S.M.A. CHELEF, H.E. ZIDANE, S. KERROUMI, M.J. YOUSFI, Service d’Urologie, Etablissement Hospitalier et Universitaire 1er Novembre 1954, Oran Faculté de médecine d’Oran.
Abstract : Paratesticular liposarcomas are relatively common sarcomas in the paratesticular region, however, the myxoïd variant is considered very rare. Due to the low specificity of clinical and radiological signs and the diversity of histopathological subtypes, no standard therapeutic strategy would be available. Multiple treatments have been reported in the literature with different results. Prognosis depends on the degree of differentiation of the tumor. We report the case of a young 17-year-old patient who was diagnosed with a scrotal right mass. A radical inguinal orchidectomy was performed. The evolution was marked by a retroperitoneal metastatic progression three months after surgery. A mono chemotherapy was administered based on doxorubicin but the patient died due to a rapid progression of the disease. Through this observation, we briefly report the data of the literature of this rare entity. Wide excision with healthy margins, whenever possible, is essential for local control of the disease. High rate of local recurrence may suggest a combined strategy combining surgery and adjuvant radiotherapy. The role of chemotherapy, although uncertain, finds its indication in metastatic cases, especially in dedifferentiated subtypes.
Key-words : Myxoïd liposarcoma, paratesticular, scrotal mass.
Résumé : Les liposarcomes paratesticulaires sont des sarcomes relativement fréquents dans la région paratesticulaire, cependant, la variante myxoïde est considérée comme très rare. Du fait de la faible spécificité des signes cliniques et radiologiques et de la diversité des sous-types histopathologiques, aucune stratégie thérapeutique standard ne serait disponible. Des traitements multiples ont été rapportés dans la littérature avec des résultats différents. Le pronostic dépend du degré de différenciation de la tumeur. Nous rapportons le cas d’un jeune patient de 17 ans, chez lequel a été décelée une masse scrotale droite. Une orchidectomie totale par voie inguinale a été réalisée. L’évolution a été marquée par une progresion métastatique rétropéritonéale trois mois après la chirurgie. Une mono chimiothérapie a été administrée à base doxorubicine ; mais le patient est décédé suite à une progression rapide de la maladie. A travers cette observation, nous rapportons brièvement les données de la littérature de cette entité rare. Une exérèse large avec des marges saines, tant que possible, est indispensable pour le contrôle local de la maladie. Le taux élevé de récidive locale pourrait inciter à envisager une stratégie combinée associant chirurgie et radiothérapie adjuvante. Le rôle de la chimiothérapie, bien qu’incertain, trouve son indication dans les cas métastatiques, surtout dans les sous types dédifférenciés.
Mots-clés : Liposarcome myxoïde, paratesticulaire, masse scrotale.
Introduction
Le liposarcome paratesticulaire est une tumeur mésenchymateuse maligne qui représente une entité pathologique extrêmement rare [1]. Environ 200 cas ont été rapportés dans la littérature. Histologiquement, il se divise en quatre sous-types : liposarcome à cellules pléomorphes, liposarcome myxoïde (et sa variante de plus haut grade à cellules rondes), liposarcome bien différenciée et liposarcome dédifférencié [2].
Le liposarcome paratesticulaire myxoïde est considéré comme très rare, représentant environ 3,3 % des liposarcomes dans cette région anatomique et survient généralement dans la cinquième décade. La littérature relative à sa prise en charge est limitée, la plupart du temps, à des petites séries ou à des rapports de cas cliniques.
Nous rapportons le cas d’un liposarcome paratesticulaire myxoïde chez un jeune adulte de 17 ans.
Observation :
Un jeune homme de 17 ans a été admis au service d’urologie pour la prise en charge d’une masse scrotale droite. Le patient n’avait pas d’antécédents pathologiques notables. L’histoire de sa maladie remontait à 5 mois par la découverte d’une masse scrotale droite augmentant progressivement de volume.
L’examen clinique mettait en évidence la présence d’une masse supratesticulaire droite de 06 cm de grand axe, de consistance molle avec des signes inflammatoires en regard et sensible à la palpation. L’examen du scrotum retrouvait des testicules palpables d’aspect normal. Les aires ganglionnaires périphériques étaient libres et l’examen abdominal était normal. Les marqueurs tumoraux (alphafœto-protéine, HCG totale, LDH) étaient normaux.
