a porté sur 49 patients avec un sex-ratio de 2,26. L’âge moyen de début était de 56 ± 12,68 ans avec des extrêmes allant de 34 à 78 ans et répartis selon des tranches d’âges (Tableau N°1).
Tableau N°1 : Répartition des patients selon les tranches d’âge, la consanguinité, et le sexe
Effectif
%
Tranches d’âge
30-39 ans
5
10,2
40-49 ans
11
22,4
50-59 ans
9
18,4
60-69 ans
15
30,6
70-79 ans
9
18,4
Consanguinité
12
24,5
Hommes
34
69,4
Femmes
15
30,6
La majorité de nos patients était originaire de la wilaya de Constantine (28,6%), suivie de la wilaya de Mila (18%) et d’Oum El Bouaghi (18%). Les facteurs de risque étaient dominés par le tabagisme (18%), les métiers astreignants étaient les plus fréquents (30,6%) (Tableau N°2).
Tableau N°2 : Professions et exposition professionnelle des patients
Profession
Effectif
%
Exposition chimique (peintres)
5
10,2
Métiers astreignants (agriculteurs, maçons)
15
30,6
Pas de facteurs de risque
4
8,2
Sans emploi
6
12,2
Non précisé
19
38,8
Le délai moyen de diagnostic était de 12 mois avec des extrêmes allant de 2 à 48 mois. La forme spinale (classique et pseudo-polynévritique) était prédominante (Tableau N°3).
Tableau N°3 : Différentes formes cliniques de SLA
Formes cliniques
Effectif
%
Classique
16
32,7
Pseudo-polynévritique
14
28,6
Bulbaire
14
28,6
Respiratoire
1
2,0
Fail arm
4
8,2
La première consultation des patients était prodiguée par le généraliste (75,5%) ou les spécialistes (ORL, rhumatologue) avant leur orientation en neurologie (Tableau N°4).
Tableau N°4 : Médecins consultés en première intention
L’ENMG avait mis en évidence dans tous les cas, une atteinte diffuse du neurone moteur périphérique. Les résultats de la Ponction Lombaire (PL) réalisée chez 18 patients étaient sans particularité. Les troubles de la déglutition étaient présents dans 61,2% des cas. Le Riluzole a été prescrit chez 28,6% des patients. 3 patients ont bénéficié d’une ventilation non invasive (VNI) à domicile.
Discussion
L’âge moyen de début de nos patients était relativement précoce (56 ans) par rapport aux données de la littérature où il est de 61,4 ans dans la cohorte coréenne (10) et il se rapproche de l’étude de Tlemcen (9).
La nette prédominance masculine, avec un sex-ratio de 2,26 ; concorde avec les données de la littérature qui le situent entre 1 et 3 (7). Ceci pourrait s’expliquer par l’implication de certains facteurs : l’activité physique, les traumatismes, l’influence hormonale et l’exposition professionnelle (5).
Les cas familiaux retrouvés dans 7,3% des cas rejoignent approximativement les 10% des SLA familiales décrits dans la littérature, avec un mode de transmission habituellement autosomal dominant (4).
Le tabagisme, comme les métiers à risque retrouvés chez la majorité de nos patients, représentent deux facteurs de risque parmi d’autres incriminés dans l’étiopathogénie de la SLA à savoir le service militaire, l’exposition aux pesticides et aux solvants et le support génétique (11).
La forme classique, débutant aux membres supérieurs et en distal était la plus fréquente (32,7%), suivie de la forme pseudo-polynévritique qui est caractérisée par un début souvent unilatéral et un déficit de type périphérique (28,6%). Ces deux formes représentant le type spinal (61,3%) étaient majoritaires rejoignant l’étude Tlemcénienne (82%) (9). La fréquence de la forme bulbaire observée chez 28,6% rejoint les données de la littérature (33%) (11).
Le délai moyen de diagnostic, de 12 mois (2-48 mois) est comparable avec celui de la littérature (5).
Les premières consultations étaient majoritairement assurées par les médecins généralistes (75,5%), et se rapprochent des résultats des travaux de Limoges (88,3%) ; suivis par les neurologues (5,2%), les rhumatologues (2,6%), les ORL (2,6%) et les psychiatres (1,3%) (13).
L’ENMG conforte le diagnostic en objectivant des signes d’atteinte du motoneurone périphérique (5).
Le Riluzole prescrit chez nos patients n’a apporté qu’un bénéfice modeste comme l’attestent les travaux de la Mayo clinique (6). L’Edaravone, un antioxydant approuvé par la Food and Drug Administration (FDA) en 2017 n’apporte également qu’un effet modeste (6).L’état nutritionnel de nos patients (15/49) évalué chaque 3 mois par son index de masse corporelle (IMC) a permis d’améliorer la qualité de vie.
La décision de gastrostomie par voie percutanée s’avère nécessaire quand les troubles bulbaires sont sévères ou quand l’état nutritionnel du patient se dégrade (5).
L’apport de la VNI (ventilation non invasive) dans notre expérience a été bénéfique. Elle a amélioré la qualité de vie des patients avec une survie prolongée pouvant aller au-delà de 40 mois.
L’insuffisance respiratoire chronique doit faire l’objet d’une prise en charge précoce par la mise en œuvre de soins adaptés : VNI intermittente, assistance à la toux, VNI en continu, voire la trachéotomie (12), ce qui suggère la généralisation de ces techniques pour tous nos patients.
Conclusion
Notre travail peut contribuer à l’enrichissement des données épidémiologiques sur la SLA dans notre pays, néanmoins, des études prospectives multicentriques s’avèrent nécessaires.
Cette étude a permis de confirmer l’hétérogénéité phénotypique de la maladie et de proposer des alternatives thérapeutiques palliatives prolongeant la médiane de survie.
Nos perspectives portent essentiellement sur l’établissement d’un registre SLA, la création de consultations pluridisciplinaires impliquant tous les intervenants de la SLA, pour une meilleure prise en charge du patient afin d’alléger le fardeau économique et psychologique aux patients et leurs proches.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
P Bouche, N Le Forestier. Sclérose latérale amyotrophique(I) Aspects cliniques. EMC1999, 17-078-A-10
F. Pradat, P. Corcia, V. Meininger. Sclérose latérale amyotrophique. EMC 2016 – Neurologie, Vol.13, n◦2, 17-078-A-10EMC – Neurologie. http://dx.doi.org/10.1016/S0246-0378(15)45800-2
Brunaud-Danel, C. Moreau, D. Devos, L. Defebvre. Les nouvelles voies de recherche thérapeutique dans la sclérose latérale amyotrophique (SLA). Pratique Neurologique – FMC 2016 ;7 :9–15
Hélène Brocq, Une réflexion sur la prise en charge des patients atteints de Sclérose Latérale Amyotrophique (2006). Med Pal ; 5 : 337-342
Épidémiologie de la sclérose latérale amyotrophique : Facteurs de risque, incidence et Phénotypes (2015), thèse, Consulté à l’adresse tel.archives-ouvertes.fr.
