A. BOUGHABA,H. KISMOUNE, Service de Chirurgie Plastique et Brûlés, CHU Djillali Bounaâma, Douéra, Alger.
Abstract : Human skin is a complex organ that provides multiple functions including protection and barrier against external aggressions. Hence the interest of ensuring a good restitution of its integrity in the event of break-in by a wound or a burn ; thus formation of the scar. Our goal is to obtain the most optimal cicatricial result possible both functionally and aesthetically and specially to have a stable scar preventing any neoplasic degeneration.
Résumé : la peau humaine est un organe complexe qui assure de multiples fonctions notamment de protection et de barrière contre les agressions externes. D’où l’intérêt d’assurer une bonne restitution de son intégrité en cas d’effraction de celle-ci par une plaie ou une brûlure ; donc formation de la cicatrice. Notre objectif est d’obtenir un résultat cicatriciel le plus optimal possible aussi bien sur le plan fonctionnel qu’esthétique et surtout d’avoir une cicatrice stable prévenant toute dégénérescence néoplasique.
Dans la société, le principe d’identité repose sur l’apparence physique ; ce qui a amené le psychanalyste Français Didier Anzieu à déterminer l’expression « moi-peau ».
Ainsi toute modification de l’intégrité de la peau peut engendrer un handicap difficile à surmonter par le patient d’où l’intérêt de bien connaitre le processus de cicatrisation, quelles sont ses formes, maitriser ses phases afin d’anticiper les conséquences et de prévoir la meilleure réparation possible.
Qu’est-ce qu’une cicatrisation :
C’est une cascade de processus physiologiques qui fait suite à toute agression cutanée dont l’objectif est d’aboutir à une restitution de l’intégrité cutanée.
« La cicatrisation est l’aventure d’une plaie, d’une nécrose, d’une brûlure, aboutissant à la cicatrice » (Vilain).
Rappel anatomique :
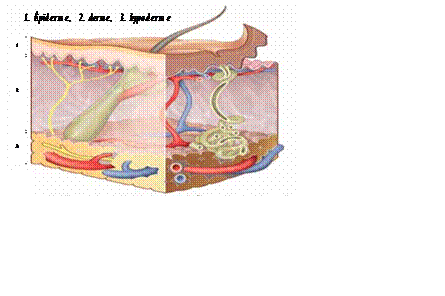
La peau est l’organe le plus étendu du corps=2 m2. Composée de trois couches qui sont de dehors en dedans
L’épiderme ;
Le derme ;
L’hypoderme. (figure 1)
Figure 1 : Schéma des couches de la peau.
Quels sont les types de la cicatrisation ?
cicatrisation primaire : elle se produit quand une suture est réalisée de façon convenable avec des berges propres, non contuses, sans tension et parfaitement affrontées. Elle se déroule en 5 phases successives. Ce processus dure 12 à 18 mois pour avoir une cicatrisation définitive.
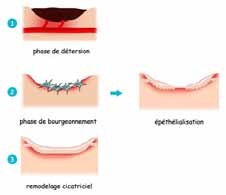
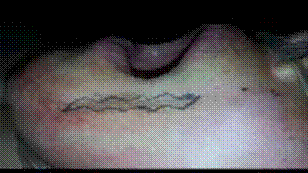
cicatrisation secondaire ou dirigée : si fermeture impossible, mauvaises conditions locales (sepsis, souillée) plaie contuse et dilacérée, brûlures cutanées. Elle se déroule en 3 phases qui se succèdent obligatoirement. Elle est souvent longue et aboutit à des cicatrices dont l’aspect cosmétique est rarement optimal (cicatrice pathologique) (figures 2, 3).
Figure 2 : Les 3 phases de la cicatrisation secondaireFigure 3 : Cicatrisation dirigée d’une PDS post infectieuse para ombilicale.
Quels facteurs influencent la cicatrisation ?
• Facteurs intrinsèques :
Siège et nature et étendue de la plaie
Caracteristiques de la plaie
Respect des règles de bases de la suture
• Facteurs extrinsèques :
Age, tabac, état général, malnutrition, diabete, éléments iatrogènes notamment la corticothérapie++.
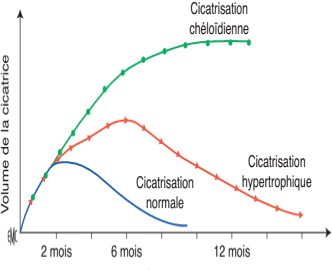
Quelles sont les formes de la cicatrisation ?
Figure 4 : Évolution cicatricielle.
• Cicatrisation normale :
C’est la restitution optimale de l’intégrité de la peau après une aggression externe par la succession des différentes phases de la cicatrisation. Néanmoins il arrive que cette cicatrisation aboutisse à un résultat cicatriciel défectueux ou qu’elle soit pathologique ; ces deux situations représentent une forte demande des patients .
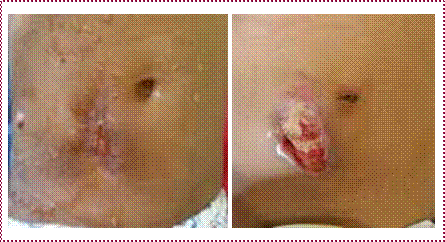
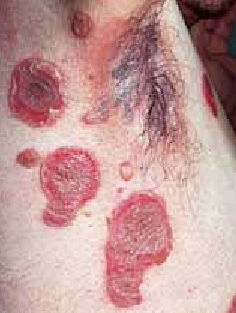
Cicatrices pathologiques : (cicatrice hypertrophiques et chéloïdes). Elles répondent à un processus évolutif, se voient surtout en cas de brûlures profondes, elles sont frustrantes (figures 5, 6).
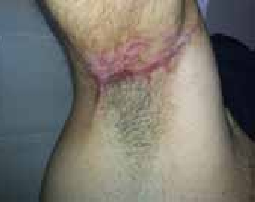
Figure 5 : Cicatrice hypertrophique post brûlure thermiqueFigure 6 : Cicatrice hypertrophique axillaire droite post traumatique
Cicatrices défectueuses : contrairement aux précédentes elles sont stables (caractère fixé). Elles se voient surtout suite à un défaut de la prise en charge initiale médicale ou chirurgicale. Elles sont de deux types soit par :
– malfaçon : décalée, adhérente, en barreau d’échelle, déprimée, tatouée, glabre et oreille cicatricielle (figures 7, 8).
– malévolution : cicatrice élargie ou déhiscente, rétractile et dyschromique (figures 9, 10).
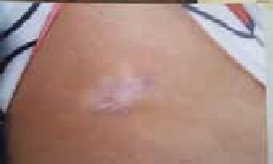
Figure 7 : Cicatrice défectueuse Décalée et déprimée
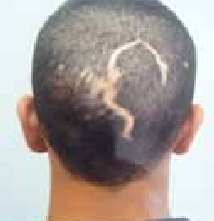
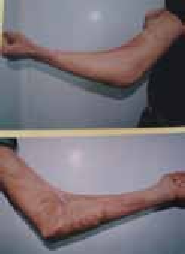
Figure 8 : Cicatrice glabre « alopécie » du cuir chevelu post chirurgicaleFigure 9 : Cicatrice déhiscenteFigure 10 : Cicatrice rétractile du coude post brûlures thermiques
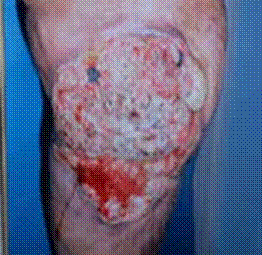
Cicatrice dégénérée post brûlures : la dégénérescence cicatricielle est une éventualité qu’il faut connaitre. Toute ulcération récidivante ou permanente survenant sur les cicatrices anciennes (20-30 ans) ou les cicatrices soumises à des traumatismes par étirement est néoplasique jusqu’à preuve du contraire (figure 11).
Figure 11 : Epithélioma spinocellulaire sur séquelle de brûlures du genou gauche – Cicatrice dégénérée
Traitement :
Cicatrice hypertrophique : essentiellement médical, s’améliore spontanément par pressothérapie.
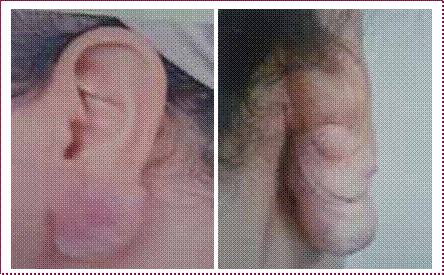
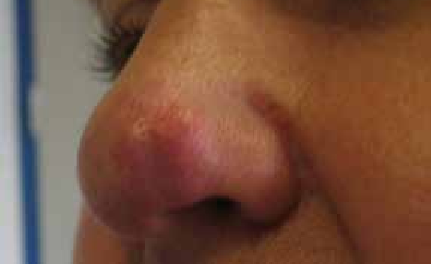
Cicatrice chéloïde : récidive souvent après exérèse chirurgicale ; répond bien à la pressothérapie, injection de corticoïdes (efficacité 40-70 %) (figure 12).
Figure 12 : Cicatrice chéloïde du lobe de l’oreille après presso thérapie rigide des clips
Autre procédés :
Radiothérapie, laser : CO2/YAG, cryothérapie 5FU, mitomycine C, acide rétinoïque, toxine botulique qui n’ont pas montré un réel avantage par rapport aux autres procédés.
Pour les cicatrices défectueuses, la prise charge chirurgicale reste le traitement de fond pour améliorer l’esthétique et le fonctionnel soit par chirurgie d’enfouissement « désépidermisation » ou par réalignement ou libération par des plasties locales « Z » « VY » (figure 13).
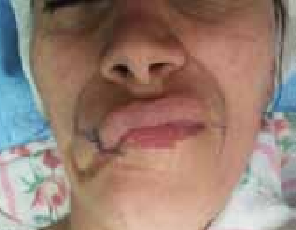
Figure 13 : Cicatrice rétractile para commissurale gauche
Armes préventives :
Pour la cicatrisation primaire : il faut
Faire un parage et nettoyage, antibiothérapie
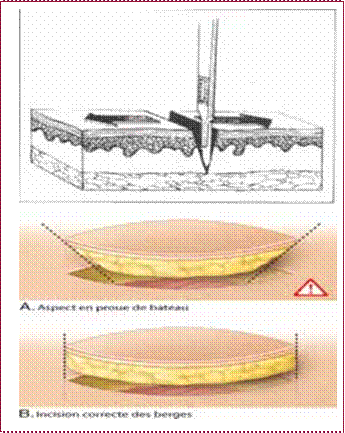
Précautions techniques, avant tout faire une incision correcte des berges avec une lame de bistouri
perpendiculaire à la peau (figure 14).
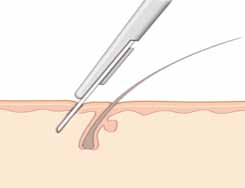
Sauf au niveau de cuir chevelu, il faut inciser parallèlement à l’implantation pilaire (figure 15).
Respecter les lignes de moindre tension de Langer, les plis de flexion et les orifices naturels,
Minimiser les manipulations des berges qui doivent être atraumatiques et utiliser un matériel adéquat (pince fines, à griffe, crochets
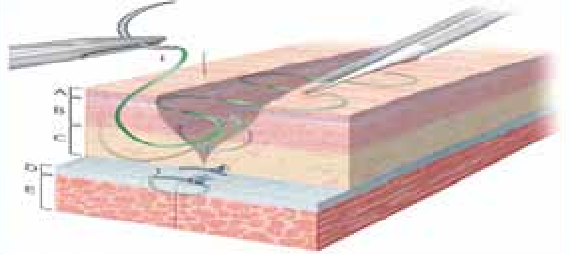
Assurer une hémostase soigneuse.Faire affrontement parfait, éviter d’avoir un espace mort : drainage +++, pas de décalage.Faire des sutures sans tension : si indication de décollement périphérique, il doit être prudent.Respect les plans de clivage, suturer plan par plan sachant que le derme est la zone de résistance de la peau, assistée par steristrips ou adhésifs (figure 16).
Choisir un bon diamètre de fils selon l’unité anatomique et faire une ablation de fils précoce. •
4ème jour au niveau de la face, 8-10ème jour au niveau de la main, 15ème jour au niveau des membres, du dos et du périnée.
Figure 14 : Techniques de l’incision cutanéeFigure 15 : L’incision au niveau du cuir chevelu FigureFigure 16 : Suture cutanée.
Particularité chez l’enfant :
Privilégier les points enfouis et steristrips.
Pour la cicatrisation secondaire et plus particulièrement les brûlures, l’arme préventive essentielle est d’avoir une cicatrisation rapide en faisant des pansements qui respectent les différentes phases de la cicatrisation, faire des recouvrements par une greffe cutanée pour les brûlures profondes avec une pressothérapie (souple ou rigide).
Au niveau des lobes de l’oreille on utilise des clips auriculaires rigides en Orlen avec plaque de gel de silicone (compression sélective). Cette pressothérapie à double objectifs thérapeutique et préventif doit être débutée immédiatement après épithélialisation.
Elle doit être > 25 mmhg pendant au moins 24 mois en double jeux avec renouvellement tous les 10 mois chez l’adulte et en fonction de la croissance chez l’enfant.
Prendre en charge toute lésion instable évoluant depuis plus de 2 mois survenant sur une cicatrice.
Conclusion :
Les bonnes pratiques de la prise en charge des plaies assurent la maitrise des séquelles fonctionnelles et esthétiques et surtout psychologiques, grâce à la connaissance de l’anatomie, des règles de réparation et des processus physiologiques de la cicatrisation.
Références
B. Chaput. Anomalie de la cicatrisation. EMC techniques chirurgicales chirurgie plastique réparatrice et esthétique 2017.
z. ELOSMANI, Y. ABI AYAD,S. HAMzAOUI, A. SERRADJ, Établissement Hospitalo-Universitaire 1er Novembre, Oran.
Abstract : Vascular abnormalities are a heterogeneous group of conditions characterized by vascular ectasia that may be of interest to all organs, but most often affect the skin. Their diagnosis is sometimes difficult and requires an accurate analysis to isolate and classify each entity. Thus, a classification of superficial vascular anomalies has been carried out thanks to a multidisciplinary approach, or there are two main categories: vascular tumors (infantile hemangiomas) and slow-flow vascular malformations, or fast-flowing malformations, arteriovenous. The objective is to facilitate diagnosis and management.
Key-words : Hemangiomas, vascular malformation.
Résumé : Les anomalies vasculaires constituent un groupe hétérogène d’affections caractérisées par des ectasies vasculaires qui peuvent intéresser tous les organes, mais touchent le plus souvent la peau. Leur diagnostic est parfois difficile et nécessite une analyse précise pour isoler et classer chaque entité. Ainsi, une classification des anomalies vasculaires superficielles a été réalisée grâce à une approche multidisciplinaire, où on retrouve deux grandes catégories : les tumeurs vasculaires (hémangiomes infantiles), et les malformations vasculaires à flux lent, ou les malformations à flux rapide, artérioveineuses. Le but étant de faciliter le diagnostic et la prise en charge.
Mots-clés : Hémangiomes, malformation vasculaire.
Introduction :
Afin d’améliorer la compréhension des anomalies vasculaires, et de ce fait, leur prise en charge thérapeutique, une classification clinique, radiologique et histopathologique, internationale et interdisciplinaire des anomalies vasculaires superficielles de la face, du tronc et des membres a été établie en 1982 par Mulliken et Glowacki et validée par l’International Society for the Study of Vascular Anomalies (ISSVA), dont la plus récente a été élaborée en 1996 [1].
Il se dégage deux grandes catégories :
Les tumeurs vasculaires (hémangiomes infantiles)
Les malformations vasculaires à flux lent (capillaires, veineuses, lymphatiques) ou les malformations à flux rapide, ( artérioveineuses).
Elles sont simples (affectant un seul secteur vasculaire) ou complexes.