L’échographie inguino-scrotale mettait en évidence une volumineuse masse scrotale droite.
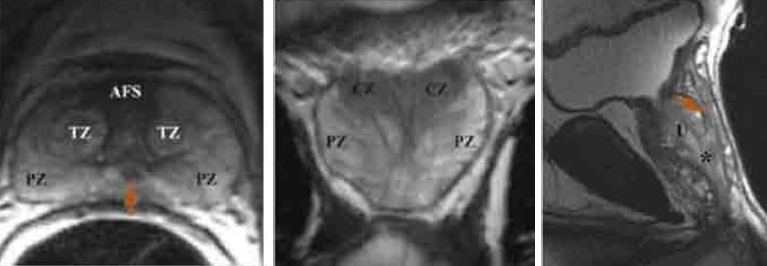
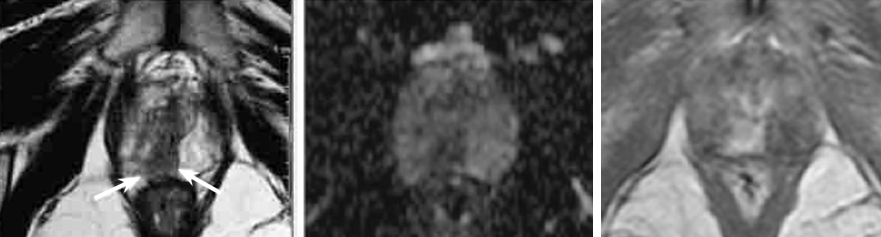
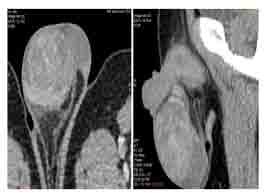
La tomodensitométrie inguino-scrotale objectivait un processus tissulaire hétérogène scrotal droit occupant la totalité de la bourse droite mesurant 56 mm x 51 mm x 72 mm de grands axes, indissociable du pôle supérieure du testicule, sans extension vers l’hémibourse controlatérale, ni vers la région inguinale (figure 1).
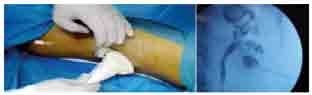
La TDM thoraco-abdominale était normale. Une orchidectomie totale droite par voie inguinale a été réalisée (figure 2). L’examen macroscopique de la pièce opératoire mettait en évidence un testicule normal avec une albuginée intacte refoulé par une masse ferme, faisant 7 x 5 x 4,5 cm, homogène, bien limitée, encapsulée, d’aspect jaunâtre, d’allure graisseuse et n’infiltrant pas le cordon spermatique.
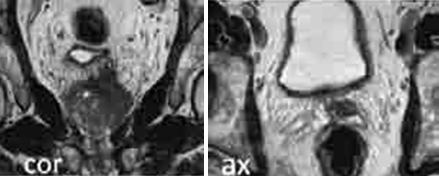
L’étude microscopique notait la présence d’une prolifération tumorale maligne de nature sarcomateuse adoptant une architecture diffuse, peu cellulaire constituée de cellules fusiformes non lipogènes avec zones myxoïdes étendues (figure 3).
La proportion des cellules rondes était inférieure à 5 %. Le stroma tumoral comportait des vaisseaux arachnoïdiens étirés au sein du stroma myxoïde comportant des éléments inflammatoires lymphoplasmocytaires.
Il n’y avait pas de nécrose, mais il y avait des microabcès au niveau de l’épididyme droit. Les limites chirurgicales étaient saines. Devant ces aspects morphologiques, le diagnostic de liposarcome paratesticulaire myxoïde de grade 1 a été posé. L’évolution a été marquée par une progression métastatique trois mois après la chirurgie. Le bilan scannographique d’extension mettait en évidence des localisations métastatiques rétropéritonéales multiples.
Après un bilan de tolérance sans anomalies, le patient a été mis sous chimiothérapie palliative à base de doxorubicine à la dose de 60 mg/m2 tous les 21 jours. Le patient est décédé au décours de la quatrième cure suite à une progression franche métastatique.