Björn Oskarsson, Amyotrophic Lateral Sclerosis: An Update for 2018, Mayo Clin Proc. n XXX 2018
Conférence de consensus, Prise en charge des personnes atteintes de sclérose latérale amyotrophique (2005), Haute Autorité de santé
Joao Costa, Michael Swash, Mamede de Carvalho, Awaji Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis, arch neurol/vol 69 (no. 11), nov 2012
Merini Selma, Profil épidémiologique de la SLA dans la région de Tlemcen. Revue neurologique 2017, 1 7 3 S
Jun KY, et al., Epidemiology of ALS in Korea using nationwide big data. Neurol Neurosurg Psychiatry, 1136/jnnp-2018-318974
The epidemiology of amyotrophic lateral sclerosis. Handbook of Clinical Neurology2016, Vol. 138 (3rd series) Neuroepidemiology
Cuvelier, H. Prigent. Particularités de la sclérose latérale amyotrophique (SLA). Session a43 : situations difficiles en ventilation pour les patients atteints de maladies neuromusculaires. Revue des Maladies Respiratoires Actualités (2012) 4, 187-189.
Torny Torny, M. Lacoste, J.M. Nguyen, M.E. Tymoczko-Nguyen, P. Couratier. Étude des causes du retard au diagnostic de la sclérose latérale amyotrophique. Mémoire. Rev Neurol (Paris) 2006; 162 : 5, 617-622
En raison du vieillissement actuel de la population, la démence représente une préoccupation sociétale majeure. Le besoin en spécialistes de la mémoire est en augmentation tout comme l’incidence des démences dans le monde et en Algérie.
Expérience du service de neurologie Annaba avec le réseau Alois mémoire Paris
Nezzal 1, R. Gnassounou 2, W. Zahi 3, B. Defontaines 4, AM. Nezzal 3, C. Belin 5, D. Maillet 5, N. Toubal 1
1 Neurologie, CHU Ibn Sina de Annaba, Algérie ; 2 Neuropsychologie, Réseau mémoire Alois, Paris ; 3 Laboratoire de recherche santé environnement, Université Badji Mokhtar, Algérie ; 4 Neurologie, Réseau mémoire Alois, Paris
5 Neurologie, Hôpital Saint-Louis AP–HP, Paris
Date de soumission : 13 Février 2020
Abstract: Due to the ageing of the general population, dementia is a major societal concern. The need for memory specialists is increasing, as is the incidence of dementia worldwide and in Algeria. Telemedicine and its use with communication and information technologies in clinical assessment and treatment have existed in different forms for over a hundred years. Its evolution is closely linked to the progress of technology. Its application in the evaluation of cognitive disorders is increasingly promoted. In this article, we report our experience in teleconsultation for the assessment of cognitive functions, between Annaba and Paris. The main objective is to reproduce this model, to cope with the lack of specialists in Algerian medical deserts, in order to allow early diagnosis and treatment of neurocognitive disorders.
Key words: memory, teleconsultation, dementia
Résumé : En raison du vieillissement actuel de la population, la démence représente une préoccupation sociétale majeure. Le besoin en spécialistes de la mémoire est en augmentation tout comme l’incidence des démences dans le monde et en Algérie. La télémédecine et son utilisation avec les technologies de la communication et d’information dans l’évaluation clinique et le traitement, existe sous différentes formes depuis une centaine d’années. Son évolution est étroitement liée au progrès de la technologie. Son application dans l’évaluation des troubles cognitifs est de plus en plus promue. Dans cet article, nous rapportons notre expérience dans la téléconsultation pour l’évaluation des fonctions cognitives, entre Annaba et Paris. L’objectif principal étant de reproduire ce modèle, pour faire face au manque de spécialistes dans les déserts médicaux algériens, afin de permettre un diagnostic et une prise en charge précoces des troubles neurocognitifs.
Mots clés : mémoire, téléconsultation, démence.
Introduction
Avec l’espérance de vie accrue, le risque de déclin cognitif devient une préoccupation croissante. Selon l’OMS, dans l’ensemble de la population, entre 5 et 8% des personnes âgées de 60 ans et plus sont atteintes de démence à un moment donné.
Le nombre total de personnes atteintes de démence devrait atteindre 82 millions en 2030 et 152 millions d’ici 2050. Cette hausse est en grande partie due à l’augmentation du nombre de cas de démence dans les pays à revenu faible ou intermédiaire.
Les nouvelles technologies de l’information et de la communication permettent des prestations de santé à distance et l’échange de l’information médicale. La téléconsultation mémoire permet de faciliter et réduire le délai de diagnostic et de prise en charge des patients déments habitant dans les zones rurales (1). L’application clinique de la technologie de télécommunication pour l’évaluation et la réhabilitation des patients âgés atteints de troubles neurodégénératifs est possible et peut améliorer les performances cognitives globales (3).
La collaboration, entre le service de neurologie du CHU (Annaba) et le réseau mémoire Alois (Paris), a débuté en 2015 pour organiser une téléconsultation neuropsychologique entre l’Algérie et la France. Le but était d’en valider la faisabilité et par la suite de reproduire ce modèle localement pour palier au manque de spécialistes dans les déserts médicaux algériens, permettant ainsi un diagnostic et une prise en charge précoce des patients atteints de troubles cognitifs.
Réalisation du projet
Plusieurs réunions ont été nécessaires pour l’organisation de la téléconsultation. Des réunions, réalisées pour la plupart en visioconférence, suivie d’une formation avec mise en situation réelle et simulations. La première téléconsultation mémoire entre Annaba et Paris a été réalisée en février 2016.
La téléconsultation mémoire se déroule dans un local calme avec la présence d’une assistante au côté du patient. Elle intervient lorsque nécessaire, à certains moments du déroulement des tests ; deux caméras sont nécessaires. La première caméra pour permettre au neuropsychologue de Paris de visualiser le patient et la seconde pour qu’il voit ce qu’écrit le patient ainsi qu’un écran sur lequel le patient peut voir le neuropsychologue. Une bonne connexion internet et un niveau sonore suffisant sont également nécessaires. Dans notre expérience, la neurologue responsable de la consultation mémoire joue le rôle d’assistante afin qu’elle puisse maitriser toutes les étapes de la démarche.