Le but de cette classification est de faciliter le diagnostic clinique et la prise en charge qui découle de celle-ci.
1. Tumeur vasculaire : l’hemangiome infantile
C’est la tumeur la plus fréquente du nourrisson (10 %), avec une nette prédominance féminine (sexe ratio : 5 f illes/1 garçon) [1]. Plusieurs facteurs prédisposants ont été mis en évidence : le sexe féminin, la peau
blanche, un contexte d’hypoxie prénatale (anomalies placentaires, pré-éclampsie maternelle), nouveau-né de petit poids de naissance. L’hémangiome infantile, correspond à une prolifération cellulaire endothéliale, qui apparaît classiquement après quelques jours ou quelques semaines de vie, cet intervalle libre représente un bon signe diagnostique [2].
on distingue trois types cliniques (figure 1, 2,3) [1,2] :
– La forme tubéreuse ou superficielle (figure 1) qui correspond à une tache rouge, saillante, à surface irrégulière et à bords nets communément appelée angiome « fraise ».
Figure 1 : Hémangiome, forme tubéreuse. (Martin A, Barbier C, Do- mengie F, Nouri M, Cottier JP, Herbreteau D. Les anomalies vasculaires Superficielles. Intérêt de la classification dans la prise en charge radiologique et thérapeutique).
La forme sous-cutanée (figure 2) (touchant le derme profond) se présente sous la forme d’une tuméfaction de consistance ferme, élastique, chaude mais non battante soulevant une peau saine légèrement bleutée ou rosée.
Figure 2 : Hémangiome sous-cutané. (Martin A, Barbier C, Domengie F, Nouri M, Cottier JP, Herbreteau D. Les anomalies vasculaires superficielles. Intérêt de la classification dans la prise en charge radiologique et thérapeutique).
La forme mixte (figure 3) réunit les deux aspects : la partie tubéreuse se développe en premier et la partie profonde apparaît quelques mois plus tard, débordant la première d’un halo bleuté.
Figure 3 : Hémangiome mixte. (Martin A, Barbier C, Domengie F, Nouri M, Cottier JP, Herbreteau D. Les anomalies vasculaires superficielles. Intérêt de la classification dans la prise en charge radiologique et thérapeutique).
Quel que soit le type, l’hémangiome est de consistance ferme et élastique, légèrement chaud à la palpation, mais non pulsatile et généralement indolore, sauf en cas d’ulcération [2].
Sa taille est très variable, allant de l’atteinte ponctiforme à l’atteinte d’un membre ou d’un hémitronc, mais, le plus souvent, elle est modérée, inférieure à 3 cm [2].
La localisation des hémangiomes est ubiquitaire, cependant, ils semblent plus fréquents sur le visage (40 %) et le cou (20 %) [2].
La croissance est accélérée pendant les trois premiers mois mais peut se prolonger jusqu’au 8ème mois pour les formes superficielles, et jusqu’au 12ème mois pour les formes à participation sous-cutanée. Dans de rares cas, cela peut aller jusqu’à deux ans [2].
Puis, il se stabilise spontanément, survient alors une régression sur plusieurs mois ou années. L’involution est lente et se traduit par l’apparition du blanchiment central des lésions superficielles et l’affaissement des composantes sous-cutanées.
La distribution des hémangiomes sur le visage est variable, il existe en effet :
Des formes focales qui sont situées sur les proéminences, et 60 % d’entre elles se concentrent en zone centrofaciale [3],
Des formes diffuses, où on retrouve une distribution segmentaire [3].
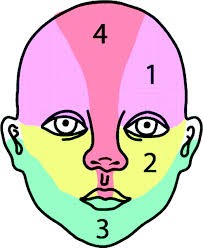
Une nouvelle classification topographique de ces formes segmentaires faciales en quatre aires de S1 à S4 a été proposée (figure 4). Ces formes segmentaires sont volontiers associées à des malformations (Syndromes PHACES et Syndrome PELVIS/SACRAL) et s’ulcèrent fréquemment [3].
Figure 4 : Segmentation S1 à S4 du du syndrome PHACES (Léauté-Labrèze C, Sans-Martin V. Hémangiome infantile, La presse médicale. tome 39 > n84 > avril 2010.doi: 10.1016/j.lpm.2009.10.015).
A. Syndrome PHACES [3] :
L’acronyme anglais PHACE(S), créé en 1996 par Frieden, regroupe les anomalies suivantes :
Hémangiome facial,
Anomalies de la fosse postérieure,
Anomalies artérielles intra et extra crâniennes,
Anomalies cardiaques congénitales et coarctation aortique,
Anomalies oculaires (eye),
Anomalies sternales et ventrales (ajoutées par la suite).
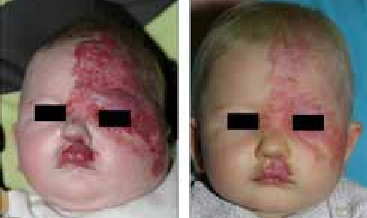
Les anomalies neurologiques intracrâniennes accompagnent dans plus de 3/4 des cas, des hémangiomes en plaques couvrant l’aire fronto palpébrale supérieure S1 + S4 (figure 5).
Figure 5 : a et b. Hémangiome dit segmentaire de type syndrome PHACES (atteinte S1 + S4) ; Fig. 5a : Hémangiome avant traitement. Fig. 5b : Hémangiome après 2 mois de propranolol. (Léauté-Labrèze C, Sans-Martin V. Hémangiome infantile. La presse médicale. tome 39 > n84 > avril 2010.doi: 10.1016/j.lpm.2009.10.015).
Les anomalies cardiaques et aortiques seraient plus particulièrement associées aux hémangiomes S3.
Ainsi, les explorations qui doivent être réalisées, sont :
l’imagerie par résonance magnétique (IRM) cérébrale,
l’échographie cardiaque et des gros vaisseaux,
l’examen ophtalmologique,
l’échographie abdominale.
A. Syndrome PELVIS ou SACRAL [3] :
L’acronyme anglais PELVIS regroupe les anomalies suivantes :
Hémangiomes périnéaux
Malformations génitales externes
Lipomyelomeningocèle
Anomalies vésicorénales
Anus imperforé
Autre marqueur cutané (skin tag).
Les explorations, qui doivent être réalisées, chez tout nouveau-né à risque sont une évaluation par IRM médullaire, et abdominopelvienne (plus sensible que l’échographie).
Traitement de l’hémangiome infantile :
L’abstention thérapeutique est parfaitement défendable dans les formes bénignes, pour lesquelles il est à prévoir peu ou pas de séquelle après régression [3].
Indications thérapeutiques [3] :
L’hémangiome infantile avec mise en jeu du pronostic vital.
L’hémangiome infantile avec mise en jeu du pronostic fonctionnel.
L’hémangiome infantile qui se complique d’ulcération,
L’hémangiome infantile qui engendre un préjudice esthétique majeur avec retentissement psychologique.
1. Le Propranolol :
«I’vebeendoingpediatricdermatologyfor25years,andthisisthefastestI’veeverseen a new therapy change the way we do things »1 (Bernard A. Cohen, M.D., Pédiatrie dermatologique Johns Hopkins Children’s Center) [3]. Le propranolol est un bêta-bloquant non cardiosélectif sans activité sympathomimétique intrinsèque.
Posologie : 2 à 3 mg/kg/j pour une durée minimum de 6 mois.
Il faut débuter le plus tôt possible, avant 3 mois (âge « idéal » probablement entre un et 2 mois).
On observe dans les heures qui suivent la prise de propranolol un affaissement et un changement de couleur de l’hémangiome, ensuite, l’effet se poursuit, mais plus lentement, aboutissant à une régression plus ou moins complète.
Les principaux effets secondaires sont : les hypoglycémies en période néonatale ou dans des situations de jeûne, des malaises avec pâleur, des épisodes de cyanose et d’hypotension. La mise en route du traitement doit se faire en milieu pédiatrique avec surveillance de la fréquence cardiaque et de la tension artérielle, puis le traitement est ambulatoire [2].
Le traitement est maintenu jusqu’à la fin de la période de croissance supposée de l’hémangiome [2].
1. La corticothérapie générale :
On utilise la prednisone le plus souvent, avec une posologie d’attaque variant entre 2 et 5 mg/kg/j pendant au moins 2 mois, puis diminuée très progressivement de façon à couvrir la période évolutive des premiers mois [2]. Le taux de réponse (régression ou simple stabilisation) n’est que de 30 à 60 %, l’effet apparaissant entre la 2ème et les 3èmes semaines de traitement.
2. La Chirurgie :
La Chirurgie précoce : en phase de croissance, est indiquée pour les hémangiomes infantiles globuleux ou
« pendulum », en particulier sur le nez, les paupières et les lèvres [2],
La Chirurgie tardive : occupe une place indispensable dans la réparation des séquelles cutanées (résidus fibro-adipeux) et structurales, après disparition de l’hémangiome [2].
3. Lasers :
Lelaseràcolorantpulsé:efficace sur la composante
superficielle en entraînant une décoloration, et aide à la cicatrisation de certains hémangiomes ulcérés. En revanche, il n’a aucun impact sur les composantes dermiques profondes [3].
Les laser CO2 ou le laser Erbium : sont indiqués en phase tardive sur des zones cicatricielles. Ils améliorent l’aspect par leur effet lissant et tenseur.
Les séances, qui sont douloureuses, sont réalisées sous anesthésie générale.
4. Autres :
Les pansements vaselinés, hydro cellulaires et hydro colloïdes sont utilisés dans les hémangiomes ulcérés. Ils ont un remarquable pouvoir antalgique [2].
2. Les malformations vasculaires :
Contrairement, aux tumeurs vasculaires, ces malformations vasculaires ne régresseront jamais [1].
Deux grands groupes se distinguent selon un critère hémodynamique [4] :
Les malformations vasculaires à flux lent.
Les malformation vasculaire à flux rapide.
A. I – Les malformations vasculaires à flux lent :
1. Les malformations capillaires :
Angiome plan (AP) :
Il s’agit d’une lésion cutanée rouge intense en période néonatale, froide et non battante, qui va pâlir lentement, sans régresser, exception faite de la forme médio frontale et cervicale qui disparaissent en un à deux ans. Cependant, il faut se méfier des faux angiomes plans, chauds, qui sont en fait la couverture cutanée d’une malformation artérioveineuse [1,4].
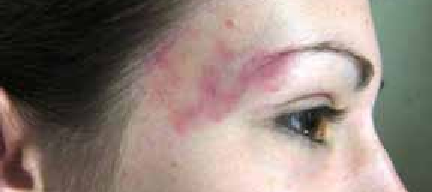
L’angiome plan n’a pas de retentissement régional ou général et constitue seulement un préjudice esthétique (figure 6), et il est dans la majorité des cas localisé, isolé, et stable [4].
Figure 6 : Angiome plan temporal – territoire V1 partiel. (Barbier C, Martin A, Papagnanaki C, Nouri M, Cottier JP, Herbreteau D. Clas- sification des anomalies vasculaires superficielles. STV. Mini-revue. Sang Thrombose Vaisseaux 2009 ;21, n° 5-6 : 248-57.doi: 10.1684/ stv.2009.0399).
Il peut, par ailleurs, être le marqueur d’une angiomatose complexe [4] :
L’angiome plan trigéminé est le témoin d’une phacomatose angiomateuse régionale appelée le syndrome de Sturge Weber ou angiomatose encéphalo-trigéminée (figure 7).
Figure 7 : Le syndrome de Sturge Weber Krabbe. (Le site des ophtalmologistes de France. Encyclopédie de la vue).
L’angiome plan situé au tronc, incite à rechercher une atteinte segmentaire cutanéo-vertébro-méningo-médullaire ou syndrome de Cobb.
L’angiome plan des membres se rencontre dans le syndrome de Klippel-Trenaunay ou dans le syndrome de Parkes Weber, associé à d’autres malformations vasculaires à flux lent dans le premier et à flux rapide dans le second.
L’angiome plan lombo-sacré s’accompagne d’autres anomalies cutanées, incite à rechercher un dysraphisme spinal. Le laser à colorant pulsé (LCP) est le traitement de référence des angiomes plans du nourrisson et de l’enfant, dont l’efficacité a été démontrée, avec une bonne tolérance (pas ou peu de risque cicatriciel), en débutant le plus tôt possible pour les surfaces étendues et l’angiome plan affichant de la face [3].
Les télangiectasies :
Il s’agit de dysplasies capillaires dermiques mais de petite taille (quelques millimètres) et de morphologie variable : lenticulaires, linéaires ou stellaire « l’angiome stellaire ». Il est nécessaire de distinguer la maladie de Rendu-Osler (télangiectasie hémorragique héréditaire) qui est une angiomatose héréditaire de transmission autosomique dominante [1,4].
2. Les malformations veineuses :
Les malformations veineuses sont constituées de tuméfactions bleutées sous-cutanées. Elles sont de consistance molle et froide au contact (Figure 8). Elles gonflent en position déclive, et se vident à la pression ou à la surélévation d’un membre. C’est un excellent signe distinctif. Les malformations veineuses intéressent tous les plans : les tissus cutanés, sous-cutanés, muqueux ou sous-muqueux, les muscles, la synoviale, l’os [4].
Lorsqu’il s’agit d’une malformations veineuses localisée, le traitement repose sur les mesures d’hygiène de vie et la sclérothérapie percutanée. En cas d’échec d’injections itératives, une chirurgie est proposée [5].
Figure 8 : Malformation Veineuse cervico-céphalique. (Barbier C, Martin A, Papagnanaki C, Nouri M, Cottier JP, Herbreteau D. Clas- sification des anomalies vasculaires superficielles. STV. Mini-revue. Sang Thrombose Vaisseaux 2009 ;21, n° 5-6 : 248-57.doi: 10.1684/ stv.2009.0399).
1. Les malformations lymphatiques kystiques :
Les malformations lymphatiques kystiques (MLK) sont hémodynamiquement inactives, et sont constituées de vaisseaux lymphatiques anormaux et de kystes. Ce sont des malformations congénitales. Elles se divisent classiquement en [1,4] :
a. Malformations lymphatiques microkystiques (forme tissulaire), formées de kystes inférieurs à 2 cm, et qui se présentent comme une nappe de petites vésicules translucides ou hématiques, non douloureuses [1,4].
b. Malformations lymphatiques macrokystiques (forme kystique), sont des tuméfactions volumineuses, formées de lésions supérieures à 2 cm, et qui apparaissent comme une tuméfaction dure, rénitente, bien limitée, localisée préférentiellement dans les régions cervico-encéphaliques et axillaires (figure 9) [1,4].
Figure 9 : Malformations lymphatiques macrokystiques (Barbier C, Martin A, Papagnanaki C, Nouri M, Cottier JP, Herbreteau D. Classification des anomalies vasculaires superficielles. STV. Mini-revue. Sang Thrombose Vaisseaux 2009 ;21, n° 5-6 : 248-57.doi : 10.1684/ stv.2009.0399).
c. Malformations lymphatiques mixtes qui associent ces deux formes.
Le traitement repose sur la sclérothérapie percutanée en première intention et s’effectue à distance d’épisodes infectieux, de ce fait, la prévention d’épisodes infectieux est primordiale dans la prise en charge au quotidien. Une chirurgie est complémentaire selon la localisation et le pronostic [5].
B. Les Malformations vasculaires à flux rapide [6] :
1. Malformations artério-veineuses :
Ce sont les malformations les plus dangereuses car hémodynamiquement actives avec des aggravations parfois dramatiques, on insistera sur la malformation artérioveineuse proprement dite qui comporte un nidus avec de multiples shunts.
L’évolution naturelle s’effectue selon 4 étapes (Schobinger 1995) [6] :
Stade I :Dormance : « pseudo-angiome plan », ce qui peut entraîner la confusion sur la prise en charge (figure 10), il faut rechercher une augmentation de la chaleur locale et un thrill (Echodoppler au moindre doute). Ici est recommandée une surveillance clinique et par Echodoppler.