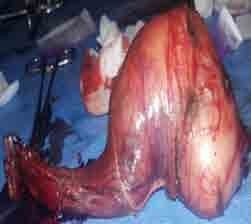
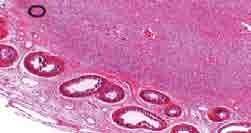
Les tumeurs malignes paratesticulaires sont rares, comprenant principalement des sarcomes provenant du mésenchyme du cordon spermatique [3].
Les liposarcomes représentent 20 % des tumeurs paratesticulaires de l’adulte et occupent la 3ème place après les léiomyosarcomes (32 %) et les rhabdomyosarcomes (24 %)[4].
Ils sont sous diagnostiqués et forment un spectre de lésions d’agressivité variable. Les liposarcomes de l’albuginé, la vaginale, le cordon spermatique, l’épididyme, la peau scrotale et les testicules sont très rares [1]. Le liposarcome paratesticulaire intéresse le sujet âgé entre 50 et 60 ans [4], avec des limites d’âge allant de 16 à 85 ans. Son origine est discutée : soit une transformation sarcomateuse d’un lipome préexistant, soit un liposarcome d’emblée malin, prenant naissance à partir de la graisse rétropéritonéale [5].
Il siège plus volontiers à droite comme le cas de notre patient. Cliniquement, les liposarcomes paratesticulaires n’ont rien de particulier par rapport aux autres tumeurs à développement intrascrotal[6]. La lésion évolue le plus souvent à bas bruit pendant plusieurs mois voire des années. Elle se manifeste par une douleur, pesanteur ou un tiraillement dans 10-15 % des cas et peut être aussi découverte de façon fortuite par la patient ou par le médecin.
Elle est ferme, irrégulière, indolore, développée à proximité ou à distance du testicule pouvant être soit intrascrotale ou au niveau de l’aine ou englober les deux. Elle peut prêter à confusion avec une hernie inguinale, une tumeur testiculaire, une hydrocèle ou une hématocèle, la taille variable allant de quelques centimètres à plus de 30 cm et peut atteindre des proportions gigantesques jusqu’à 13,5 kg.
Il n’y a pas de corrélation entre le volume de la tumeur et sa malignité. Le caractère limité, indolore peut être faussement rassurant, et souvent le diagnostic n’est pas affirmé en pré-opératoire. Il n’y a pas de marqueurs tumoraux pouvant aider au diagnostic. Sur le plan radiologique il n’existe pas de signes radiologiques caractéristiques.
La tomodensitométrie ne semble pas supérieure à l’échographie dans l’exploration locale. L’échographie inguino-scrotale met en évidence typiquement des lésions solides, hyperéchogènes et hétérogènes mais celles-ci peuvent se présenter comme des nodules indurés de petite taille au sein d’un tissu adipeux de consistance proche de la normale, ne permettant pas ainsi la distinction entre les lésions bénignes et malignes.
La tomodensitométrie apporte la preuve de la localisation, de l’étendue, et des rapports de la masse intrascroTale avec le testicule, l’épididyme, et le cordon spermatique. Les liposarcomes sont de faible densité et peuvent être bien délimités.
Il n’y a pas de caractéristiques pathognomoniques pour la différenciation des masses bénignes et malignes définies dans la littérature [7]. Les liposarcomes ont tendance à apparaître bien circonscrits et lobulés sur les IRM [8]. La prise de contraste dépend du niveau de différenciation. Les liposarcomes myxoïdes, montrent une hétérogénéité dans la prise de contraste[8].
Le FDG-PET scan peut être utile dans les cas récurrents mais son utilisation en routine n’est pas indiquée [9].
Sur le plan histologique, le liposarcome paratesticulaire ne diffère pas des autres liposarcomes [8]. Il peut contenir des contingents fibroblastiques et myxomateux.
Ces tumeurs ont été classées en fonction de la prédominance tissulaire en 5 types :
- Le liposarcome bien différencié,
- Le liposarcome myxoïde,
- Le liposarcome à cellules rondes,
- Le liposarcome pléomorphe et
- Le liposarcome indifférencié.