Des patients d’expression francophone présentant des plaintes mnésiques sont présélectionnés, ils ont un MMSE supérieur à 20.
Une évaluation globale des fonctions cognitives est réalisée grâce à 10 tests :
MMSE : est un questionnaire de trente questions permettant de tester les facultés cognitives, il touche aux sphères de la mémoire, de l’apprentissage, du langage, du calcul, des repères spatio-temporels, de la transcription et de l’attention. Il permet le dépistage et le suivi des patients souffrant de troubles cognitifs,
Empans de chiffres : il consiste à présenter oralement des listes croissantes de chiffres que le sujet doit rappeler dans l’ordre. L’empan est le nombre maximum d’éléments que le sujet peut rappeler immédiatement. Il permet d’explorer la mémoire de travail,
RLRI 16 : Rappel Libre et Rappel Indicé à 16 items : RL/RI-16 (Van Der Linden et al. 2004). Il s’agit de 16 mots à mémoriser, présentés sur des fiches par groupes de 4. C’est une épreuve de mémoire verbale très utilisée en pratique courante ; sa procédure, singulière, fondée sur le contrôle et le renforcement de l’encodage et la distinction entre la récupération spontanée et la réactivité à l’indiçage, permet une certaine maitrise des facteurs attentionnels et exécutifs impliqués dans la mémorisation. Il est jugé très sensible au syndrome amnésique de type temporal interne dès le stade précoce de la maladie d’Alzheimer (11),
TNI 93 : Ce test de mémoire se base sur le principe de spécificité de l’encodage et utilise des images représentant des objets de la vie quotidienne plutôt que des mots à lire, utilisé chez les patients analphabètes. Une phase d’encodage simultané de neuf images est proposée au sujet, avant d’effectuer (après une courte tâche interférente) une phase de rappel libre et de rappel indicé (12),
Figure de Rey : La figure de Rey est composée de 18 éléments organisés en trois parties : une forme globale (le grand rectangle), des éléments externes (carrés, croix, triangles), et des éléments internes à la forme globale (lignes, ronds). C’est un test qui permet l’évaluation des fonctions exécutives, telles que les capacités visuospatiales et visuoconstructives, la mémoire non verbale et la mémoire de travail, l’attention et la planification,
DO 40 : La dénomination orale d’images est une épreuve incontournable dans le bilan de langage. Elle permet l’évaluation des troubles du langage,
BREF : C’est une batterie rapide d’évaluation frontale (ou FAB en anglais : Frontal Assessment Battery at Bedside), mise au point par Dubois et al., en 2000 (1) pour évaluer rapidement la présence ou non d’un syndrome dysexécutif cognitif et comportemental (13),
Fluences verbales : C’est la capacité à évoquer des mots en rapport avec une consigne donnée (catégorielle, sémantique, littérale), pendant un temps donné,
TMT A et B : C’est une épreuve de mesure de la flexibilité mentale, elle se déroule en deux temps. Dans un premier temps, le sujet doit relier des chiffres dans l’ordre croissant le plus rapidement possible (1-2-3-4, etc.), et dans un second temps, il doit procéder de la même manière, mais en alternant des chiffres et des lettres (1-A-2-B-3-C, etc.),
Évaluation des praxies : La batterie comprend trois dimensions différentes : exécution sur commande verbale de gestes symboliques, de mimes d’action et imitation de gestes abstraits.
Déroulement d’une téléconsultation mémoire
Un manuel guide est remis à l’assistante lui expliquant ses différentes taches : vérification du matériel avant le début de l’évaluation, accueil du patient. 2-15 minutes avant l’évaluation : explication du déroulement du bilan et description rapide du matériel (informatique et non informatique) qui sera utilisé. Les supports nécessaires à la passation des tests : cahier de passation patient, classeur de tests parties assistante, sont en possession de l’assistante. Elle est formée et habilitée au préalable.
Les subtests qui nécessitent l’intervention ‘active’ de l’assistante :
Empan en modalité auditivo/verbale (en entier), BREF (subtests), Figures complexes REY/TAYLOR (optionnel), batterie brève d’évaluation des praxies gestuelles (subtest gestes abstraits), TMT A et B (dans la partie ‘exemples’).
La téléconsultation débute par un entretien entre le neuropsychologue et le patient, il commence par se présenter et explique au patient le déroulement de la consultation, il le rassure, car souvent les patients sont un peu anxieux à l’idée de passer des tests pour leur mémoire. Il interroge le patient sur ses antécédents, son histoire médicale et celle de sa famille et sur ses plaintes (Fig. 1).
La passation des tests se fait dans un ordre précis et le même pour tous les patients pendant 2 à 3 heures. Il évalue la mémoire, le langage, les praxies, les gnosies et les fonctions exécutives. L’évaluation peut être modifiée en fonction des performances du patient et de l’hypothèse diagnostique, au moment de la réalisation des tests.
À la fin de l’évaluation, le patient est remercié et libéré, un nouveau rendez-vous lui est fixé, pour lui communiquer les résultats de son évaluation.
Le neuropsychologue rédige et envoie le compte rendu de son évaluation par mail, dans un délai de 24 heures (annexe 1).
Fig. 1 : Séance de téléconsultation
Résultats
Quinze téléconsultations ont été réalisées, de février 2016 à novembre 2018, chez des patients dont 4 ont des antécédents familiaux de démence. L’âge moyen des patients est de 64,6 ans avec des extrêmes de 51 ans à 75 ans. La téléconsultation a concerné 6 femmes et 9 hommes avec un MMSE moyen de 23,61.
Sur les quinze patients évalués, nous avons pu diagnostiquer : quatre patients avec un stade précoce (mild cognitive impairement, MCI), trois patients avec une démence à corps de Lewy (DCL), trois avec une maladie d’Alzheimer, un avec une aphasie progressive primaire (APP), un avec une démence mixte (DM), deux troubles anxio-dépressifs, un patient avec des antécédents familiaux de démence a été évalué et ne présentait aucun trouble (Tableau 1).