Figure 10 : MAV du nez en phase de dormance. (Barbier C, Martin A, Papagnanaki C, Nouri M, Cottier JP, Herbreteau D. Classification des anomalies vasculaires superficielles. STV. Mini-revue. Sang Throm- bose Vaisseaux 2009 ;21, n° 5-6 : 248-57.doi : 10.1684/stv.2009.0399).
Stade II : Expansion : la malformation s’étend, devient rouge, chaude et battante. Des veines de drainage tortueuses apparaissent à la surface.
Stade III :Destruction, s’accompagne habituellement de complications tissulaires comme l’ischémie, l’ulcération, la nécrose et les hémorragies.
Stade IV : Stade III avec décompensation cardiaque.
Pour les Stades II, III et IV : on réalise une embolisation endo-vasculaire sélective ou par l’association embolisation-chirurgie.
Conclusion :
Cette classification clinique permet d’améliorer la prise en charge des malformations vasculaires superficielles qui sont rares, et donc souvent méconnues ; au sein de groupes multidisciplinaires, permettant ainsi un diagnostic rapide et un traitement adéquat.
Références :
Barbier C, Martin A, Papagnanaki C, Nouri M, Cottier JP, Herbreteau D. Classification des anomalies vasculaires superficielles. STV. Mini-revue. Sang Thrombose Vaisseaux 2009 ;21, n° 5-6 : 248-57.doi : 10.1684/stv.2009.0399.
Léauté-Labrèze C, Sans-Martin V. Hémangiome infantile. La presse médicale. tome 39 > n84 > avril 2010.doi: 10.1016/j.lpm.2009.10.015.
Léauté-Labrèze C. Les anomalies vasculaires : Classification, diagnostic, traitement. Diplôme inter universitaire de lasers dermatologiques. Bordeaux. Février 2016.
Martin A, Barbier C, Domengie F, Nouri M, Cottier JP, Herbreteau D. Stratégies d’exploration et traitement des anomalies vasculaires superficielles. STV. Mini-revue. Sang Thrombose Vaisseaux 2009 ;21, n° 5-6 : 259-67. doi : 10.1684/stv.2009.0400.
Dompmartin A. Classification des anomalies vasculaires. Annales de dermatologie et de vénéréologie (2013) 140, 337—339.doi. org/10.1016/j.annder.2013.02.002.
Bonnetblanc J.-M. Angiomes cutanés. Annales de dermatologie et de vénéréologie (2008) 135S, F181—F187. doi: 10.1016/j.annder.2008.07.001.
z. ELOSMANI, Y. ABI AYAD,S. HAMzAOUI, A. SERRADJ, Établissement Hospitalo-Universitaire 1er Novembre, Oran.
Abstract : Vascular abnormalities are a heterogeneous group of conditions characterized by vascular ectasia that may be of interest to all organs, but most often affect the skin. Their diagnosis is sometimes difficult and requires an accurate analysis to isolate and classify each entity. Thus, a classification of superficial vascular anomalies has been carried out thanks to a multidisciplinary approach, or there are two main categories: vascular tumors (infantile hemangiomas) and slow-flow vascular malformations, or fast-flowing malformations, arteriovenous. The objective is to facilitate diagnosis and management.
Key-words : Hemangiomas, vascular malformation.
Résumé : Les anomalies vasculaires constituent un groupe hétérogène d’affections caractérisées par des ectasies vasculaires qui peuvent intéresser tous les organes, mais touchent le plus souvent la peau. Leur diagnostic est parfois difficile et nécessite une analyse précise pour isoler et classer chaque entité. Ainsi, une classification des anomalies vasculaires superficielles a été réalisée grâce à une approche multidisciplinaire, où on retrouve deux grandes catégories : les tumeurs vasculaires (hémangiomes infantiles), et les malformations vasculaires à flux lent, ou les malformations à flux rapide, artérioveineuses. Le but étant de faciliter le diagnostic et la prise en charge.
Mots-clés : Hémangiomes, malformation vasculaire.
Introduction :
Afin d’améliorer la compréhension des anomalies vasculaires, et de ce fait, leur prise en charge thérapeutique, une classification clinique, radiologique et histopathologique, internationale et interdisciplinaire des anomalies vasculaires superficielles de la face, du tronc et des membres a été établie en 1982 par Mulliken et Glowacki et validée par l’International Society for the Study of Vascular Anomalies (ISSVA), dont la plus récente a été élaborée en 1996 [1].
Il se dégage deux grandes catégories :
Les tumeurs vasculaires (hémangiomes infantiles)
Les malformations vasculaires à flux lent (capillaires, veineuses, lymphatiques) ou les malformations à flux rapide, ( artérioveineuses).
Elles sont simples (affectant un seul secteur vasculaire) ou complexes.
Le but de cette classification est de faciliter le diagnostic clinique et la prise en charge qui découle de celle-ci.
1. Tumeur vasculaire : l’hemangiome infantile
C’est la tumeur la plus fréquente du nourrisson (10 %), avec une nette prédominance féminine (sexe ratio : 5 f illes/1 garçon) [1]. Plusieurs facteurs prédisposants ont été mis en évidence : le sexe féminin, la peau
blanche, un contexte d’hypoxie prénatale (anomalies placentaires, pré-éclampsie maternelle), nouveau-né de petit poids de naissance. L’hémangiome infantile, correspond à une prolifération cellulaire endothéliale, qui apparaît classiquement après quelques jours ou quelques semaines de vie, cet intervalle libre représente un bon signe diagnostique [2].
on distingue trois types cliniques (figure 1, 2,3) [1,2] :
– La forme tubéreuse ou superficielle (figure 1) qui correspond à une tache rouge, saillante, à surface irrégulière et à bords nets communément appelée angiome « fraise ».
Figure 1 : Hémangiome, forme tubéreuse. (Martin A, Barbier C, Do- mengie F, Nouri M, Cottier JP, Herbreteau D. Les anomalies vasculaires Superficielles. Intérêt de la classification dans la prise en charge radiologique et thérapeutique).
La forme sous-cutanée (figure 2) (touchant le derme profond) se présente sous la forme d’une tuméfaction de consistance ferme, élastique, chaude mais non battante soulevant une peau saine légèrement bleutée ou rosée.
Figure 2 : Hémangiome sous-cutané. (Martin A, Barbier C, Domengie F, Nouri M, Cottier JP, Herbreteau D. Les anomalies vasculaires superficielles. Intérêt de la classification dans la prise en charge radiologique et thérapeutique).
La forme mixte (figure 3) réunit les deux aspects : la partie tubéreuse se développe en premier et la partie profonde apparaît quelques mois plus tard, débordant la première d’un halo bleuté.
Figure 3 : Hémangiome mixte. (Martin A, Barbier C, Domengie F, Nouri M, Cottier JP, Herbreteau D. Les anomalies vasculaires superficielles. Intérêt de la classification dans la prise en charge radiologique et thérapeutique).
Quel que soit le type, l’hémangiome est de consistance ferme et élastique, légèrement chaud à la palpation, mais non pulsatile et généralement indolore, sauf en cas d’ulcération [2].
Sa taille est très variable, allant de l’atteinte ponctiforme à l’atteinte d’un membre ou d’un hémitronc, mais, le plus souvent, elle est modérée, inférieure à 3 cm [2].
La localisation des hémangiomes est ubiquitaire, cependant, ils semblent plus fréquents sur le visage (40 %) et le cou (20 %) [2].
La croissance est accélérée pendant les trois premiers mois mais peut se prolonger jusqu’au 8ème mois pour les formes superficielles, et jusqu’au 12ème mois pour les formes à participation sous-cutanée. Dans de rares cas, cela peut aller jusqu’à deux ans [2].
Puis, il se stabilise spontanément, survient alors une régression sur plusieurs mois ou années. L’involution est lente et se traduit par l’apparition du blanchiment central des lésions superficielles et l’affaissement des composantes sous-cutanées.
La distribution des hémangiomes sur le visage est variable, il existe en effet :
Des formes focales qui sont situées sur les proéminences, et 60 % d’entre elles se concentrent en zone centrofaciale [3],
Des formes diffuses, où on retrouve une distribution segmentaire [3].
Une nouvelle classification topographique de ces formes segmentaires faciales en quatre aires de S1 à S4 a été proposée (figure 4). Ces formes segmentaires sont volontiers associées à des malformations (Syndromes PHACES et Syndrome PELVIS/SACRAL) et s’ulcèrent fréquemment [3].
Figure 4 : Segmentation S1 à S4 du du syndrome PHACES (Léauté-Labrèze C, Sans-Martin V. Hémangiome infantile, La presse médicale. tome 39 > n84 > avril 2010.doi: 10.1016/j.lpm.2009.10.015).
A. Syndrome PHACES [3] :
L’acronyme anglais PHACE(S), créé en 1996 par Frieden, regroupe les anomalies suivantes :
Hémangiome facial,
Anomalies de la fosse postérieure,
Anomalies artérielles intra et extra crâniennes,
Anomalies cardiaques congénitales et coarctation aortique,
Anomalies oculaires (eye),
Anomalies sternales et ventrales (ajoutées par la suite).
Les anomalies neurologiques intracrâniennes accompagnent dans plus de 3/4 des cas, des hémangiomes en plaques couvrant l’aire fronto palpébrale supérieure S1 + S4 (figure 5).
Figure 5 : a et b. Hémangiome dit segmentaire de type syndrome PHACES (atteinte S1 + S4) ; Fig. 5a : Hémangiome avant traitement. Fig. 5b : Hémangiome après 2 mois de propranolol. (Léauté-Labrèze C, Sans-Martin V. Hémangiome infantile. La presse médicale. tome 39 > n84 > avril 2010.doi: 10.1016/j.lpm.2009.10.015).
Les anomalies cardiaques et aortiques seraient plus particulièrement associées aux hémangiomes S3.
Ainsi, les explorations qui doivent être réalisées, sont :
l’imagerie par résonance magnétique (IRM) cérébrale,
l’échographie cardiaque et des gros vaisseaux,
l’examen ophtalmologique,
l’échographie abdominale.
A. Syndrome PELVIS ou SACRAL [3] :
L’acronyme anglais PELVIS regroupe les anomalies suivantes :
Hémangiomes périnéaux
Malformations génitales externes
Lipomyelomeningocèle
Anomalies vésicorénales
Anus imperforé
Autre marqueur cutané (skin tag).
Les explorations, qui doivent être réalisées, chez tout nouveau-né à risque sont une évaluation par IRM médullaire, et abdominopelvienne (plus sensible que l’échographie).
Traitement de l’hémangiome infantile :
L’abstention thérapeutique est parfaitement défendable dans les formes bénignes, pour lesquelles il est à prévoir peu ou pas de séquelle après régression [3].
Indications thérapeutiques [3] :
L’hémangiome infantile avec mise en jeu du pronostic vital.
L’hémangiome infantile avec mise en jeu du pronostic fonctionnel.
L’hémangiome infantile qui se complique d’ulcération,
L’hémangiome infantile qui engendre un préjudice esthétique majeur avec retentissement psychologique.
1. Le Propranolol :
«I’vebeendoingpediatricdermatologyfor25years,andthisisthefastestI’veeverseen a new therapy change the way we do things »1 (Bernard A. Cohen, M.D., Pédiatrie dermatologique Johns Hopkins Children’s Center) [3]. Le propranolol est un bêta-bloquant non cardiosélectif sans activité sympathomimétique intrinsèque.
Posologie : 2 à 3 mg/kg/j pour une durée minimum de 6 mois.
Il faut débuter le plus tôt possible, avant 3 mois (âge « idéal » probablement entre un et 2 mois).
On observe dans les heures qui suivent la prise de propranolol un affaissement et un changement de couleur de l’hémangiome, ensuite, l’effet se poursuit, mais plus lentement, aboutissant à une régression plus ou moins complète.
Les principaux effets secondaires sont : les hypoglycémies en période néonatale ou dans des situations de jeûne, des malaises avec pâleur, des épisodes de cyanose et d’hypotension. La mise en route du traitement doit se faire en milieu pédiatrique avec surveillance de la fréquence cardiaque et de la tension artérielle, puis le traitement est ambulatoire [2].
Le traitement est maintenu jusqu’à la fin de la période de croissance supposée de l’hémangiome [2].
1. La corticothérapie générale :
On utilise la prednisone le plus souvent, avec une posologie d’attaque variant entre 2 et 5 mg/kg/j pendant au moins 2 mois, puis diminuée très progressivement de façon à couvrir la période évolutive des premiers mois [2]. Le taux de réponse (régression ou simple stabilisation) n’est que de 30 à 60 %, l’effet apparaissant entre la 2ème et les 3èmes semaines de traitement.
2. La Chirurgie :
La Chirurgie précoce : en phase de croissance, est indiquée pour les hémangiomes infantiles globuleux ou
« pendulum », en particulier sur le nez, les paupières et les lèvres [2],
La Chirurgie tardive : occupe une place indispensable dans la réparation des séquelles cutanées (résidus fibro-adipeux) et structurales, après disparition de l’hémangiome [2].
3. Lasers :
Lelaseràcolorantpulsé:efficace sur la composante
superficielle en entraînant une décoloration, et aide à la cicatrisation de certains hémangiomes ulcérés. En revanche, il n’a aucun impact sur les composantes dermiques profondes [3].
Les laser CO2 ou le laser Erbium : sont indiqués en phase tardive sur des zones cicatricielles. Ils améliorent l’aspect par leur effet lissant et tenseur.
Les séances, qui sont douloureuses, sont réalisées sous anesthésie générale.
4. Autres :
Les pansements vaselinés, hydro cellulaires et hydro colloïdes sont utilisés dans les hémangiomes ulcérés. Ils ont un remarquable pouvoir antalgique [2].
2. Les malformations vasculaires :
Contrairement, aux tumeurs vasculaires, ces malformations vasculaires ne régresseront jamais [1].
Deux grands groupes se distinguent selon un critère hémodynamique [4] :
Les malformations vasculaires à flux lent.
Les malformation vasculaire à flux rapide.
A. I – Les malformations vasculaires à flux lent :
1. Les malformations capillaires :
Angiome plan (AP) :
Il s’agit d’une lésion cutanée rouge intense en période néonatale, froide et non battante, qui va pâlir lentement, sans régresser, exception faite de la forme médio frontale et cervicale qui disparaissent en un à deux ans. Cependant, il faut se méfier des faux angiomes plans, chauds, qui sont en fait la couverture cutanée d’une malformation artérioveineuse [1,4].
L’angiome plan n’a pas de retentissement régional ou général et constitue seulement un préjudice esthétique (figure 6), et il est dans la majorité des cas localisé, isolé, et stable [4].
Figure 6 : Angiome plan temporal – territoire V1 partiel. (Barbier C, Martin A, Papagnanaki C, Nouri M, Cottier JP, Herbreteau D. Clas- sification des anomalies vasculaires superficielles. STV. Mini-revue. Sang Thrombose Vaisseaux 2009 ;21, n° 5-6 : 248-57.doi: 10.1684/ stv.2009.0399).
Il peut, par ailleurs, être le marqueur d’une angiomatose complexe [4] :
L’angiome plan trigéminé est le témoin d’une phacomatose angiomateuse régionale appelée le syndrome de Sturge Weber ou angiomatose encéphalo-trigéminée (figure 7).
Figure 7 : Le syndrome de Sturge Weber Krabbe. (Le site des ophtalmologistes de France. Encyclopédie de la vue).
L’angiome plan situé au tronc, incite à rechercher une atteinte segmentaire cutanéo-vertébro-méningo-médullaire ou syndrome de Cobb.
L’angiome plan des membres se rencontre dans le syndrome de Klippel-Trenaunay ou dans le syndrome de Parkes Weber, associé à d’autres malformations vasculaires à flux lent dans le premier et à flux rapide dans le second.