La nouvelle classification de l’organisation mondiale de la Santé OMS fusionne le liposarcome myxoïde et le liposarcome à cellules rondes, puisque ce dernier est simplement une variante de haut grade du premier, et qu’il est commun de voir une transition entre l’un et l’autre dans la même tumeur. Le liposarcome myxoïde survient classiquement dans les tissus mous profonds des extrémités. La localisation rétropéritonéale est exceptionnellement rare, et représente plus probablement une métastase ou une forme très myxoïde de liposarcome bien différencié. D’un point de vue morphologique, le liposarcome myxoïde est constitué de cellules fusiformes, dans une matrice myxoïde, abondante, peu cellulaire, caractérisée par un riche réseau capillaire, grêle. Il est généralement facile de repérer des lipoblastes uni ou multivacuolaires. Il est également possible de retrouver des foyers de tissu adipeux plus mature dans un liposarcome myxoïde, et ceci ne doit pas empêcher de poser le diagnostic. Le liposarcome myxoïde possède un risque de métastase variant de 20 à 40 %, et qui croît avec la proportion de morphologie « à cellules rondes ». Les métastases peuvent survenir à des sites inhabituels, comme les os, le rétropéritoine ou d’autres sites anatomiques dans les tissus mous des extrémités ou du tronc. Contrairement au liposarcome bien différencié, le risque de métastase n’est jamais négligeable. En immunohisto-chimie, un marqueur réputé sensible et spécifique a récemment été décrit (NY-ESO-1) [10], mais son utilisation est peu répandue et sa fiabilité reste à démontrer.
Suivant les principes généraux du traitement du sarcome, l’orchidectomie radicale et la résection large de la tumeur avec des marges négatives microscopiques, représentent la pierre angulaire du traitement à visée curative[11].
L’hémiscrotectomie avec ablation de la peau et du tissu sous-cutané, voire une partie de la paroi abdominale autour de l’orifice inguinal peut être justifiée en cas de tumeur extra capsulaire avec envahissement des structures adjacentes.
L’objectif carcinologique est la résection en monobloc de la tumeur en marge saine microscopique (R0). La qualité d’exérèse est le facteur pronostique le plus significatif, le caractère R2 prédit indépendamment le risque de mortalité spécifique. Il n’y pas de consensus clair concernant l’intérêt du curage ganglionnaire rétropéritonéal, il doit être réservé aux patients chez lesquels des adénopathies ont été identifiées.
Le rôle de la radiothérapie est encore incertain. Alors que Coleman [7] (47 patients) a rapporté que la radio-thérapie adjuvante ne diminue pas significativement le taux de récidives locales et n’améliorait pas la survie globale, d’autres auteurs ont noté un contrôle plus durable après la chirurgie et la radiothérapie combinée. Selon ces auteurs, un traitement combiné est une modalité qui devrait être envisagée, surtout en cas de tumeurs de haut grade, d’invasion lymphatique, de marge de résection envahie, ou en cas de rechute [12].
La zone irradiée doit comprendre la partie proximale du scrotum, et le trajet du canal inguinal, ainsi que les tissus avoisinants et les ganglions pelviens homolatéraux. La dose d’irradiation doit être de 60 à 65 Gy, durant six semaines.
Le rôle de la chimiothérapie dans le traitement des liposarcomes est controversé et également incertain, elle peut être proposée au cas par cas. La plupart des études sont des rapports de cas ou des séries d´un petit nombre de patients. Pour les patients atteints de la maladie métastatique non résécable, une utilisation judicieuse de la chimiothérapie offre une palliation des symptômes et évite une progression rapide de la maladie.
Le choix du protocole de chimiothérapie est souvent calqué sur ceux de liposarcome des tissus mous. Les liposarcomes myxoïdes et à cellules rondes sont parmi
les sous-types les plus sensibles à la chimiothérapie. Les sous types dédifférenciés présentent une grande variabilité individuelle de chimiosensibilité [13]. La chimiothérapie la plus utilisée dans la littérature pour cette localisation rare est la doxorubicine à des doses de 60 à 80 mg/m2 [7].
La récidive locale est le problème principal des sarcomes paratesticulaires. Cette récidive survient dans 30 à 50% des cas [7].