Tableau 1 : Diagnostics des patients évalués par téléconsultation
Diagnostics
Effectifs
Pourcentage
Mild cognitive impairement
4
26,7
Démence à corps de Lewy
3
20
Maladie d’Alzheimer
3
20
Aphasie progressive primaire
1
6,6
Démence mixte
1
6,6
Trouble anxio-dépressif
2
13,4
Pas de troubles
1
6,7
Total
15
100
Discussion
La télémédecine est de plus en plus utilisée pour prodiguer des consultations et des soins dans les zones rurales (5), notamment dans la pathologie neurocognitive. Plusieurs chercheurs se sont intéressés à la faisabilité et l’acceptabilité de ce mode d’évaluation et à la possibilité de poser un diagnostic fiable (5,9,10). Il a été démontré à titre d’exemple qu’il n’y avait pas de différence significative entre les scores MMSE réalisés en face à face et par vidéo télémédecine (8). Également, les patients ne sont pas gênés par ce nouveau mode d’évaluation (10). Une étude a aussi démontré que l’utilisation de la téléconsultation mémoire permettrait aussi de réduire le coût financier du diagnostic et de prise en charge des patients des habitats ruraux (4).
La collaboration, réseau « Alois mémoire » (Paris) et service de neurologie (Annaba) a permis le lancement d’une téléconsultation mémoire, il s’agit du premier dispositif de ce genre : téléconsultation neurologique et neuropsychologique en Algérie.
L’Algérie est un vaste pentagone de 2.381.741 km2 (le plus grand pays d’Afrique), divisé en 48 wilayas et 1.541 communes pour une population résidente de 39 millions d’habitants (recensement de 2015) avec une espérance de vie à la naissance de plus de 76 ans (6).
Cette collaboration nous a permis d’acquérir un savoir-faire reproductible, afin de permettre aux patients victimes de désertification médicale en Algérie d’accéder à une méthode d’évaluation de la fonction cognitive, sans se déplacer et sans déplacer le médecin, permettant ainsi un diagnostic précoce des maladies neurocognitives (notamment la maladie d’Alzheimer). Un diagnostic précoce permet de ralentir l’évolution de la maladie par le traitement des symptômes et la rééducation cognitive ; et aussi de retarder la perte d’autonomie du patient et la préparation des aidants à l’évolution de la maladie sur le plan psychologique et logistique.
Conclusion
La télémédecine est devenue incontournable dans le domaine de la santé globale et de la collaboration médicale internationale décentralisée. Ces nouvelles pratiques rendues nécessaires par le manque de spécialistes dans certaines zones sont possibles grâce à l’utilisation des nouvelles technologies. Elles apportent un vrai service aux populations éloignées, notamment dans l’évaluation cognitive en réduisant le délai d’attente pour une consultation et en permettant ainsi un diagnostic et une prise en charge précoce. Cela a été le cas pour notre expérience, qui va nous permettre de reproduire, en l’adaptant, la prise en charge à distance, des patients confrontés à un manque de spécialistes en neurologie et en neuropsychologie. La formation de neuropsychologues ouverts aux nouvelles technologies de l’information et de la communication s’avère ainsi une nécessité et une urgence.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références :
Providing Dementia Consultations to Veterans Using Clinical Video Telehealth: Results from a Clinical Demonstration Project. Dang S, Gomez-Orozco CA, van Zuilen MH, Levis S. Telemed J E Health. 2018 Mar;24(3):203-209. doi: 10.1089/tmj.2017.0089. Epub 2017 Jul 7.
A Multidisciplinary Model of Dementia Care in an Underserved Retirement Community, Made Possible by Telemedicine. Tso JV, Farinpour R, Chui HC, Liu CY. Front Neurol. 2016 Dec 23;7:225. doi: 10.3389/fneur.2016.00225. eCollection 2016.
Feasibility and efficacy of cognitive telerehabilitation in early Alzheimer’s disease: a pilot study. Jelcic N, Agostini M, Meneghello F, Bussè C, Parise S, Galano A, Tonin P, Dam M, Cagnin A. Clin Interv Aging. 2014 Sep 24;9:1605-11. doi: 10.2147/CIA.S68145. eCollection 2014.
A break-even analysis of delivering a memory clinic by videoconferencing. Comans TA, Martin-Khan M, Gray LC, Scuffham PA. J Telemed Telecare. 2013 Oct;19(7):393-6. doi: 10.1177/1357633X13506532.
Video-telemedicine in a memory disorders clinic: evaluation and management of rural elders with cognitive impairment. Barton C, Morris R, Rothlind J, Yaffe K. Telemed J E Health. 2011 Dec;17(10):789-93. doi: 10.1089/tmj.2011.0083. Epub 2011 Oct 24.
Les déserts médicaux ou inégalités territoriales en matière de répartition de l’offre de soins Larbi Abid ; santé maghreb
Comparison of the accuracy of patients’ recall of the content of telephone and face-to-face consultations: an exploratory study. McKinstry B, Watson P, Elton RA, Pinnock H, Kidd G, Meyer B, Logie R, Sheikh A. Postgrad Med J. 2011 Jun;87(1028):394-9. doi: 10.1136/pgmj.2010.101287. Epub 2011 Mar
Reliability of the MMSE administered in-person and by telehealth. McEachern W1, Kirk A, Morgan DG, Crossley M, Henry C. Can J Neurol Sci. 2008 Nov;35(5):643-6.
Cognitive intervention for community-dwelling older persons with memory problems: telemedicine versus face-to-face treatment. Poon P1, Hui E, Dai D, Kwok T, Woo J. Int J Geriatr Psychiatry.2005 Mar;20(3):285-6.
Feasibility of neuropsychological testing of older adults via videoconference: implications for assessing the capacity for independent living. Hildebrand R, Chow H, Williams C, Nelson M, Wass P. J Telemed Telecare. 2004;10(3):130-4.
Le RL/RI 16 items : comment lire les scores en pratique clinique ? Quels sont les pièges à éviter ? V. Hahn-Barma* La Lettre du Neurologue • Vol. XV – n° 8 – octobre 2011
Évaluation cognitive des patients illettrés et de bas niveau d’éducation, Didier Maillet, Catherine Belin, Rev Neuropsychol 2014 ; 6 (3) : 201-6
La BREF, une batterie rapide d’évaluation frontale FAB, Frontal assessment battery at bedside Author links open overlay panelDartinet1O.MartinaudNPG Neurologie – Psychiatrie – Gériatrie Volume 5, Issue 29, October 2005, Pages 43-46
La prévalence des crises d’épilepsie est plus élevée chez les sujets atteints de maladie d’Alzheimer par rapport à la population générale du même âge. L’objectif de la présente étude est de déterminer la fréquence et les facteurs de risque d’épilepsie chez les patients atteints de la maladie d’Alzheimer suivis au service de neurologie du CHU d’Oran
Chentouf1,2,3, M.L. Oubaiche 2,3
1 LABSIS : laboratoire des systèmes d’information en santé ; 2 Faculté de Médecine d’Oran, Université Oran 1 ;
3 Service de Neurologie, CHU Benaouda Benzerdjeb, Oran.
Date de soumission : 09 Février 2020.