L’angiome plan lombo-sacré s’accompagne d’autres anomalies cutanées, incite à rechercher un dysraphisme spinal. Le laser à colorant pulsé (LCP) est le traitement de référence des angiomes plans du nourrisson et de l’enfant, dont l’efficacité a été démontrée, avec une bonne tolérance (pas ou peu de risque cicatriciel), en débutant le plus tôt possible pour les surfaces étendues et l’angiome plan affichant de la face [3].
Les télangiectasies :
Il s’agit de dysplasies capillaires dermiques mais de petite taille (quelques millimètres) et de morphologie variable : lenticulaires, linéaires ou stellaire « l’angiome stellaire ». Il est nécessaire de distinguer la maladie de Rendu-Osler (télangiectasie hémorragique héréditaire) qui est une angiomatose héréditaire de transmission autosomique dominante [1,4].
2. Les malformations veineuses :
Les malformations veineuses sont constituées de tuméfactions bleutées sous-cutanées. Elles sont de consistance molle et froide au contact (Figure 8). Elles gonflent en position déclive, et se vident à la pression ou à la surélévation d’un membre. C’est un excellent signe distinctif. Les malformations veineuses intéressent tous les plans : les tissus cutanés, sous-cutanés, muqueux ou sous-muqueux, les muscles, la synoviale, l’os [4].
Lorsqu’il s’agit d’une malformations veineuses localisée, le traitement repose sur les mesures d’hygiène de vie et la sclérothérapie percutanée. En cas d’échec d’injections itératives, une chirurgie est proposée [5].
Figure 8 : Malformation Veineuse cervico-céphalique. (Barbier C, Martin A, Papagnanaki C, Nouri M, Cottier JP, Herbreteau D. Clas- sification des anomalies vasculaires superficielles. STV. Mini-revue. Sang Thrombose Vaisseaux 2009 ;21, n° 5-6 : 248-57.doi: 10.1684/ stv.2009.0399).
1. Les malformations lymphatiques kystiques :
Les malformations lymphatiques kystiques (MLK) sont hémodynamiquement inactives, et sont constituées de vaisseaux lymphatiques anormaux et de kystes. Ce sont des malformations congénitales. Elles se divisent classiquement en [1,4] :
a. Malformations lymphatiques microkystiques (forme tissulaire), formées de kystes inférieurs à 2 cm, et qui se présentent comme une nappe de petites vésicules translucides ou hématiques, non douloureuses [1,4].
b. Malformations lymphatiques macrokystiques (forme kystique), sont des tuméfactions volumineuses, formées de lésions supérieures à 2 cm, et qui apparaissent comme une tuméfaction dure, rénitente, bien limitée, localisée préférentiellement dans les régions cervico-encéphaliques et axillaires (figure 9) [1,4].
Figure 9 : Malformations lymphatiques macrokystiques (Barbier C, Martin A, Papagnanaki C, Nouri M, Cottier JP, Herbreteau D. Classification des anomalies vasculaires superficielles. STV. Mini-revue. Sang Thrombose Vaisseaux 2009 ;21, n° 5-6 : 248-57.doi : 10.1684/ stv.2009.0399).
c. Malformations lymphatiques mixtes qui associent ces deux formes.
Le traitement repose sur la sclérothérapie percutanée en première intention et s’effectue à distance d’épisodes infectieux, de ce fait, la prévention d’épisodes infectieux est primordiale dans la prise en charge au quotidien. Une chirurgie est complémentaire selon la localisation et le pronostic [5].
B. Les Malformations vasculaires à flux rapide [6] :
1. Malformations artério-veineuses :
Ce sont les malformations les plus dangereuses car hémodynamiquement actives avec des aggravations parfois dramatiques, on insistera sur la malformation artérioveineuse proprement dite qui comporte un nidus avec de multiples shunts.
L’évolution naturelle s’effectue selon 4 étapes (Schobinger 1995) [6] :
Stade I :Dormance : « pseudo-angiome plan », ce qui peut entraîner la confusion sur la prise en charge (figure 10), il faut rechercher une augmentation de la chaleur locale et un thrill (Echodoppler au moindre doute). Ici est recommandée une surveillance clinique et par Echodoppler.
Figure 10 : MAV du nez en phase de dormance. (Barbier C, Martin A, Papagnanaki C, Nouri M, Cottier JP, Herbreteau D. Classification des anomalies vasculaires superficielles. STV. Mini-revue. Sang Throm- bose Vaisseaux 2009 ;21, n° 5-6 : 248-57.doi : 10.1684/stv.2009.0399).
Stade II : Expansion : la malformation s’étend, devient rouge, chaude et battante. Des veines de drainage tortueuses apparaissent à la surface.
Stade III :Destruction, s’accompagne habituellement de complications tissulaires comme l’ischémie, l’ulcération, la nécrose et les hémorragies.
Stade IV : Stade III avec décompensation cardiaque.
Pour les Stades II, III et IV : on réalise une embolisation endo-vasculaire sélective ou par l’association embolisation-chirurgie.
Conclusion :
Cette classification clinique permet d’améliorer la prise en charge des malformations vasculaires superficielles qui sont rares, et donc souvent méconnues ; au sein de groupes multidisciplinaires, permettant ainsi un diagnostic rapide et un traitement adéquat.
Références :
Barbier C, Martin A, Papagnanaki C, Nouri M, Cottier JP, Herbreteau D. Classification des anomalies vasculaires superficielles. STV. Mini-revue. Sang Thrombose Vaisseaux 2009 ;21, n° 5-6 : 248-57.doi : 10.1684/stv.2009.0399.
Léauté-Labrèze C, Sans-Martin V. Hémangiome infantile. La presse médicale. tome 39 > n84 > avril 2010.doi: 10.1016/j.lpm.2009.10.015.
Léauté-Labrèze C. Les anomalies vasculaires : Classification, diagnostic, traitement. Diplôme inter universitaire de lasers dermatologiques. Bordeaux. Février 2016.
Martin A, Barbier C, Domengie F, Nouri M, Cottier JP, Herbreteau D. Stratégies d’exploration et traitement des anomalies vasculaires superficielles. STV. Mini-revue. Sang Thrombose Vaisseaux 2009 ;21, n° 5-6 : 259-67. doi : 10.1684/stv.2009.0400.
Dompmartin A. Classification des anomalies vasculaires. Annales de dermatologie et de vénéréologie (2013) 140, 337—339.doi. org/10.1016/j.annder.2013.02.002.
Bonnetblanc J.-M. Angiomes cutanés. Annales de dermatologie et de vénéréologie (2008) 135S, F181—F187. doi: 10.1016/j.annder.2008.07.001.
A. CHIALI,A. KHELIL,N.H. MAHMOUDI, Service de Dermatologie, CHU Benaouda Benzerdjeb, Oran.*
Abstract : Atopic dermatitis (AD) is a chronic, inflammatory and pruriginous dermatosis occurring in the form of acute eczema flares on a cutaneous xerosis background. It is common in children and young adults. AD affects genetically predisposed subjects and is favored by environmental factors. It is determined by the association of an innate functional impairment of the cutaneous barrier, the development of a cutaneous inflammatory reaction involving innate and adaptive immunity and the action of an anomaly of the digestive and cutaneous microbiomes diversity. The diagnosis of AD is clinical, facilitated by the use of certain criteria. Clinical aspects vary according to age. In infants, they are represented by outbreaks of acute and itchy inflammatory lesions starting on the face. In children over 2 years old, adolescents and adults, cutaneous lesions are lichenified and chronic, affecting folds. The management of a patient with AD must include therapeutic education and potential aggravating factors eviction (food allergy, superinfection, psychological factors and contact eczema). The objective of the treatment is to improve patient’s quality of life. Topical corticosteroids are used as first-line treatment for AD. Tacrolimus can be used as a second line. Photo- therapy may be indicated especially in adults. Indications of systemic treatments (immuno-suppressive agents and targeted therapies) are very rare. The only preventive treatment is based on the administration of cow’s milk protein hydrolysates to newborns at risk of atopy, and probiotics and/or prebiotics.
Résumé : La dermatite atopique (DA) est une dermatose chronique, inflammatoire et prurigineuse survenant sous forme de poussées d’eczéma aigu sur un fond de xérose cutanée. Elle est fréquente chez l’enfant et l’adulte jeune. La DA touche des sujets génétiquement prédisposés et elle est favorisée par des facteurs environnementaux. Elle est déterminée par l’association d’une altération fonctionnelle innée de la barrière cutanée, d’un développement d’une réaction inflammatoire cutanée impliquant l’immunité innée et adaptative et de l’action d’une anomalie de diversité des microbiomes digestif et cutané. Le diagnostic de la DA est clinique, facilité par le recours à certains critères. Les aspects cliniques varient selon l’âge. Chez le nourrisson, ils sont représentés par des poussées de lésions inflammatoires aiguës et prurigineuses débutant sur le visage. Chez l’enfant de plus de 2 ans, l’adolescent et l’adulte, les lésions cutanées sont lichénifiées et chroniques touchant les plis. La prise en charge d’un patient atteint de DA doit comporter une éducation thérapeutique et l’éviction de facteurs aggravants potentiels (allergie alimentaire, surinfection, facteurs psychologiques et eczéma de contact). L’objectif du traitement est d’améliorer la qualité de vie du patient. Les dermocorticoïdes sont utilisés en première intention en traitement d’attaque de la DA. Le tacrolimus peut être utilisé en seconde intention. La photothérapie peut être indiquée surtout chez l’adulte. Les indications des traitements systémiques (immunosuppresseurs et thérapies ciblées) sont très rares. Le seul traitement préventif est basé sur l’administration d’hydrolysats de protéines de lait de vache chez les nouveau-nés à risque d’atopie et des probiotiques et/ou des prébiotiques.
La dermatite atopique (DA) (ou eczéma constitutionnel) est une dermatose inflammatoire, prurigineuse et chronique, se développant préférentiellement chez le nourrisson et le jeune enfant mais parfois présente chez l’adolescent et l’adulte. C’est une pathologie polymorphe dans son expression clinique variant en fonction de l’âge du sujet atteint. Elle constitue une des manifestations d’hyper-sensibilité et s’intègre dans le cadre de la maladie atopique au même titre que l’asthme ou la rhinite [1]. La DA débute dans les premiers mois de vie généralement vers 3 mois, mais parfois dès le premier mois [2,3]. La prise en charge de la DA de l’enfant est multidisciplinaire impliquant les pédiatres, les allergologues et les dermatologues en fonction de la gravité de la maladie et de l’âge des enfants.
Épidémiologie :
En Algérie, la prévalence de la DA est de 5 % [4]. Par ailleurs, cette affection se rencontre chez 10 à 20 % des enfants dans les pays industrialisés à niveau socio- économique élevé où il existe un doublement de sa prévalence en une vingtaine d’années [5]. Une étude chinoise a mis en évidence un gradient de prévalence de la DA urbain/ rural (10,2 % vs 4,6 %) [6]. Cette diminution du risque d’atopie en milieu rural peut s’expliquer par le fait qu’une plus grande biodiversité environnementale en milieu rural est très significativement associée à une plus grande diversité des bactéries commensales digestives et cutanées [7,8].
Physiopathologie :
La DA est une maladie multifactorielle due à l’association de facteurs génétiques et environnementaux [9]. Sa physiopathologie fait intervenir plusieurs mécanismes dont une altération fonctionnelle innée de la barrière cutanée liée en partie à une mutation du gène de la filag plus élevée chez le sujet atteint de DA), tend à polariser l’activation lymphocytaire T vers un profil de type Th2 (associé à la production d’interleukines 4, 5, 9, 13, 31) et Th22 (avec production d’interleukine 22) [13]. À la phase chronique, une activation lymphocytaire T de type Th1 (associée à la production d’interféron gamma, de GM- CSF [Granulocyte macrophage colony-stimulating factor] et d’interleukine 12) s’associe à la réponse Th2. La sti- mulation des neurones par une cytokine proinflammatoire (thymic stromal lymphopoietin : TSLP), secrétée par les kératinocytes est responsable de la sensation de prurit [14].
Réaction inflammatoire induite
L’état défectueux de la fonction de la barrière cutanée permet la pénétration dans la peau d’allergènes ainsi que des irritants chimiques externes. Ce phénomène va stimuler l’inflammation [12]. L’immunité adaptative contribue à initier le processus inflammatoire. À la phase aiguë, l’activation des cellules de Langerhans (cellules dendritiques épidermiques dont la concentration est plus élevée chez le sujet atteint de DA), tend à polariser l’activation lymphocytaire T vers un profil de type Th2 (associé à la production d’interleukines 4, 5, 9, 13, 31) et Th22 (avec production d’interleukine 22) [13]. À la phase chronique, une activation lymphocytaire T de type Th1 (associée à la production d’interféron gamma, de GM- CSF [Granulocyte macrophage colony-stimulating factor] et d’interleukine 12) s’associe à la réponse Th2. La stimulation des neurones par une cytokine proinflammatoire (thymic stromal lymphopoietin : TSLP), secrétée par les kératinocytes est responsable de la sensation de prurit [14].
Rôle du microbiome digestif
Le microbiome digestif correspond à l’ensemble des bactéries commensales colonisant le tube digestif et constituant un écosystème complexe qui intervient dans la maturation du système immunitaire. Des anomalies de diversification précoce du microbiote intestinal ont été observées chez les enfants et les nouveau-nés à risque d’atopie [15].
Rôle du microbiome cutané
En dehors des poussées, on observe dans la DA une grande diversité de souches bactériennes commensales à la surface de la peau (microbiome cutané). Cette diversité décroît au cours des poussées de la maladie au profit des souches de staphylocoques [16]. La colonisation de la peau par le staphylocoque doré est très fréquente au cours de la DA (90 % vs 5 % des sujets sains). Un déficit de l’immunité innée cutanée semble être en partie en cause [17].
Clinique :
Le diagnostic de DA est aisé et s’effectue sur des données cliniques. Les critères diagnostiques le plus souvent utilisés sont ceux de l’UK Working Party [18] (Tableau 1).
Critère majeur : dermatose prurigineuse chronique
Associée à moins 3 des critères suivants : Eczéma visible des plis de flexion (ou des joues et/ou des faces d’extension des membres avant l’âge de 18 mois) Antécédent personnel d’eczéma des plis de flexion (ou des joues et/ou des faces d’extension des membres avant l’âge de 18 mois) Antécédent personnel de peau sèche au cours de la dernière année Antécédent personnel d’asthme ou de rhinite allergique (ou antécédent familial direct d’atopie chez l’enfant de moins de 4 ans) Apparition des lésions avant 2 ans (critère utilisé chez les enfants de plus de 4 ans)
Tableau 1 : Dermatite atopique : critères diagnostiques (UK Working Party’s for diagnostic criteria for AD)
Aspects cliniques
Les aspects cliniques varient avec l’âge :
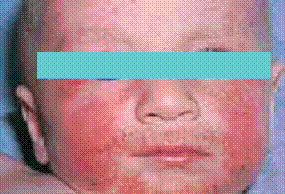
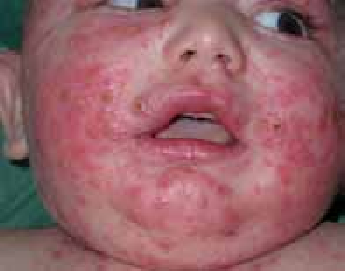
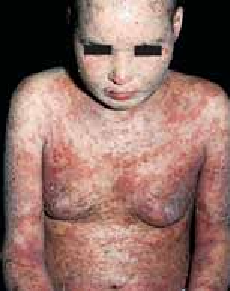
Chez le nourrisson, les lésions débutent sur les joues, le front et le cuir chevelu. Secondairement, elles s’étendent sur les faces d’extension des membres et le tronc. Le siège et la région médiofaciale (en particulier la pointe du nez) sont épargnés (figure 1). Ce sont des lésions aiguës, érythémateuses, mal limitées, suintantes, croûteuses et prurigineuses. Les excoriations cutanées dues au grattage sont fréquentes. L’évolution est marquée par des poussées entrecoupées de rémissions.