Sa fréquence augmente avec le grade histologique. Elle peut survenir après des délais pouvant dépasser cinq ans. Une surveillance sur plusieurs années est donc requise dans tous les cas. Le pronostic du liposarcome serait meilleur que celui des autres tumeurs malignes paratesticulaires, il dépend fortement du degré de différenciation. Datta pense que ces tumeurs seraient d’autant moins différenciées que le sujet est plus jeune et de ce fait, leur pronostic serait plus mauvais [6].
La survie à 5 ans est de 85 % pour les formes bien différenciées et myxoïdes, 20 % pour les formes lipoblastiques à cellules rondes et les pléomorphes. Les liposarcomes dédifférenciés sont par contre de mauvais pronostic.
Conclusion :
Les liposarcomes paratesticulaires myxoïdes sont des tumeurs malignes extrêmement rares, et se confondent facilement avec une hernie inguinale, une hydrocèle ou des tumeurs testiculaires.
L’absence de signes cliniques et radiologiques spécifiques rend le diagnostic pré-opératoire difficile malgré l’apport des moyens d’imagerie récente.
Le traitement idéal est une orchidectomie totale par voie inguinale élargie, si besoin aux structures adjacentes. Ces tumeurs ont une tendance à la récidive locale, mais la survenue de métastases n’est pas fréquente.
La stratégie la plus probable, pour réduire ce taux de récidive locale élevé, serait une combinaison de chirurgie et de radiothérapie.
Néanmoins, aucune série n’est suffisamment importante pour évaluer définitivement l´efficacité de cette stratégie, et des études prospectives sont exclues du fait de la rareté de cette pathologie.
Références :
- John T, Portenier D, Auster B, Mehregan D, Drelichman A, Telmos A. Leiomyosarcoma of scrotum-case report and review of literature. Urology. 2006; 67(2): 424.
- Evans H.L. Liposarcoma. A study of 55 cases with a reassessment of its classification. Am. J. Surg. Pathol., 1979, 3, 507-523.
- Schwartz SL, Swierzewski SJ 3rd, Sondak VK, Grossman HB. Liposarcoma of the spermatic cord: report of 6 cases and review of the literature. J Urol. 1995 Jan;153(1):154–7.
- Khoubehi B, Mishra V, Ali M, Motiwala H, Karim O. Adult paratesticular tumors. BJU int. 2002; 90 (7): 707-715.
- Cuzzocrea D.E., Loconte G., Aiello E., Menniti D.B., Bertoni F., Fornarola V. Liposarcome: review of the literature and des- cription of two cases. Acta Urol. Ital., 1998, 12, 237-239.
- Datta N.S., Singh S.M., Bapna B.C. Liposarcoma of the spermatic cord: report of a case and review of the literature. J. Urol., 1971, 106, 888-889.
- Coleman J, Brennan MF, Alektiar K, Russo P. Adult sperma- tic cord sarcomas: management and results. Ann Surg Oncol. 2003 Jul; 10(6): 669-75.
- Arkun R, Memis A, Akalin T et al. Liposarcoma of soft tis- sue: MRI findings with pathologic correlation. Skeletal Radio- logy. 1997 Mar; 26(3): 167-72.
- Goodman FR, Staunton MD, Rees HC. Liposarcoma of the spermatic cord. J R Soc Med. 1991 Aug;84(8):499–500.
- Hemminger JA, Iwenofu OH. NY-ESO-1 is a sensitive and specific immuno-histo-chemical marker for myxoid and round cell liposarcomas among related mesenchymal myxoïd neoplasms. Mod Pathol. 2013 Sep; 26(9): 1204-10.
- Rabbani F, Wright J, Mcloughlin M. Sarcomas of the sperma- tic cord: signifiance of wide local excision. Canad J Urol. 1997; 4(2): 366-376.
- Hassan JM , Quisling SV, Melvin WV et al. «Liposarcoma of the Spermatic Cord Masquerading as an Incarcerated In- guinal Hernia.» The American Surgeon. 2003; 69(2): 163-165.
- Jones RL, Fisher C, Al-Muderis O, Judson IR. Differential sensitivity of liposarcoma subtypes to chemotherapy. Eur J Cancer. 2005; 41:2853.