Abstract: Purpose: The prevalence of epilepsy is higher in people with Alzheimer’s disease compared to the general population of the same age. The objective of this study is to determine the frequency and risk factors of epilepsy in patients with Alzheimer’s disease followed in the neurology department of Oran University Hospital. Materials and methods: This is a retrospective descriptive study conducted at the neurology department of Oran University Hospital over a period of five years (from January 2015 to December 2019). The variables studied were: sociodemographic, metabolic, cardiovascular, cognitive and therapeutic characteristics. Results: From the files of 441 patients referred to the neurology department of Oran University Hospital for cognitive disorders, 105 patients with Alzheimer’s disease were identified, of which 11 (10.5%) had experienced at least one epileptic seizure after the onset of cognitive impairment. The risk factors identified were: duration of Alzheimer’s disease, severity of cognitive impairment and high cholesterol. Conclusions: Epileptic seizures are common in Alzheimer’s disease and often overlooked. Thorough questioning and rigorous clinical and paraclinical examination are essential in any patient with Alzheimer’s disease in order to identify patients at high risk.
Résumé : Objectif : La prévalence des crises d’épilepsie est plus élevée chez les sujets atteints de maladie d’Alzheimer par rapport à la population générale du même âge. L’objectif de la présente étude est de déterminer la fréquence et les facteurs de risque d’épilepsie chez les patients atteints de la maladie d’Alzheimer suivis au service de neurologie du CHU d’Oran Matériels et Méthodes : Il s’agit d’une étude descriptive rétrospective menée au service de neurologie du CHU d’Oran sur une période de cinq ans (de janvier 2015 à Décembre 2019). Les variables étudiées étaient : les caractéristiques sociodémographiques, métaboliques, cardiovasculaires, cognitives et thérapeutiques. Résultats : À partir des dossiers de 441 patients référés au service de neurologie du CHU d’Oran pour plainte mnésique, 105 patients atteints de maladie d’Alzheimer ont été identifiés, dont 11 (10,5%) avaient présenté au moins une crise d’épilepsie après l’installation des troubles cognitifs. Les facteurs de risque identifiés étaient la durée d’évolution de la maladie d’Alzheimer, la sévérité des troubles cognitifs et l’hypercholestérolémie. Conclusions : Les crises d’épilepsie sont fréquentes dans la maladie d’Alzheimer et souvent méconnues. Un interrogatoire minutieux et un examen clinique et paraclinique rigoureux s’imposent chez tout patient atteint de maladie d’Alzheimer afin d’identifier les patients à haut risque.
Mots clés : Maladie d’Alzheimer, démence, épilepsie, crise, facteurs de risque.
Introduction
Épilepsie et démences sont deux pathologies fréquentes chez le sujet âgé. La prévalence de la démence dans les pays européens est estimée à 6-8% après l’âge de 65 ans [1]. Elle augmente avec l’âge et double tous les 5 ans pour atteindre un taux de 30 % après l’âge de 85 ans [2,3].
Les démences dégénératives sont responsables d’épilepsie tardive dans 10 à 20% des cas [4], une grande partie de la recherche à ce jour se concentre sur la maladie d’Alzheimer.
La maladie d’Alzheimer (MA) est un trouble neuro-dégénératif caractérisé par une détérioration progressive et irréversible de la mémoire et de la cognition, et est la principale cause de démence sénile. Actuellement, aucun traitement curatif n’est disponible pour cette maladie dévastatrice.
Le risque de développer une épilepsie tardive chez les patients atteints de MA après 65 ans est multiplié par 10 [5]. Les mécanismes physiopathologiques incriminés sont l’accumulation des lésions amyloïdes, les dégénérescences neurofibrillaires ou encore la perte neuronale. D’autres facteurs peuvent intervenir en particulier les comorbidités, les désordres métaboliques, la polymédication, ainsi que les troubles psycho-comportementaux incitant la prescription de molécules qui abaissent le seuil épileptogène [6].
Les crises d’épilepsie peuvent modifier l’histoire naturelle de la maladie d’Alzheimer et influencer son pronostic par l’accentuation des troubles cognitifs (effets indésirables des médicaments antiépileptiques), et la perte d’autonomie occasionnée par les évènements indésirables liés aux crises tels que les chutes avec fractures et les traumatismes crâniens [5,7]. Le diagnostic précoce des crises d’épilepsie chez les patients atteints de MA, ainsi que leur prise en charge appropriée s’avèrent essentiels pour améliorer le pronostic de ces patients.
L’impact considérable de l’association épilepsie-Alzheimer sur la santé publique et l’absence de données algériennes, nous ont incité à mener une étude épidémiologique visant à évaluer la fréquence des crises d’épilepsie chez les patients atteints de MA fréquentant le service de neurologie du CHU d’Oran ; et à identifier les facteurs de risque de survenue de crises.
Matériels et méthodes
Il s’agit d’une étude observationnelle descriptive rétrospective menée au service de neurologie du CHU d’Oran sur une période de 05 ans : du 1er janvier 2015 au 31 décembre 2019. Tous les registres de consultation de 2015 à 2019 ont été examinés. Nous avons répertorié tous les patients dont le motif de la première consultation était une plainte mnésique. Après examen des dossiers, tous les patients répondant aux critères de MA probable du DSM-V [8] ont été inclus.
Tous les patients ont été évalués sur le plan cognitif par le test MMSE (Mini Mental State Examination), sur le plan fonctionnel par l’IADL (Instrumental Activities of Daily Living), comportemental par la NPI (Neuro Psychiatrie Inventory) ; et global par la GDSR (Global Dementia Scale de Reisberg). Le stade de sévérité clinique (CDR) était apprécié de manière indépendante après entretien avec le patient et l’aidant principal.
Les données sociodémographiques collectées comprenaient l’âge, le sexe, le niveau d’instruction et le statut matrimonial.
Les données cliniques exploitées étaient la durée d’évolution de la MA (nombre d’années à partir du début des symptômes cognitifs signalés), la sévérité du syndrome démentiel évaluée par la version française de la CDR (Clinical Dementia Rating Scale), et les résultats du test MMSE (Mini Mental State Examination).