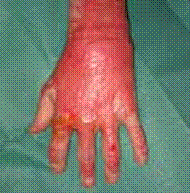
Figure 1 : Eczéma atopique du nourrisson (atteinte du visage). (Iconographie adresse URL : Eczema Picture Image on MedicineNet.com)
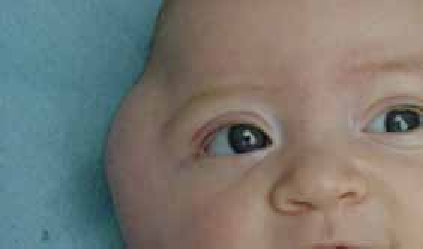
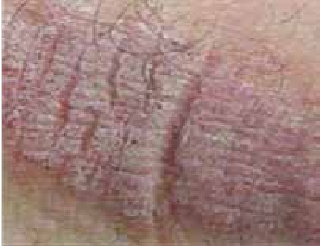
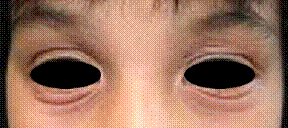
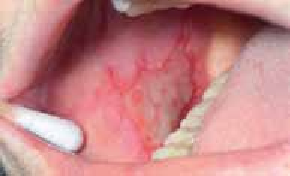
Chez l’enfant de plus de 2 ans, les lésions cutanées siègent au niveau des plis (cou, coudes, genoux, fissures sous et rétroauriculaires) chroniques. Elles se manifestent par l’aspect d’une lichénification (épaississement de la peau) (figure 2). D’autres signes s’ajoutent parfois tels qu’un prurigo, un double pli palpébral inférieur (signe de Dennie-Morgan) (figure 3) et une xérose cutanée (sécheresse).
Chez l’adolescent et l’adulte, le tableau clinique est similaire à celui de l’enfant de plus de 2 ans avec en plus des lésions qui se localisent au visage et au cou et sont franchement lichénifiées sur les membres.
Certaines localisations sont rares mais caractéristiques : l’atteinte des mamelons, l’atteinte des lèvres (chéilite atopique) et des paupières [9].
Des formes cliniques existent [9] :
L’eczéma nummulaire est caractérisé par des lésions rondes, inflammatoires, résistantes aux traitements,
L’atteinte des mains peut être observée au cours de la DA. Elle se caractérise par les lésions périunguéales avec parfois une dystrophie unguéale associée
L’atteinte pulpaire entretenue par un facteur aggravant de contact ;
une érythrodermie [5].
D’autres manifestations d’atopie sont souvent associées (asthme, rhinite allergique, allergie alimentaire).
Examens complémentaires
Le diagnostic de la DA est clinique et anamnestique. Une hyperéosinophilie sanguine et une augmentation des IgE sériques sont fréquentes [7].
Histopathologie
L’aspect histologique comporte une atteinte épidermique prédominante avec un afflux de lymphocytes T (exocytose) qui s’accompagne d’un œdème intercellulaire (spongiose) réalisant des vésicules. En cas de lichénification, l’épiderme s’épaissit. Le derme superficiel comporte un infiltrat mononucléé périvasculaire.
La dilatation des capillaires superficiels est responsable de l’érythème et l’extravasation de protéines plasmatiques de l’œdème cutané (papules œdémateuses) [3].
Immunohistochimie
De façon caractéristique, des éosinophiles dégranulés sont objectivables (présence de major basic protein) [3].
Explorations allergologiques
Explorations allergologiques (prick-tests cutanés, dosage des IgE sériques spécifiques, test de provocation) justifiées devant l’association d’une allergie respiratoire (asthme, rhinite ou rhinoconjonctivite), ou d’une allergie alimentaire suspectée devant sa survenue après ingestion ou contact avec un aliment, devant une DA grave ou devant une stagnation ou une cassure de la courbe pondérale [5]. Tests épicutanés ou patch tests en cas d’association avec un eczéma de contact évoqué devant l’apparition d’un eczéma de zones inhabituelles et devant une DA qui ne répond pas au traitement ou s’aggrave [5].
Évolution :
La DA comporte des formes étendues, mais son évolution est bonne avec une rémission complète dans la majorité des cas. Les formes qui persistent chez l’enfant sont plus localisées. Une recrudescence peut se produire à l’occasion de conflits psychoaffectifs.
La survenue de manifestations respiratoires (asthme ou rhinite) est d’autant plus fréquente qu’il y a des antécédents familiaux atopiques au premier degré [3].
Complications infectieuses :
La surinfection par le staphylocoque doré (SA) : Le SA colonise constamment la peau des atopiques, même en dehors des zones cliniquement atteintes. Cette surinfection se manifeste cliniquement par la présence d’un écoulement purulent, de lésions vésiculo-bulleuses et de croûtes jaunes [9].
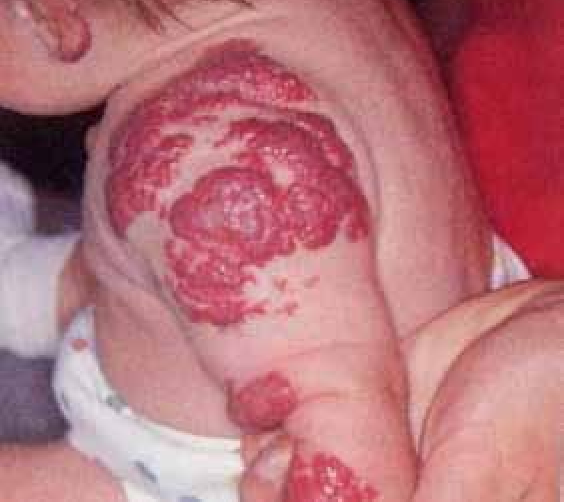
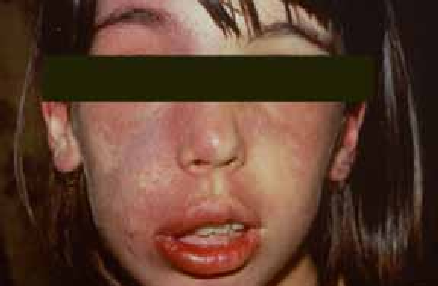
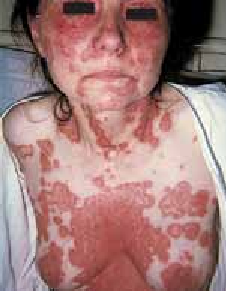
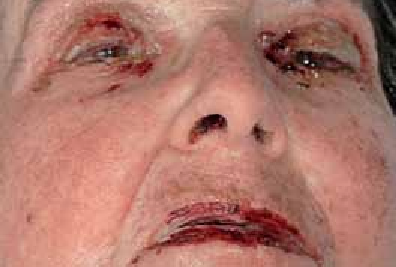
La surinfection herpétique (HSV1) ou syndrome de Kaposi-Juliusberg : elle se traduit par une aggravation brutale de la maladie avec apparition de vésicules hémorragiques et de pustules ombiliquées rapidement extensives (figure 4) [9].
Figure 4 : Syndrome de Kaposi Juliusberg (surinfection herpétique) (Iconographie Item 164 – Infections à herpès cutané et muqueux (ANAES 2001)). Adresse URL: smartfiches.fr/dermatologie/item-164-infections-a-herpes…/infections-a-herpes.
Autres complications :
Dermatite (ou eczéma) de contact : il existe un risque important de sensibilisation chez l’enfant atteint de DA [5].
Retard de croissance : rare mais peut être observé dans les cas de DA graves. Il se corrige habituellement quand la DA est traitée efficacement. Il doit faire rechercher une allergie alimentaire [5].
Complications ophtalmologiques : rares à type de kératoconjonctivite, cataracte [5].
Associations morbides :
Il a été rapporté une augmentation de la fréquence de maladies auto-immunes telles que les maladies inflammatoires chroniques de l’intestin, pelade, polyarthrite rhumatoïde et lupus érythémateux, et en particulier chez les sujets jeunes [19]. Des cas de DA associant des troubles psychiatriques en particulier le déficit de l’attention et l’hyperactivité. Le prurit chronique, les troubles de sommeil et l’altération de la qualité de vie peuvent expliquer cette association [20].
Prise en charge thérapeutique de la DA :
Elle dépend de :
L’évaluation de la gravité de la maladie. Plusieurs scores cliniques composites ont été validés (SCORAD, EASI [21], SASSAD).
La qualité de vie : dans les formes modérées et graves, la qualité de vie des sujets atteints et de leur famille est souvent très altérée en raison du prurit, des perturbations du sommeil et du caractère affichant de la maladie. La prise en charge thérapeutique comporte :
Les soins d’hygiène non agressifs et adaptation de l’environnement :
Éviter le savon et utiliser un gel ou un pain sans savon doux non parfumé
Réduire la fréquence des lavages de la peau et éviter les bains chauds prolongés
Privilégier les textiles vestimentaires doux (éviter la laine)
Éviter l’exposition à la chaleur (intolérance à la sueur).
L’éducation thérapeutique qui a pour objectif d’apprendre au patient à vivre de manière optimale avec une maladie chronique
Les moyens thérapeutiques :
Dermocorticoïdes
Les dermocorticoïdes (DC) ont une triple action : anti-inflammatoire, immunosuppressive et antimitotique. Ils sont très efficaces à court terme sur les poussées de DA. Paradoxalement, la corticothérapie locale (sans antibiothérapie associée) est un moyen efficace de réduire la colonisation cutanée par le staphylocoque doré [22,23].
Les DC sont classés selon leur puissance et leur forme galénique (crème, pommade, lotions et gels). Ils représentent le traitement topique de référence [24].
Le choix est fait en fonction de l’âge, de la sévérité de la DA, du site et de l’étendue à traiter.
Les DC d’activité très forte (classe IV) sont contre-indiqués chez le nourrisson et le jeune enfant, sur le visage, les plis, et le siège. En règle générale :
Les formes « pommade » sont réservées aux zones cutanées lichénifiées et sèches ;
Les formes « crème » sont réservées aux zones suintantes, aux plis et aux grandes surfaces cutanées ;
Les formes « lotion » sont réservées aux zones pileuses et aux plis ;
Les formes « gel » sont réservées au cuir chevelu ; Une seule application par jour est suffisante.
Le « wet wrapping »
Cette technique de double bandage (une couche de bandes humidifiées, une couche de bande sèche) associée à une corticothérapie locale semble être plus efficace sur le prurit à court terme que la corticothérapie seule [19].
La corticophobie est un phénomène fréquent, ayant pour conséquence une faible adhésion thérapeutique dans la DA. La prise en charge de la corticophobie s’impose afin d’améliorer l’adhésion au traitement et diminuer les échecs thérapeutiques.
Le tacrolimus topique sous forme de pommade est indiqué après l’âge de 2 ans chez l’enfant (tacrolimus 0,03%) et chez l’adulte (tacrolimus 0,1 %) dans la DA modérée à sévère, en cas d’échec ou de contre-indications aux DC. Prescrit en traitement d’entretien, il prévient les poussées [26]. Une application 2 fois par jour est préconisée jusqu’à disparition des lésions puis la posologie peut être diminuée à 1 fois par jour. Il est impératif d’éviter la photo-exposition après l’application afin de réduire les risques d’intolérance immédiate transitoires, de brûlures et d’exacerbation du prurit au cours des premières applications [9].
Antihistaminiques oraux (anti-H1)
Seuls, ils sont insuffisants pour traiter une poussée de DA. Les antihistaminiques de 1ère génération peuvent être utilisés pour le confort du patient. Ils diminuent le prurit et les réveils nocturnes (du fait de leur effet sédatif).
Ils n’ont pas d’intérêt au long cours en prévention des poussées de DA [27].
Antiseptiques et antibiotiques locaux ou systémiques utiles en cas de surinfection des lésions [27].
Photothérapie
La photothérapie UVA-UVB et UVB à spectre étroit est un traitement de seconde ou de troisième ligne de la DA à partir de l’âge de 8-10 ans. Elle n’est indiquée que dans des DA sévères, résistantes aux autres thérapeutiques, mais pendant des périodes courtes (risques carcinogènes potentiels à long terme).
Traitements immunosuppresseurs systémiques
Les immunosuppresseurs systémiques sont des traitements de deuxième ou troisième ligne de la DA chez l’adolescent et l’adulte, exceptionnellement chez l’enfant.
Le traitement systémique de première ligne chez l’enfant et l’adulte est la ciclosporine. Les traitements de deuxième ligne sont représentés par l’azathioprine et le méthotrexate.
La ciclosporine est prescrite par voie orale à la dose de 3 à 5 mg/kg/j. La durée du traitement est de 6 à 9 mois. L’efficacité est souvent rapide (une semaine) mais les rechutes sont fréquentes à l’arrêt du traitement. Cette thérapeutique nécessite une surveillance de la tension artérielle et de la fonction rénale [28].
Le délai d’action du méthotrexate (hors AMM) est de l’ordre de 4 semaines.
Des thérapies ciblées sont en cours d’évaluation chez l’adulte.
Le rituximab (anti-CD20), les anti-TNFα et le tocilizu- mab (anti-IL6) ont montré une efficacité mais au prix d’effets indésirables sévères pour le tocilizumab [29,30].
La thérapie ciblée la plus intéressante actuellement est le dupilumab. Cet anticorps monoclonal humanisé anti- IL4/IL13 efficace dans l’asthme, a donné une réponse favorable dans la DA modérée à sévère avec une meilleure tolérance [31].
Les mesures préventives
Laits artificiels et allaitement maternel
L’instauration d’un lait artificiel contenant un hydrolysat de protéines de lait de vache (seul ou en complément de l’allaitement maternel) entraine une réduction modérée du risque de DA dans les premières années de vie dans la population à risque d’atopie [32]. Aujourd’hui, il n’y a pas d’argument convaincant pour suggérer que l’allaitement maternel exclusif protège du risque de DA [33].
Probiotiques et prébiotiques
Instaurés à un âge très précoce, les probiotiques et/ou les prébiotiques per os pourraient avoir un effet préventif sur la DA. Les souches lactobacillus semblent être actuellement les plus efficaces.
Mesures contre la sensibilisation L’immunothérapie spécifique (désensibilisation). Des résultats récents ont démontré son intérêt dans la DA de l’adulte, chez des malades porteurs d’une forme sévère avec sensibilisations aux acariens et aux pollens [34].
L’utilisation de topiques potentiellement à risque (émollients ou topiques contenant des conservateurs très sensibilisants, des parfums, de la néomycine) est à limiter afin de prévenir la survenue de l’eczéma de contact.
Conclusion :
La DA s’intègre dans le cadre des maladies allergiques. C’est une maladie inflammatoire secondaire à plusieurs facteurs (altération de la fonction de la barrière épidermique et dysbiose intestinale et cutanée). Elle est plus fréquente dans les pays développés. Généralement, la DA débute dans la petite enfance Elle est caractérisée par une prédisposition génétique et une évolution récurrente avec des manifestations cliniques, en fonction de l’âge du patient. Concernant la prise en charge, le traitement local consiste toujours en l’usage des émollients et des dermocorticoïdes qui représentent toujours les al- ternatives de choix. Par contre, le traitement systémique s’est enrichi grâce aux progrès de la biotechnologie.
De nouvelles molécules de biothérapie visant à inhiber des mécanismes effecteurs clés dans la physiopathologie de la DA représentent une perspective thérapeutique très prometteuse.
Références :
Irvine AD, Mclean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med 2011; 365: 1315-27.
Queille-Roussel C, Raynaud F, Saurat JH: A prospective computerized study of 500 cases of atopic dermatitis in childhood. Acta Derm Venereol Suppl (Stockh) 1986; 114:87–92.
Taïeb A, Hennino A, Bérard F, Nicolas JF. Dermatite atopique, Eczémas et dermatoses Spongiformes. In: Saurat JH, Lachapelle JM, Lipsker D, Thomas L. Dermatologie et infections sexuellement trans- missibles. Paris: Elsevier; 2009. p.67-80.
Baghou S, Bensaad D, Taieb A, Ammar-Khodja A. Prévalence et profil clinique de la dermatite atopique en Algérie Ann Dermatol Vé- néréol 2012;139(12S):B140. Doi : 10.1016/j.annder.2012.10.200
Auteurs et membres du Collège des enseignants en dermatolo- gie de France. Allergies cutanéo-muqueuses chez l’enfant et l’adulte : dermatite (ou eczéma) atopique. Item 114. Ann dermatol vénéréol 2008; 135S : F80—F87. Consultable sur : https://doi.org/10.1016/j. annder.2008.07.001
Xu F, Yan S, Li F Et al. Prevalence of childhood atopic dermatitis: an urban and rural community-based study in Shanghai, China. PLoS One, 2012;7: e36174.
Barbarot S. Physiopathologie de la dermatite atopique et perspec- tives thérapeutiques systémiques. Mises au point interactives, éd. Réalités Thérapeutiques en Dermato-Vénérologie- Performances Médicales. Cahier 1 2016 (256). p.48-49. Consultable sur : jird.info/ wp-content/uploads/2017/01/MOP4.pdf
Barbarot S, Aubert H. Physiopathologie de la dermatite atopique. Ann Dermatol Vénéréol 2017;144 Suppl 1: S14-S20.
Stalder JF, Barbarot S & Aubert H. Dermatite atopique. In : L. Thé- rapeutique Dermatologique. Un manuel de référence en dermatologie 27 juillet 2015. Consultable sur : http:// www.therapeutique-dermato- logique.org/spip.php?article1068
Palmer CN, Irvine AD , Terron-Kwiatkowski A et al. Common loss- of- function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 2006; 38:441-6.
Strachan DP. Family size, infection and atopy: the first decade of the «hygiene hypothesis». Thorax 2000;55 Suppl 1:S2-S10.
Garn H, Neves JF, Blumberg RS et al. Effect of barrier microbes on organ-based inflammation. J Allergy Clin Immunol 2013; 131:465-78.
Gittler JK, Shemer A, Suarez-Farinas M Et al. Progressive activa- tion of T(H)2/T(H)22 cytokines and selective epidermal proteins cha- racterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol 2012; 130:1344-54.
Wilson SR, The L, Batia LM et al. The Epithelial Cell-Derived Ato- pic Dermatitis Cytokine TSLP Activates Neurons to Induce Itch. Cell 2013; 155:285-95.
Ismail IH, Oppedisano F, Joseph SJ et al. Reduced gut microbial diver- sity in early life is associated with later development of eczema but not atopy in high-risk infants. Pediatr Allergy Immunol 2012; 23:674-81.
Kong HH, Oh J, Deming C Et al. Temporal shifts in the skin micro- biome associated with disease flares and treatment in children with atopic dermatitis. Genome research 2012; 22:850-9.
Kuo IH, Yoshida T, De Benedetto A et al. The cutaneous innate immune response in patients with atopic dermatitis. J Allergy Clin Immunol 2013; 131:266-78.
Williams HC, Burney PG, Hay RJ et al. The U. K. Working Party’s Diagnostic Criteria for Atopic Dermatitis. I. Derivation of a minimum set of discriminators for atopic dermatitis. Br J Dermatol 1994; 131:383-96.
Chiavreini C. Quoi de neuf en dermatologie pédiatrique. Ann Dermatol Vénéréol 2017;144 suppl 4:IVS29-IVS39.
Strom MA, Fishbein AB, Paller AS, Silverberg JI. Association between atopic dermatitis and attention deficit hyperactivity disorder in U.S. children and adults. Br J Dermatol 2016; 175:920.
Barbier N, Paul C, Luger T Et al. Validation of the Eczema Area and Severity Index for atopic dermatitis in a cohort of 1550 patients from the pimecrolimus cream 1% randomized controlled clinical trials program. Br J Dermatology 2004; 150:96-102.
Stalder JF, Fleury M, Sourisse M Et al. Local steroid therapy and bacte- rial skin flora in atopic dermatitis. Br J Dermatol 1994; 131:536-40.
Bath-Hextall FJ, Birnie AJ, Ravenscroft JC et al. Interventions to reduce Staphylococcus 23. aureus in the management of atopic ecze- ma: an updated Cochrane review. Br J Dermatol 2011; 164:228.
Leloup P, Stalder JF, Barbarot S. Outpatient home-based wet wrap dressings with topical steroids with children with severe recal- citrant atopic dermatitis: A Feasibility Pilot Study. Pediatr Dermatol 2015;32:e177-8.
Lucky AW, Leach AD, Laskarzewski P, Wenck H. Use of an emol- lient as a steroid-sparing agent in the treatment of mild to moderate atopic dermatitis in children. Pediatr Dermatol 1997;14:321-4.
Schmitt J, Von Kobyletzki L, Svensson A, Apfelbacher C. Efficacy and tolerability of proactive treatment with topical corticosteroids and calci- neurin inhibitors for atopic eczema: systematic review and meta-analysis of randomized controlled trials. Br J Dermatol 2011;164:415-28.
Barbarot S, Aubert H, Stalder JF, Bernier C. Dermatite atopique. Ency- cl Med Chir, Dermatologie 2016;11(3):1-20 [Article 98-150-A-10].
Schmitt J, Schmitt N, Meurer M. Cyclosporin in the treatment of patients with atopic eczema – a systematic review and meta-analysis. J Eur Acad Dermatol Venereol 2007;21:606-19.
Simon D, Hösli S, Kostylina G Et al. Anti-CD20 (rituximab) treatment improves atopic eczema. J Allergy Clin Immunol 2008; 121:122-8.
Jacobi A, Antoni C, Manger B Et al. Infliximab in the treatment of mo- derate to severe atopic dermatitis. J Am Acad Dermatol 2005; 52:522-6.
Simpson EL, Bieber T, Guttman-Yassky E et al. SOLO 1 and SOLO 2 Investigators. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N Engl J Med 2016; 375(24):2335-2348. ISSN 0028-4793 Consultable sur: https://doi.org/10.1056/NEJMoa1610020.
*Von Berg A, Filipiak-Pittroff B, Kramer U et al. Allergies in high- risk schoolchildren after early intervention with cow’s milk protein hydrolysates: 10-year results from the German Infant Nutritional Intervention (GINI) study. J Allergy Clin Immunol 2013;131:1565-73.
Flohr C, Nagel G, Weinmayr G Et al. Lack of evidence for a pro- tective effect of prolonged breastfeeding on childhood eczema: lessons from the International Study of Asthma and Allergies in Childhood (ISAAC) Phase Two. Br J Dermatol 2011;165:1280-9.
Bae JM, Choi YY, Park CO et al. Efficacy of allergen-specific immunotherapy for atopic dermatitis: a systematic review and me- ta-analysis of randomized controlled trials. J Allergy Clin Immunol 2013;132:110-7.
S. ZOBIRI, Service de Dermatologie, CHU Mustapha Bacha, Alger.
Abstract : Acne is a chronic inflammatory dermatosis of the pilosebaceous unit affecting frequently adolescents. Physio pathogenesis of acne is multifactorial involving mainly hyperseborrhoea, inflammation and genetics. Clinically acne is retentional and/or inflammatory. In most cases, acne lesions disappear after adolescence. Treatment depends on the severity of the acne using local and general treatments.
Résumé : l’acné est une dermatose inflammatoire chronique du follicule pilo-sébacé touchant fréquemment l’adolescent. Sa physio pathogénie est multifactorielle faisant intervenir principalement l’hyperséborrhée, l’inflammation et la génétique. Elle se présente cliniquement sous formes de lésions rétentionnelles et/ou inflammatoires. L’évolution est le plus souvent favorable, la prise en charge dépend de la sévérité de l’acné et fait appel à des traitements locaux et généraux.
L’acné est l’affection la plus fréquemment rencontrée en pratique dermatologique quotidienne, elle est définie comme une maladie inflammatoire chronique du follicule pilo-sébacé. C’est une affection qui survient le plus souvent durant la période pubertaire, elle est d’ailleurs considérée comme une affection de l’adolescence même si elle peut se voir chez l’adulte [1].
Épidémiologie :
La prévalence de l’acné chez l’adolescent est difficile à évaluer et diffère en fonction des études mais la plupart des séries s’accordent sur une prévalence de 80 % chez les 12-20 ans. La prévalence globale en fonction du sexe est de 72,5 % chez les filles et de 71,9 % chez les filles [2].
En Algérie, une prévalence de 81,7% a été rapportée chez les 11-19 ans dans une étude menée dans la commune de Constantine en 2015 [3].
L’âge de survenue est noté entre 14-16 ans pour les filles et vers 16-17 ans pour les garçons, ceci correspond à la stimulation de la glande sébacée par les androgènes lors de la puberté.
Physiopathologie de l’acné :
La physiopathologie de l’acné fait intervenir les 4 facteurs suivants : l’hyper séborrhée, les troubles de la kératinisation, l’inflammation et le terrain génétique.
Au cours de l’acné il existe des modifications quantitatives et qualitatives du sébum. Concernant les modifications quantitatives, il existe une augmentation de 59% de la quantité du sébum par rapport au sujet sain entrainant une modification de l’aspect de la peau acnéique qui apparait luisante. Cette hyper séborrhée est considérée comme une réponse exagérée du follicule pilo-sébacé aux androgènes [4].
Sur le plan qualitatif, la proportion des différents lipides composant le sébum est modifiée avec une augmentation des squalènes et des triglycérides à l’origine de la formation du comédon [5].
Le deuxième facteur impliqué dans la physiopathologie de l’acné est représenté par les troubles de la kératinisation de l’infra infundibulum. En effet, il existe des anomalies de la prolifération, de la différenciation et de l’adhésion des kératinocytes de l’infra infundibulum sous l’action de l’IL1 alpha principalement. L’hyper cornification empêche l’évacuation normale du sébum entrainant une rétention et une dilatation du follicule pilo-sébacé aboutissant au micro comédon [6].
L’inflammation du follicule pilo-sébacé, 3ème facteur étiopathogénique est responsable de différentes lésions cliniques. Elle a longtemps été liée à une bactérie : Cutibacterium acnes (C. acnes). C’est une bactérie anaérobie, à gram positif, immobile, appartenant au genre Propionibacterium, retrouvée au niveau du follicule pilo-sébacé, en peau saine et en peau acnéique [7].
Le C. acnes agit dans le processus inflammatoire par des actions enzymatiques et immunologiques, et non par une prolifération bactérienne excessive. Son action s’exerce par une activation du complément, la stimulation de la production de cytokines pro-inflammatoires par les kératinocytes et les sébocytes ; et par l’induction de la formation d’espèces réactives de l’O2 [8].
Le facteur génétique est un facteur important puisqu’une concordance de 98 % est retrouvée chez les jumeaux monozygotes atteints d’acné alors qu’elle est de 46 % chez les dizygotes. Le mode de transmission reste inconnu car il n’a pas été identifié de gènes précis cependant un polymorphisme génétique est de plus en plus rapporté. Les gènes incriminés sont les gènes de l’inflammation, les gènes du récepteur aux androgènes et les gènes des enzymes de la Stéroïdogenèse [9].
Aspects cliniques :
Les lésions élémentaires de l’acné sont représentées par la séborrhée, les lésions rétentionnelles et les lésions inflammatoires.
La séborrhée donne un aspect gras, brillant et luisant de la peau, plus marqué sur les zones médio faciales (front,nez, joues et menton). Cette prédominance médio faciale est liée à la plus forte concentration des glandes sébacées dans ces zones [10].
Les lésions rétentionnelles sont représentées par le comédon ouvert ou point noir et le comédon fermé ou point blanc. Ces lésions correspondent à l’accumulation de sébum et de kératine dans l’orifice infundibulaire. Les comédons sont faciles à extraire par la pression des doigts ou par un tire comédon et apparaissent sous forme de filaments gras, jaunâtres.
Les lésions inflammatoires se distinguent en papules et en pustules qui sont des lésions superficielles, et en nodules (lésions profondes) [11].
L’acné inflammatoire est la forme la plus fréquente (figure 1, 2), cependant selon le type lésionnel prédominant, l’acné peut être rétentionnelle, surtout en pré pubertaire, ou mixte, associant des comédons ouverts et fermés à des lésions inflammatoires.
Figure 1 : Acné inflammatoire du visage : pustules et papulesFigure 2 : Acné inflammatoire du visage : papules
L’acné juvénile touchant l’adolescent est la forme la plus fréquente, néanmoins d’autres tranches d’âge peuvent être touchées. En effet, 20 % des nouveaux nés dans les trois premiers mois de vie peuvent présenter une acné néonatale. Celle ci est due à une stimulation sébacée par les androgènes maternels. Son évolution est spontanément favorable en 2 à 3 mois [12]. A coté de ces formes de l’adolescent et du nouveau né, il existe une entité de plus en plus rapportée dans la littérature, il s’agit de l’acné de la femme adulte, touchant 25 % des femmes, survenant entre 25 et 45 ans, c’est une acné le plus souvent inflammatoire touchant la partie inférieure du visage (menton), de sévérité modérée. La recherche de signes cliniques d’hyper androgénie s’impose (hirsutisme, alopécie androgénique) [13]. Si dans la majorité des cas, l’acné est d’évolution bénigne, il existe deux formes graves : l’acné conglobata et l’acné fulminans.
L’acné conglobata est rare, plus fréquente chez l’homme et réalise une acné suppurative chronique avec de multiples nodules volumineux et douloureux sur les épaules, le dos, les fesses et le visage. L’évolution est constamment cicatricielle en brides et en chéloïdes (figure 3).
Figure 3 : Acné conglobata : nodules étendus sur le dos
L’acné fulminans est la forme la plus grave. Elle est rare, à prédominance masculine et se manifeste par une évolution ulcéro-nécrotique brutale des lésions d’acné. Des signes généraux sont présents à type de fièvre avec altération de l’état général, des douleurs musculaires et articulaires, et des signes biologiques d’inflammation. L’acné fulminans peut survenir spontanément ; des cas ont été rapportés après prescription d’isotrétinoine. Évaluer la sévérité de l’acné est important car le traitement dépend du grade de sévérité, pour cela, il existe plusieurs méthodes d’évaluation permettant un suivi clinique et thérapeutique du patient. L’échelle GEA (Global Evaluation of Acne) est la plus utilisée [14], elle divise l’acné en six grades : du grade 0 au grade 5. C’est une échelle facile, simple et d’utilisation rapide (tableau 1).
Tableau 1 : Cotation de la sévérité de l’acné par l’échelle GEA
Évolution / Complications :
L’acné juvénile évolue spontanément vers l’amélioration vers l’âge de 20-25 ans, même si certains cas évoluent vers une acné de l’adulte.
Les complications sont dominées par le risque cicatriciel, les cicatrices peuvent être hypertrophiques (figure 4), atrophiques (figure 5) ou pigmentées.
Certains facteurs favorisent cette évolution cicatricielle notamment le terrain génétique, le début précoce de l’acné et l’évolution prolongée.
L’acné a également des répercussions psychologiques (honte, frustration) et psychosociales (emploi, sorties, sport), c’est une affection qui retentit sur la qualité de vie au même titre que les maladies chroniques comme l’asthme. La dépression et l’anxiété sont considérées comme des comorbidités psychiatriques plus souvent associées à l’acné [15].
Figure 4 : Acné cicatricielle : chéloïdes sur le visageFigure 5 : Acné cicatricielle : cicatrices déprimées sur le visage.
Traitement de l’acné :
Le but du traitement est de guérir les lésions d’acné et d’améliorer la qualité de vie des patients. On dispose de traitements locaux et de traitements généraux.
Les traitements locaux ayant l’AMM dans l’acné sont le peroxyde de benzoyle, les rétinoïdes locaux et les antibiotiques topiques.