Les données sur les comorbidités ont été relevées. Il s’agissait en particulier du diabète, de l’hypertension artérielle et de l’hypercholestérolémie. Tous les patients diabétiques étaient sous traitement antidiabétique oral ou sous-cutané. Les patients hypertendus bénéficiaient de médicaments antihypertenseurs pour maintenir des valeurs de la pression artérielle systolique et diastolique <140/80 mmHg. Des statines ont été prescrites aux patients atteints d’hypercholestérolémie (> 240 mg / L).
Les données biologiques comprenaient la glycémie à jeun, l’HbA1C, la FNS, le bilan hépatique, le bilan rénal, le bilan thyroïdien, le dosage de la vitamine B12 et les sérologies.
Tous les patients présentant une épilepsie possible, probable ou certaine ont bénéficié d’un enregistrement électroencéphalographique (EEG) de base et d’enregistrements de contrôle tous les 6 mois.
Tous les patients avaient au moins une IRM cérébrale (1,5 Tesla) réalisée à n’importe quel moment de leur suivi.
Les données thérapeutiques ont également été prises en considération, en particulier les médicaments susceptibles d’abaisser le seuil épileptogène tels que les traitements spécifiques de la MA (inhibiteurs de la cholinestérase et mémantine), les antidépresseurs (inhibiteurs sélectifs de la recapture de la sérotonine [ISRS] et antidépresseurs tricycliques) ; et les agents antipsychotiques.
Analyses statistiques
Les variables quantitatives ont été exprimées en moyennes ± écart type et les variables qualitatives en fréquences et pourcentages. Afin d’identifier les facteurs de risque d’épilepsie, nous avons comparé les deux groupes (MA avec épilepsie, et MA sans épilepsie) ; par le test de Chi-deux lorsque les variables indépendantes étaient qualitatives, et le test T de Student lorsqu’il s’agissait de moyennes. Des valeurs de p égales ou inférieures à 0,05 ont été considérées comme statistiquement significatives.
Résultats
L’analyse des registres de consultation entre le 1er janvier 2015 et le 31 décembre 2019 a permis d’identifier 441 patients dont le motif de la première consultation était une plainte mnésique.
Après examen rétrospectif des dossiers médicaux, le diagnostic de syndrome démentiel était posé chez 367 patients (83,2%) alors que 74 patients (16,8%) n’ont manifesté aucun signe de déficience cognitive.
Parmi les 367 patients déments, 105 sujets (28,6%) répondaient aux critères de MA, 121 individus (33,0%) répondaient aux critères de démence vasculaire [9] et 91 patients (24,8%) présentaient des syndromes démentiels potentiellement réversibles (carence en vitamine B12, thyroïdite d’Hashimoto).
Nous avons exclu 16 dossiers (4,3%) qui étaient incomplets et 34 patients (9,3%) étaient perdus de vue (Figure 1).
Caractéristiques sociodémographiques de la population d’étude
Nous avons colligé 105 patients répondant aux critères de MA, dont 62 femmes (59,05%) et 43 hommes (40,95%). L’âge moyen était de 77,4 ± 8,1 ans avec des extrêmes allant de 55 à 94 ans. Le nombre moyen d’années d’études était de 7,5 ± 5,2 ans avec des extrêmes allant de 0 à 17 ans. Concernant le statut matrimonial, 83 patients (79,0%) étaient mariés, 13 patients (12,4%) étaient veufs, 7 patients (6,7%) étaient divorcés et deux patientes (1,9%) étaient célibataires.
Caractéristiques cliniques de la population d’étude
La durée d’évolution de la MA était de 5,6 ± 3,2 ans avec des extrêmes allant de 1 à 12 ans. Le score moyen du Mini Mental State Examination (MMSE) était de 19,4 ± 5,9 points avec des extrêmes allant de 3 à 22. Selon l’échelle d’évaluation de la sévérité de la démence (CDR), 33 patients (31,4%) avaient une démence légère (stade 1), 54 patients (51,4%) avaient une démence modérée (stade 2) et 18 patients (17,2%) souffraient d’une démence sévère (stade 3).
Fréquence des crises d’épilepsie
Parmi les 105 patients atteints de MA, 11 patients (10,5%) dont 7 hommes et 4 femmes avaient présenté au moins une crise convulsive depuis l’installation de leurs troubles cognitifs. La fréquence des crises était deux fois plus élevée chez les hommes que chez les femmes, mais la différence n’était pas significative (Tableau 1). Sept patients (63,6%) avaient des crises focales complexes et deux patients (18,2%) présentaient des crises généralisées myocloniques. Chez les deux derniers patients qui présentaient des crises exclusivement morphéiques, il nous a été difficile de déterminer si les crises étaient généralisées d’emblée, ou à point de départ focal avec généralisation secondaire.
Pour tous les patients souffrant de crises focales complexes, les témoins des crises ont signalé des signes moteurs initiaux, notamment une déviation de la tête ou des automatismes oraux et une suspension de la conscience, suivis de crises tonico-cloniques généralisées avec confusion postcritique prolongée. Aucun des dossiers ne mentionnait d’antécédents familiaux d’épilepsie ou d’antécédents de convulsions avant le début des troubles cognitifs.
Les anomalies EEG étaient deux fois plus fréquentes dans le groupe épileptique que dans le groupe sans crises. Les enregistrements EEG ont montré des anomalies chez 100% des patients MA avec épilepsie. Des pointes ondes ont été retrouvées chez 9 patients alors que chez les deux autres, l’enregistrement a mis en évidence un simple ralentissement du tracé.
Dans notre série, le délai moyen entre le diagnostic de MA et la survenue de la première crise était de 4,2 ± 1,9 ans. Tous les patients ayant présenté des crises ont été traités avec des médicaments antiépileptiques.
Comparaison des groupes MA avec crises et MA sans crises
Concernant les caractéristiques sociodémographiques, aucune différence d’âge, de sexe ou de niveau d’instruction n’a été observée entre les deux groupes (Tableau 1).
Une association significative a été retrouvée entre la survenue de crises d’épilepsie et la durée d’évolution ainsi que la sévérité des troubles cognitifs (Tableau 1).
Concernant les facteurs de risque vasculaires, nous n’avons retrouvé aucune association entre l’épilepsie et l’hypertension artérielle ou le diabète. En revanche, l’hypercholestérolémie semble être un facteur de risque significatif de survenue de crises d’épilepsie dans notre cohorte.