Le peroxyde de benzoyle : il est doté d’une activité antibactérienne et anti inflammatoire et se présente sous forme de gel ou crème à 2 %, 5 % et 10 %. Il est indiqué dans l’acné mixte à prédominance inflammatoire [16].
Les rétinoïdes locaux : indiqués dans l’acné rétentionnelle (microkystes et comédons) et inflammatoire. Les principaux effets indésirables sont une irritation locale et un effet asséchant. Leur prescription n’est pas recommandée durant la grossesse [17].
Les antibiotiques topiques : Deux antibiotiques sont disponibles dans le traitement de l’acné : l’érythromycine et la clindamycine en gel. Leur principal risque est l’induction d’une résistance bactérienne de P. acnes estimée à 60 %, entrainant une diminution de l’efficacité du traitement, il est recommandé de ne pas dépasser un mois de traitement et de toujours associer l’antibiotique local à un rétinoïde topique ou à un peroxyde de benzoyle [16,18].
Les traitements locaux peuvent être utilisés seuls ou en association.
Traitements systémiques :
Les cyclines : Les cyclines utilisées sont les tétracyclines et la doxycycline à 100 mg/j. Les effets secondaires les plus fréquents sont les troubles digestifs et la photosensibilité.
Les contre-indications des cyclines sont principalement la femme enceinte et l’enfant de moins de 8 ans (coloration des dents de lait).
Le problème majeur posé par les antibiotiques est l’induction de résistance bactérienne, en effet, la large utilisation des antibiotiques par voie locale et générale pendant des durées prolongées a conduit à l’apparition de résistance bactérienne. De ce fait, il est recommandé de prescrire les antibiotiques topiques toujours en association à peroxyde de benzoyle ou un rétinoïde local et de prescrire les antibiotiques par voie générale sur des durées courtes ne dépassant pas 3 mois [19].
Isotrétinoine : Dérivé de la vitamine A qui entraine une atrophie de la glande sébacée et une réduction de l’inflammation locale. Cette molécule est indiquée dans
les acnés graves et en deuxième intention après échec des cyclines et des topiques. Les doses utilisées sont de 0,5-1 mg/kg/j avec une dose cumulée de 120 à 150 mg/kg. Les effets secondaires sont temporaires à type de chéilite constante avec une sécheresse cutanée, des douleurs musculo-articulaires. L’isotrétinoïne peut provoquer des perturbations du bilan lipidique et hépatique avec élévation du cholestérol, des triglycérides et des transaminases. Il est donc recommandé de faire un bilan avant le début du traitement et un mois après [20].
Le risque tératogène est estimé entre 20 à 35 % des cas nécessitant des précautions d’emploi : une contraception orale ou au stérilet doit être instaurée un mois avant le début du traitement et poursuivie jusqu’à un mois après l’arrêt du traitement [21].
A ce jour, le lien entre la prise d’isotrétinoïne et les troubles psychiatriques n’est pas établi au niveau d’une population (le risque individuel n’est pas éliminé).
Il faudra évaluer chaque patient lors de la consultation [22].
Gluconate de zinc : molécule anti inflammatoire à la dose de 30mg/j à jeun avant le repas.
Hormonothérapie : L’acétate de cyprotérone associé à l’éthinyloestradiol (EE) est disponible sous forme de Diane 35® (2 mg d’acétate de cyprotérone et 35 mg d’EE), cette association est la seule possédant l’AMM dans le traitement de l’acné cependant son effet contraceptif n’est pas validé.
Les œstroprogestatifs (OP) trouvent leurs indications dans l’acné légère à modérée avec des signes d’hyper androgénie clinique ou biologique, dans le syndrome des ovaires polykystiques, en l’absence de réponse à un traitement conventionnel et comme contraceptifs [23].
Traitements physiques : représentés par l’exérèse mécanique des comédons, les peelings et le laser.
Prise en charge psychologique : elle consiste à mieux expliquer les causes de l’acné pour améliorer l’estime du soi et à expliquer les différents traitements et surtout leurs délais d’action.
Les conseils cosmétiques : Les soins d’hygiène doivent être adaptés avec une toilette non détergente et non agressive avec des produits à base de détergents synthétiques doux.
Des crèmes hydratantes sont utilisées pour diminuer la xérose cutanée qui accompagne le traitement par isotrétinoine.
Les produits de maquillage, notamment les fonds de teint doivent être testés non comédogènes.
Indications/Stratégie thérapeutique :
Un algorithme a été proposé par un groupe d’experts avec un traitement d’attaque et un traitement d’entretien.
Traitement d’attaque : Selon les recommandations de bonne pratique dans la prise en charge de l’acné du 10 Juin 2015 [24], il est préconisé en fonction de chaque grade une thérapeutique précise (Tableau 2).
Antibiothérapie orale (Doxycycline 100mg/J, Lymecyclyne 300mg/J) traitement local (PBO, rétinoïdes) Si échec : Isotrétinoine per os.
Grade 4
Antibiothérapie orale + traitement local Si échec : Isotrétinoine per os.
Grade 5
Isotrétinoine per os d’emblée
Tableau 2 : Thérapeutique en fonction des grades.
Conclusion :
L’acné est une dermatose fréquente de l’adolescent ; dont la prise en charge précoce est importante du fait du retentissement psychologique de cette affection sur la qualité de vie et du fait du risque cicatriciel.
Références
B. Dreno, corrélations cliniques et psychologiques dans l’acné. Ann Dermatol Venereol 2007 ;134 :451-5
N. Freton-Danou, Impact psychologique de l’acné. Ann Dermatol Venereol 2010 ;137 : S62-S65
Rod Tucker, The role of benzoyl peroxide in the management of acne vulgaris. The Pharmaceutical Journal 2007 ; (Vol 279): pp. 48-53.
Albert M. Kligman The treatment of acne with topical retinoids: One man’s opinions J Am Acad Dermatol 1997, Volume 36, Issue 6, Supple- ment, Pages S92-S95.
A. S. CHEHAD, Service de Dermatologie, CHU Lakhdar Benbadis, Constantine, Algérie.
Abstract : Biologics or biopharmaceuticals derived from living cells, are becoming in the last 15 years a common treatment for psoriasis. Currently their use spans many dermatologic conditions beyond psoriasis and psoriatic arthritis. Some indications such as psoriasis, atopic dermatitis, melanoma, hidradenitis suppurative and chronic urticaria, have already been approved by the main drugs agencies like the European Medicines Agency or the Food and Drug Administration. In recent years, there has been considerable interest in their use for other inflammatory skin diseases.
Résumé : Les biothérapies ou biomédicaments dérivés de cellules vivantes, sont devenues en une quinzaine d’années un traitement courant du psoriasis. Actuellement, leur utilisation s’est étendue à de nombreuses affections dermatologiques autres que le psoriasis et le rhumatisme psoriasique. Certaines indications ont déjà reçu l’approbation des agences de médicaments (Food and Drug Administration, Agence Européenne des Médicaments), telles que le psoriasis, la dermatite atopique, le mélanome, l’hidrosadénite suppurée et l’urticaire chronique. Ces dernières années, leurs indications dans d’autres affections inflammatoires dermatologiques suscitent un intérêt considérable.
Depuis l’utilisation du premier anti-tumor necrosis factor α (TNFα) dans la maladie de Crohn, fin des années 1990, les biothérapies sont devenues des options thérapeutiques incontournables dans plusieurs maladies cutanées modérées à sévères. Avec les avancées majeures dans la compréhension de la physiopathologie de diverses maladies dermatologiques, de nombreuses nouvelles molécules, issues de la biotechnologie sont développées ou en cours de développement. Les biothérapies déjà approuvées ou à l’étude sont dirigées contre le psoriasis, la dermatite atopique, le mélanome, l’hidrosadénite suppurée, le pemphigus vulgaire et l’urticaire chronique ainsi que dans plusieurs autres dermatoses le plus souvent hors autorisation de mise sur le marché (AMM).
Nomenclature :
Par rapport aux petites molécules, les biothérapies sont des protéines de masse moléculaire élevée qui doivent être injectées, car elles se dégraderaient dans le tractus gastro-intestinal si elles étaient administrées par voie orale. Alors que les petits médicaments moléculaires sont capables de traverser la membrane cellulaire, les bio-médicaments agissent à l’extérieur ou à la surface des cellules. De plus, les biothérapies nécessitent une ingénierie à partir de cellules vivantes spécialisées (1).
La nomenclature des protéines de fusion et des anticorps monoclonaux suit les règles établies par les dénominations communes internationales établies par l’Organisation Mondiale de la Santé. «-Cept» est utilisé comme suffixe pour les protéines de fusion (exp, Etanercept), et «-mab» (pour « monoclonal antibodies ») est utilisé comme suffixe pour désigner les anticorps monoclonaux (AcM). L’origine de l’AcM est indiquée en rajoutant un «-u» pour l’origine entièrement humaine (exp, le sécukinu- mab). Les AcM provenant à la fois de l’homme et de la souris sont humanisés, «-zu-» (exp, ixekizumab) ou chimériques, «-xi-» (exp, infliximab) (2).
Psoriasis :
Le psoriasis est une affection cutanée à médiation immunitaire, phénotypiquement hétérogène et qui suit souvent une évolution par poussées-rémissions (3). Les traitements systémiques traditionnels de la maladie sont représentés par l’acitrétine, la cyclosporine et le méthotrexate (4). Avec la découverte des cellules-clés et des médiateurs impliqués dans la maladie (figure 1) [5], les biothérapies sont devenues des options thérapeutiques importantes dans les formes modérées à sévères. Initialement, le TNF-ɒ était considéré comme la principale cytokine impliquée dans la physiopathologie du psoriasis, d’où les premières AMM pour les trois Anti- TNF-ɒ,
les seuls disponibles actuellement en Algérie, l’Etanercept (Enbrel®), l’Infliximab (Remicade®) et l’Adalimu-mab (Humira®) respectivement en 2004, 2006 et 2008 (3). Néanmoins, l’axe IL-23 / IL-17A a récemment été reconnu comme jouant également un rôle important dans le psoriasis en plaques (7). Cela a conduit au développe- ment de nouveaux médicaments, tels que l’AcM ciblant IL-12/23 l’ustekinumab (Stelara®) et les inhibiteurs de l’IL-17A. Trois AcM anti-IL-17 ont obtenu l’AMM pour le psoriasis en plaques : le secukinumab (Cosentyx®) en 2015 ; l’ixekizumab (Taltz®) en 2016 et le brodalumab (Kyntheum®) en 2017 (8-11). Les dernières biothérapies approuvées dans le psoriasis sont les anti-IL 23 : le gu- selkumab (Tremfya®) en Septembre 2017, et le Tildraki- zumab (Ilumya®) en Mars 2018 (11).
Le succès des biothérapies susmentionnées a entrainé leur large prescription ce qui a augmenté le fardeau financier inhérent à leur utilisation. Cependant, l’approbation officielle, depuis 2016, des 3 biosimilaires des anti-TNF-ɒ Etanercept (Erelzi®), Adalimumab (Amje- vita®) et Infliximab (Inflectra®) représente une épargne économique non négligeable (12-14).
Figure 1 : La pathogénie du psoriasis est subdivisée en deux phases : la phase d’initiation et la phase de maintenance. Le « stress » des kératinocytes entraine la libération de l’acide désoxyribonucléique (ADN). L’ADN libéré et le peptide antimicrobien LL-37 (cathélicidine) forment un complexe qui va se lier à son site récepteur présent sur les cellules dendritiques plasmacytoïdes (CP) du derme. Ces dernières sécrètent des interférons de type 1 (IFN-ɒ et -ẞ), Tumor necrosis factor-ɒ (TNF-ɒ), Interleukines (IL) IL-6 et IL-1 qui stimulent la migration des cellules dendritiques myéloïdes (CD) vers les ganglions lymphatiques de drainage. Au contact des Lymphocytes T naïves (TH0), les CD sécrètent TNF-ɒ, IL-12 et IL-23 qui in- duisent la différentiation des TH0 en Th1, Th17 et Th22 matures. Lors du retour à la peau via les vaisseaux sanguins, ces lymphocytes T spécifiques libèrent du TNF-ɒ, IFN-γ, IL-17A et -F, et IL-22 qui stimulent la prolifération et altèrent la différentiation des kératinocytes. Ceux-ci vont produire eux-mêmes des cytokines (TNF-ɒ, IL-6, IL-1) et chimiokines (CXCL8, CXCL10 et CCL20) pro-inflammatoires qui augmentent l’activation et la maturation des cellules T déjà présentes et ferment la boucle du cycle inflammatoire. Les biomédicaments approuvés jusqu’à maintenant vont agir sur les différents médiateurs inflammatoires.
La dermatite atopique :
La dermatite atopique (DA) est la plus fréquente des maladies inflammatoires chroniques cutanées touchant 2 à 10% des adultes et jusqu’à 15 à 30 % des enfants dans les pays développés (3).
De nombreux patients atteints de DA, essentiellement modérée à sévère, peuvent ne pas répondre ou sont mal contrôlés par les thérapeutiques standards voire même la photothérapie (15). Les traitements systémiques actuels sont principalement hors AMM, ont une efficacité limitée et des effets secondaires non négligeables (16).
Une meilleure compréhension de la pathogenèse de la DA a permis le développement de biomédicaments, qui inhibent sélectivement certaines voies-clés de la maladie. Le Dupilumab, un antagoniste de l’IL-4 et de l’IL-13, qui s’oppose à la réaction inflammatoire médiée par les lymphocytes T de type 2 (Th2), est le médicament biologique le plus avancé de ce groupe (16). Dans les essais cliniques, il a démontré son efficacité en réduisant l’activité et les symptômes de la maladie et en améliorant son phénotype génomique par la réduction significative des chimiokines Th2 et l’inversion des marqueurs épidermiques-clés de la DA. Il a également un profil de sécurité favorable. Après son approbation par la FDA, la Commission Européenne a accordé, en Septembre 2017, une autorisation de mise sur le marché à Dupilumab (Dupixent®) dans le traitement de la DA modérée à sévère de l’adulte candidat à un traitement systémique (16).
La nouvelle ère a seulement commencé avec le développement des douzaines de nouveaux agents biologiques ciblant une variété de voies cytokinéiques (en particu- lier l’IL-17, IL-22, IL-31 et TSLP) (17).
Mélanome :
Le mélanome est un problème majeur de santé publique dans les pays occidentaux avec une incidence et une mortalité en constante augmentation (32). Bien que l’exérèse chirurgicale des mélanomes diagnostiqués à un stade précoce, soit associée à un taux de survie de 95% à 5 ans, près de 20 % des patients atteints de mélanome développeront une maladie métastatique dont le pronostic est extrêmement sombre (33). Les molécules classiquement utilisées au stade métastatique (dacarbazine, fotémustine) ont une efficacité limitée et sans impact sur la survie globale (32).
Les anti-BRAF vemurafenib (Zelboraf®), le Dabrafenib (Tafinlar®) et l’anti-MEK trametinib (Mekinist®) ont été approuvés dès 2011. Le taux de réponses à ces thérapies ciblées est élevé (45% à 53%), mais la durée médiane de celle-ci est inférieure à 1 an (35).
Quoique l’association anti-BRAF / anti-MEK ait montré de meilleurs taux et durées de réponse, leur indication est limitée à 40% de patients présentant cette mutation (35,37).
Il est bien montré que le mélanome est une tumeur immunogène, et cette hypothèse est appuyée par des observations cliniques, des études histologiques et l’efficacité partielle des immunothérapies, comme les activateurs immunitaires IFN-α2b et IL-2, tous deux approu- vés dans le traitement du mélanome (41).
Les AcM capables de bloquer les points de contrôle immunitaires « checkpoints » de l’antigène-4 des lymphocytes-T cyto- toxiques (cytotoxic T-lymphocyte antigen-4 = CTLA-4), de la protéine de mort cellulaire programmée 1 (programmed cell death protein 1= PD-1) et son ligand (PD-L) ont démontré une grande efficacité dans le mélanome métastatique et autres tumeurs solides (43).