Sur le plan thérapeutique, aucun traitement (spécifique ou symptomatique) de la MA ne semble conférer une vulnérabilité aux crises
Discussion
Dans cette étude rétrospective basée sur les dossiers de patients atteints de MA fréquentant le service de neurologie du CHU d’Oran entre Janvier 2015 et décembre 2019, la fréquence des crises d’épilepsie était de 9,7%. Nos résultats rejoignent ceux de plusieurs études qui rapportent une fréquence, variant entre 10 et 20 % [10,11]. Par ailleurs, d’autres études publiées décrivent des fréquences plus importantes allant jusqu’à 26% [12,13].
En revanche, d’autres études rapportent des fréquences plus faibles allant de 1,5 à 3,5% [14-16]. Cette divergence peut être expliquée par les différences entre les critères d’inclusion.
En général, le risque de crises est maximal après 3,5 à 6 ans d’évolution de la maladie [17,18]. Ce risque est cumulatif et estimé à 11 % après 10 ans d’évolution et 26 % après 15 ans [19].
Dans notre série, les crises étaient focales complexes dans 63,6% des cas, et généralisées de type myocloniques dans 18,2% des cas. Les données de la littérature concernant le type de crises au cours de la MA sont mitigées, probablement en raison des différences des méthodes d’échantillonnage. En effet, du fait de la difficulté de mise en évidence de certains types de crises non motrices chez les patients déments tels que les crises dysmnésiques ou les crises avec phénomènes végétatifs, certaines études n’ont pris en considération que les crises généralisées ou les crises avec manifestations motrices [11,20].
Concernant les facteurs de risque de survenue de crises épileptiques chez les patients atteints de MA, seuls trois facteurs ont été significativement associés à l’épilepsie en l’occurrence la durée d’évolution du syndrome démentiel, la sévérité du déclin cognitif et l’hypercholestérolémie.
La MA étant une maladie neuro-dégénérative, il est communément admis que la durée d’évolution de la MA est étroitement liée à l’aggravation du déclin cognitif. Nos résultats sont compatibles avec ceux de l’étude prospective d’Amatniek et al., qui rapportent un risque relatif de 4,15 (IC 95% : 1,06–16,27), avec un suivi de 6 mois [12]. Par ailleurs, dans une étude ayant inclus 81 patients avec MA et épilepsie, Hauser et al., ont rapporté une fréquence de 11% de crises épileptiques chez les sujets au cours des 10 premières années de la maladie [19]. Cette fréquence passe à 26% après 15 ans d’évolution. Une autre étude cas-témoins ayant porté sur des sujets atteints de MA avec déclin cognitif sévère (CDR = 3), les auteurs ont rapporté une fréquence de 23% des crises [21]. Dans l’étude de Förstl et al., (1992), 11% des patients ont développé des crises motrices généralisées aux stades avancés de la maladie [10]. Cette fréquence élevée pourrait être expliquée par l’accumulation de lésions cérébrales aux stades avancés de la MA. En effet, des études neuropathologiques post-mortem de patients atteints de MA ayant présenté des crises convulsives ont mis en évidence des modifications gliales et une dégénérescence neuronale dans des zones spécifiques du cerveau (hippocampe et néocortex) [5,7]. Par conséquent, la formation de plaques séniles diffuses dans ces zones cruciales peut conduire au développement de crises. Par ailleurs, la mort neuronale peut affecter les circuits inhibiteurs GABAergiques et altérer l’équilibre entre l’excitation et l’inhibition, favorisant ainsi la survenue de crises [10].
Toutefois, il faut signaler que les crises ne sont pas spécifiques de la MA mais peuvent se voir dans d’autres types de démences. En effet, dans une étude portant sur tous types de démences confondus, Hersdoffer et al., ont rapporté un risque d’épilepsie dans tous les groupes [11]. Cette constatation suggère que tout processus pathologique suffisamment grave pour entraîner un déclin cognitif peut être associé à un risque accru de crises. La même étude rapporte un risque de crises huit fois plus élevé chez les patients déments non atteints de MA [11].
Nous avons testé la relation entre l’épilepsie et trois caractéristiques sociodémographiques chez nos patients atteints de MA à savoir l’âge, le sexe et le niveau d’instruction mais aucune association significative n’a été mise en évidence. Dans une étude rétrospective ayant inclus 145 patients MA dont 14 avec épilepsie tardive, Bernardi et al., ont identifié le sexe masculin comme facteur de risque d’épilepsie [22]. Bien que cette constatation soit difficile à interpréter compte tenu du faible effectif, les mêmes résultats ont été rapportés par d’autres équipes [7,18]. Néanmoins, sur la base de la conception rétrospective de notre étude et du petit échantillon inclus, nous pensons qu’une étude prospective sur un plus grand échantillon s’impose pour vérifier ces résultats.
L’analyse des comorbidités cardiovasculaires potentiellement traitables a trouvé une association significative entre épilepsie et hypercholestérolémie mais pas avec l’hypertension artérielle ni avec le diabète. Une étude Algérienne menée à Skikda ayant exploré la relation entre la MA et certains paramètres biologiques suggère que les patients atteints de MA présentent une hypercholestérolémie totale modérée mais permanente quel que soit l’âge du sujet ou le stade de gravité de la démence [23]. Une autre étude finlandaise suggère qu’un taux élevé de cholestérol à un âge moyen est un facteur de risque de MA [24]. Plusieurs études expérimentales ont démontré le rôle du cholestérol dans la production anormale de fragments aß dans les dépôts amyloïdes [25]. Bien que nous n’ayons trouvé aucune donnée publiée à l’appui de cette hypothèse, un dépôt anormal de fragments aß dans des sites épileptogènes, tels que l’hippocampe et les zones corticales, pourrait éventuellement favoriser la survenue de crises [10].
Dans notre étude, l’hypertension artérielle ne semble pas influencer le risque de survenue de crises. Dans leur étude, Amatniek et al., ont démontré que le traitement de l’hypertension artérielle avait un effet protecteur contre l’épilepsie [12], tandis que Hersdoffer et al., ont suggéré que l’HTA non traitée pourrait favoriser les crises chez les personnes âgées atteintes de MA [11]. Le traitement antihypertenseur pourrait jouer un rôle protecteur contre la dégénérescence cellulaire, réduisant ainsi le risque de crises. En effet, des études in vitro récentes ont identifié une classe de médicaments antihypertenseurs qui ralentissent le dépôt de plaques bêta-amyloïdes [26].
Nous n’avons trouvé aucune relation entre l’épilepsie et la consommation de médicaments antipsychotiques ou antidépresseurs, connus pour abaisser le seuil épileptogène. Ceci pourrait être expliqué par le fait que nos patients utilisaient des médicaments de nouvelle génération et à faibles doses. Nous n’avons pas non plus trouvé d’association entre la prise de médicaments anti-démence et les crises.
Limites de l’étude
Les limites de notre étude doivent être soulignées. D’abord, l’étude était rétrospective, sujette à des biais de mémorisation. De plus, les données sur le début des crises ont été collectées à partir de dossiers de patients suivis en consultation externe ; ces dossiers n’étant pas destinés à la recherche clinique. Par ailleurs, les symptômes des crises épileptiques sont difficiles à reconnaître et rarement signalés, en particulier pour les patients souffrant de troubles cognitifs sévères. Enfin, la faible taille de notre échantillon ne permet pas d’extrapoler les résultats à grande échelle.
Conclusion
L’épilepsie est fréquente chez les patients atteints de maladie d’Alzheimer suivis au service de neurologie du CHU d’Oran. Les facteurs de risque identifiés chez nos patients sont la durée d’évolution de la démence, la sévérité des troubles cognitifs et l’hypercholestérolémie. Les crises d’épilepsie influencent de façon péjorative l’évolution naturelle de la maladie d’Alzheimer en aggravant les troubles du comportement et le déclin cognitif. Un dépistage systématique ainsi qu’un traitement adapté sont le seul garant d’une meilleure prise en charge de ces patients.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
Fratiglioni L, Launer LJ, Andersen K, Breteler MM, Copeland JR, Dartigues JF, et al., Incidence of dementia and major subtypes in Europe: A collaborative study of population based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000; 54:S10-S15.
De Ronchi D, Berardi D, Menchetti M, Ferrari G, Serretti A, Dalmonte E, et al., Occurrence of cognitive impairment and dementia after the age of 60: a population-based study from Northern Italy. Dement Geriatr Cogn Disord 2005; 19:97-105.
Ramaroson H, Helmer C, Barberger-Gateau P, Letenneur L, Dartigues JF. Prevalence of dementia and Alzheimer’s disease among subjects aged 75 years or over: updated results of the PAQUID cohort. Rev Neurol 2003; 159:405-11.
Stefan H. Epilepsy in the elderly: facts and challenges. Acta Neurol Scand 2011; 124: 223–37.
Hommet C, Mondon K, Camus V, De Toffol B, Constans T. Epilepsy and dementia in the elderly. Dement Geriatr Cogn Disord 2008; 25: 293-300.
Pandis D, Scarmeas N. Seizures in Alzheimer disease: clinical and epidemiological data. Epilepsy Curr 2012; 12: 184-7.
Volicer L, Smith S, Volicer BJ. Effect of seizures on progression of dementia of the Alzheimer type. Dementia 1995; 6: 258–63.
DSM-5, Manuel diagnostique et statistique des troubles mentaux (“Diagnostic and Statistical Manual of Mental Disorders“), publié par l’American Psychiatric Associationen 2013.
Tris Oman GC, Tatemichi TK, Erkinjuntti T, et al., Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology 1993; 43: 250-60.
Forstl H, Burns A, Levy R, Cairns N, Luthert P, Lantos P. Neurologic signs in Alzheimer’s disease. Results of a prospective clinical and neuropathologic study. Arch Neurol 1992; 49:1038-42.
Hersdoffer DC, Hauser WA, Annegers JF, Kokmen E, Rocca WA. Dementia and adult onset unprovoked seizures. Neurology 1996;46: 727-30.
Amatniek JC, Hauser WA, Del Castillo-Castaneda C, etal., Incidence and Predictors of Seizure in Patients with Alzheimer’s Disease. Epilepsia 2006; 47: 867-72.
Lozsadi DA, Larner AJ. Prevalence and Causes of Seizures at the Time of Diagnosis of Probable Alzheimer’s Disease. Dementia and Geriatric Cognitive Disorders 2006; 22: 121-4.
Hommet C, Hureaux R, Barre J, Constans T, Berrut G. Epileptic seizures in clinically diagnosed Alzheimer’s disease: report from a geriatric medicine population. Aging Clin Exp Res 2007; 19:430-1.
Rao SC, Dove G, Cascino GD, Petersen RC. Recurrent seizures in patients with dementia: frequency, seizure types, and treatment outcome. Epilepsy Behav 2009; 14:118-20.
Scarmeas N, Honig LS, Choi H, Cantero J, Brandt J, Blacker D, et al., Seizures in Alzheimer disease: who, when, and how common? Arch Neurol 2009; 66:992-7.
Mendez M, Lim G. Seizures in elderly patients with dementia: epidemiology and management. Drugs Aging 2003; 20:791-803.
Risse SC, Lampe TH, Bird TD, Nochlin D, Sumi SM, Keenan T, et al., Myoclonus, seizures, and paratonia in Alzheimer disease. Alzheimer Dis Assoc Disord 1990; 4:217-25.
Hauser WA, Morris ML, Heston LL, Anderson VE. Seizures and myoclonus in patients with Alzheimer’s disease. Neurology 1986;36: 1226-30.
Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia 1993; 34:453–468
Romanelli MF, Morris JC, Ashkin K, Coben LA. Advanced Alzheimer’s Disease is a Risk Factor for Late Onset Seizures. Archives of Neurology 1990; 47: 847-50.
Silvia Bernardi, Nicola Scaldaferri, Nicola Vanacore, Alessandro Trebbastoni, Ada Francia, Aurelio D’Amico, Massimiliano Prencipe. Seizures in Alzheimer’s disease: a retrospective study of a cohort of outpatients. Epileptic Disord 2010; 12 (1): 16-21
Said Ramdane, Doria Daoudi-Gueddah. Mild Hypercholesterolemia, Normal Plasma Triglycerides, and Normal Glucose Levels Across Dementia Staging in Alzheimer’s Disease: A Clinical Setting-Based Retrospective Study. American Journal of Alzheimer’s Disease & Other Dementias 2011; 26(5) 399-405
Sjögren M, Mielke M, Gustafson D, Zandi P, Skoog I. Cholesterol and Alzheimer Disease: is there a relation. Mechanism of aging and development 2006; 127: 138-47.
Hoglund, K., Blennow, K. Effect of HMG-CoA Reductase Inhibitors on beta-amyloid Peptide Levels. CNS Drugs 2007; 21, 449-462
Wang J, Ho L, Chen L, et al., Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. Journal of Clinical Investigation 2007; 117: 3393-402.