L’Ipilimumab (Yervoy®) est le premier et le seul médicament anti-CTLA-4 approuvé depuis 2010 ; le taux de réponses est seulement de 10,9 % mais avec une rémission prolongée assez élevée de 60 % à 2 ans. Cependant, 23 % des patients présenteront des effets indésirables d’origine immunitaire de grade 3 ou 4 (45).
Les anticorps anti-PD-1 nivolumab (Opdivo®) et pembrolizumab (Keytruda®), approuvés depuis 2015, ont démontré un taux de réponses de 30 à 40 %, avec une majorité de réponses prolongées (49).
Plusieurs études sont actuellement en cours pour d’autres AcM ciblant le PD-1 ou son ligand (51), ainsi que de nombreuses associations, également à l’essai, afin d’élever la barre d’efficacité de l’immunothérapie : anti- PD-1 avec anti-BRAF, anti- MEK ou anti-CTLA-4, et anti-PD-L1 avec anti-BRAF (52).
Hidrosadénite suppurée :
L’hidrosadénite suppurée (HS) est une maladie cutanée inflammatoire chronique, récidivante et délabrante du follicule pileux qui se manifeste par des lésions inflammatoires des zones des glandes apocrines du corps (53).
Plusieurs médiateurs inflammatoires ont été impliqués dans la pathogenèse de l’HS, mais seulement certains sont des cibles des bio-médicaments existants (59). Les biopsies de lésions de l’HS ont révélé une abondance de PNN, Th1, Th17, macrophages et cellules dendritiques. Les cytokines impliquées comprennent le TNF-α, l’IFN-γ et les interleukines (IL-lß, IL-10, IL-17A et IL-23) (60).
Les niveaux du TNF-α sont cinq fois plus élevés dans les lésions de l’HS par rapport à d’autres dermatoses inflammatoires, en particulier le psoriasis (60).
En 2015, l’adalimumab est devenu le premier et le seul AcM approuvé dans l’indication : HS modérée à sévère (62). Son efficacité a été initialement observée chez des patients atteints de la maladie de Crohn associée à une HS où il a été constaté une résolution concomitante des lésions de l’HS, en plus d’une diminution des symptômes de la MICI (64). Bien que l’adalimumab soit le seul médica- ment ayant une AMM, plusieurs autres biothérapies ont démontré une efficacité potentielle en ciblant les diffé- rentes cytokines inflammatoires impliquées dans l’HS, notamment le TNF-α (infliximab), IL-1 (anakinra), IL-7A (sécukinumab) et IL-12/23 (ustekinumab) (62).
Le pemphigus :
Le pemphigus est une dermatose bulleuse auto-immune potentiellement mortelle affectant la peau et les muqueuses. Il se caractérise par la production d’auto- anticorps pathogènes dirigés contre les molécules d’adhésion desmogléine 1 et desmoglycine 3 responsables de la cohésion entre les kératinocytes de la peau et des muqueuses (65). Les corticoïdes systémiques associés ou non aux immunosuppresseurs sont considérés comme le traitement standard de la maladie (69).
Le Rituximab (Mabtera®) est un anticorps monoclonal dirigé contre l’antigène CD20 des lymphocytes B. Bien qu’il n’ait pas reçu d’AMM dans les maladies bulleuses auto-immunes, certaines indications sont reconnues comme acceptables : pemphigus corticorésistant, corticodépendant ou en cas de contre-indication aux corticoïdes ou aux immunosuppresseurs (70). Cependant, vu que le traitement au long cours par les corticoïdes pourrait induire des effets secondaires graves et même potentiellement mortels, certaines études ont suggéré que l’utilisation du rituximab en première intention dans le pemphigus permettrait un sevrage rapide de la corticothérapie (65).
Urticaires chroniques :
L’urticaire chronique (UC) est définie comme la survenue d’enflure (urticaire superficielle), angiœdème ou les deux pendant plus de 6 semaines (74). La libération de l’histamine et autres facteurs pro-inflammatoires est considérée comme le « résultat final » justifiant l’utilisation des antihistaminiques H1, en tant que traitement de première intention de l’UC (75). Le concept d’un rôle central de l’IgE et du FcεRI dans la stimulation initiale des mastocytes (ou basophiles) aboutissant à leur dégranulation a conduit à l’avènement de nouvelles thérapies ciblées. L’omalizumab (Xolair®, anti-IgE) s’est montré très efficace et sûr, même au long cours, ou sur les rechutes, dans l’UC (spontanée ou induite) avec amélioration substantielle de la qualité de vie des patients (76).
Conclusion :
Les biothérapies sont devenues des médicaments largement utilisées en dermatologie. Avec la recherche fondamentale toujours en cours, de nombreux autres bio-médicaments prometteurs sont également à l’étude ainsi que des biosimilaires, afin de diminuer le fardeau financier des produits originaux.
Référence :
Morrow T, Felcone, L.H. Defining the difference : What makes biologics unique. Biotechnol. Healthc. 2004, 1,24–29.
World Health Organization. International Nonproprietary Names (INN) for Biological and Biotechnological Substances. http://www. webcitation.org/6rGAZvnd2.
Nestle FO, Kaplan DH, Barker J. Mechanisms of disease; psoriasis. N Engl J Med 2009; 361:496-509.
Yiu ZZ, Warren RB. Novel oral therapies for psoriasis and psoriatic arthritis. Am J Clin Dermatol 2016; 17:191-200.
Kofoed K, Skov L, Zachariae C. New drugs and treatment targets in psoriasis. Acta Derm Venereol 2015; 95:133-9.
Veilleux MS, Shear NH. Biologics in patients with skin diseases. J Allergy Clin Immunol. 2017 May;139(5):1423-1430. doi: 10.1016/j. jaci.2017.03.012.
Torres T, Romanelli M, Chiricozzi A. A revolutionary therapeutic approach for psoriasis: bispecific biological agents. Expert Opin Inves- tig Drugs 2016;25: 751-4.
AbuHilal M, Walsh S, Shear N. The role of IL-17 in the pathogenesis of psoriasis and update on IL-17 inhibitors for the treatment of plaque psoriasis. J Cutan Med Surg 2016; 20:509-16.
Habashy J, Robles DT, James WD. Psoriasis Medication, Updated: Mar 27, 2018. https://emedicine.staging.medscape.com/ article/1943419-medication#12.
FDA approves Inflectra, a biosimilar to Remicade. 2016. http://www. fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm494227.
FDA approves Erelzi, a biosimilar to Enbrel. 2016. http://www.fda. gov/NewsEvents/Newsroom/PressAnnouncements/ucm518639.
FDA approves Amjevita, a biosimilar to Humira. 2016. http://www. fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm522243. htm.
Simpson EL, Bruin-Weller M, Flohr C et al. When does atopic dermatitis warrant systemic therapy? Recommendations from an expert panel of the International Eczema Council. J Am Acad Dermatol 2017; 77:623-33.
Osinka K, Dumycz K, Kwiek B, Feleszko W. Novel Therapeutic Approaches to Atopic Dermatitis. Arch. Immunol. Ther. Exp. Published online : 31 Aug 2017.
R. BOUSSAID, Service de Dermatologie, CHU Dr Lakhdar Benbadis, Constantine
Abstract : Chronic urticaria (UC) is a common pathology whose etiology is unknown. Its impact on quality of life is not negligible. Antibodies directed to mast cell and basophilic IgE expressed receptors (FcεRI) are found in approximately 30% of cases and could be associated to more severe and prolonged forms. Many pathologies can be expressed by a UC. However, in the absence of evocative clinical signs, an extensive etiological assessment rarely leads to an underlying pathology diagnosis. In this case, only a minimal etiological assessment is indicated. Latest-generation systemic antihistamines are the first-line treatment. In refractory cases, other molecules are available but there are few controlled studies on the topic.
Key-words : Histamine, urticaria, antihistamines
Résumé : L’urticaire chronique (UC) est une pathologie fréquente dont l’étiologie est inconnue. Son impact sur la qualité de vie est non négligeable. Des anticorps dirigés contre les récepteurs des IgE exprimés par les mastocytes et les basophiles (FcεRI) sont retrouvés dans environ 30% des cas et seraient associés à des formes plus sévères et prolongées. De nombreuses pathologies peuvent s’exprimer par une UC. Toutefois, en l’absence d’une clinique évocatrice, un bilan étiologique extensif permet rarement de diagnostiquer une pathologie sous-jacente. Dans ce cas, seul un bilan étiologique à minima est indiqué. Les antihistaminiques systémiques de dernière génération constituent le traitement de première intention. Dans les cas réfractaires, d’autres molécules sont à disposition mais il existe peu d’études contrôlées à leur sujet.
Mots-clés : Histamine, urticaria, antihistamines.
Introduction :
Dans la nouvelle classification 2013, l’urticaire chronique (UC) regroupe désormais les UC inductibles (par un stimulus donné avec des tests de provocation standardisés) et les UC spontanées, et exclut les éruptions pseudo-urticariennes avec lésions cutanées superficielles fixes durant plus de 24 heures et/ou les œdèmes durant plus de 72 heures. La sévérité et l’impact psychosocial de cette maladie chronique peuvent être mesurés par plusieurs scores spécifiques, pas encore tous disponibles en français : urticaria et Angioedema Activity Score, Urticaria Control Test, CU-Q2OL, AE-QOL. Bien qu’une origine allergique soit reconnue comme rarissime dans l’UC.
Définitions
Urticaire : éruption cutanée prurigineuse et fugace d’apparition rapide, secondaire à un œdème vasculaire circonscrit du derme superficiel et caractérisée par des papules érythémateuses ou anémiques (blanches) entourées d’un érythème.
Angioedème : œdème localisé aigu des tissus sous-cutanés ou sous-muqueux d’apparition subite, provoquant souvent une douleur plutôt qu’un prurit.
Urticaire chronique : urticaire spontanée lors de laquelle les lésions sont récidivantes ou apparaissent de manière permanente durant plus de six semaines. La taille, le nombre et la forme des lésions varient considérablement.
On la différencie de l’urticaire aiguë dont l’évolution est de moins de six semaines, 10% évoluant en urticaire chronique.
Physiopathologie
Les processus physiopathologiques menant à l’UC sont multiples et sont résumés dans la figure 1. Ils aboutissent à la libération des médiateurs des mastocytes (histamine, prostaglandines, leucotriènes, bradykinine, substance P) à l’origine des manifestations de l’urticaire et/ou de l’angioedème.
Figure 1 : Physiopathologie des urticaires chroniquesSource des photos : Pr Fréderic Bérard, Service d’Allergologie Hôpital Pierre Bénite, Lyon-Sud
Environ 30% des patients présentent des anticorps sériques dirigés contre le récepteur des IgE exprimés à la surface des mastocytes et des basophiles (FceRI), induisant leur dégranulation (3-5).
Ces anticorps suggèrent un mécanisme auto-immun. Le complément et des isotypes d’immunoglobulines G fixant le complément (IgG1 et 3) semblent être requis pour que ces autoanticorps agissent. Ce type d’UC, appelée aussi UC auto-immune, est probablement une forme d’UC plus sévère et de plus longue durée. On a identifié récemment des anticorps anti-IgE et des anticorps dirigés contre le récepteur IgE de faible affinité (FceRII), libérant des médiateurs des éosinophiles (major basic protein principalement) et, secondairement, des mastocytes. Le test au sérum autologue n’est pas important pour poser le diagnostic d’UC et peut être difficile à interpréter par des non-spécialistes.
Une composante d’urticaire physique associée à l’UC est souvent retrouvée, en particulier l’urticaire à la pression et l’urticaire factice (urticaire dermographique).
De nombreux facteurs peuvent aggraver l’UC. Les facteurs aggravant ne peuvent être attribués à une exacerbation de l’UC que si le temps écoulé entre l’exposition et l’aggravation des lésions est court (moins de 2 heures). Certains médicaments peuvent exacerber une UC (AINS, morphiniques).
De même, certains aliments (tableau 1) contiennent des amines vaso-actives (dont l’histamine) et d’autres constituants dont le rôle dans l’exacerbation d’une UC (réaction pseudo-allergique) est controversé.
Une allergie alimentaire vraie (IgE-médiée) est rarement à l’origine d’une UC.
Tableau 1 : Principaux aliments riches en histamine et histamino- libérateurs
Il est difficile de différencier une origine pseudo-allergique d’une allergie vraie. Toutefois, de manière générale, l’éviction d’un aliment suspect permet une résolution des symptômes en 24-48 heures lors d’allergie vraie (IgE-médiée), alors qu’un bénéfice n’est rapporté qu’après deux à trois semaines d’éviction d’aliments riches en amines vaso-actives.
En cas de doute concernant le mécanisme en jeu, seul un bilan allergologique (tests cutanés, IgE spécifiques, test de provocation) permet de trancher.
Le rôle de certains agents infectieux est controversé. Le traitement d’une infection (abcès dentaire, sinusite, cystite, cholécystite) n’induit pas plus de rémission de l’UC que l’absence de traitement. Il n’existe pas de bonnes preuves concernant le rôle d’une infection à Helicobacter pylori, de l’hépatite B et C et de la candidose. Une parasitose peut occasionnellement être à l’origine d’une UC.
Tableau 2 : Pathologies pouvant se manifester par des lésions cutanées mimant une urticaire
Rarement, une UC ou une vasculite urticarienne peuvent être l’expression d’une maladie systémique ou d’un syndrome sous-jacent (tableau 2) et ces pathologies doivent être recherchées en présence d’une clinique extra cutanée évocatrice.
Dans la vasculite urticarienne, les lésions cutanées peuvent être atypiques : persistance durant plus de 24 heures au même endroit, induisant une douleur ou une brûlure plutôt qu’un prurit, pouvant provoquer des lésions pétéchiales ou pigmentées. Des anticorps anti-thyroïdiens et/ou une dysthyroïdie sont retrouvés de manière significativement plus élevée chez les patients souffrant d’UC que dans la population générale.
Les patientes souffrant d’UC peuvent présenter des exacerbations avant leurs menstruations et durant la grossesse. Dans des cas anecdotiques, l’éruption est reproductible par l’administration de progestérone et, plus rarement, d’œstrogène.
Traitement
1. Traitements disponibles :
a. Les antihistaminiques :
– Les antihistaminiques anti-H1 :
Les antihistaminiques anti-H1 de 2e génération sont le traitement de première intention de l’UC.
Entre 45 et 80 % des urticaires chroniques sont améliorées par les antihistaminiques, selon les études. Une méta-analyses récente sur les antihistaminiques anti-H1 au cours de l’UC ne retrouve pas de supériorité d’un antihistaminique par rapport aux autres compte tenu du peu d’études et des différentes méthodologies.
Une conférence d’experts internationaux récente (2014) propose de majorer jusqu’à 4 fois les doses d’anti-H1 de 2e génération en cas d’inefficacité à posologie classique (1 comprimé par jour).
Les antihistaminiques anti-H2
Les antihistaminiques anti-H2 (type ranitidine, cimetidine) n’ont pas leur place dans le traitement de l’UC.
b. Les anti-leucotriènes :
L’efficacité propre du montélukast est difficile à évaluer car il est le plus souvent associé à un antihistaminique anti-H1 dans les études : néanmoins, il serait efficace dans environ 50 % des cas.
c. Les immunomodulateurs :
Corticothérapie générale :
Elle n’est pas recommandée et n’a pas sa place dans le traitement de l’urticaire chronique spontanée (UCS), d’après la conférence de consensus française de 2003. Elle aurait des effets délétères favorisant la corticodépendance, l’escalade thérapeutique et l’aggravation
de l’UC lors de son arrêt. La conférence d’experts internationaux de 2014, la préconise en cures courtes de 3 à 7 jours dans les exacerbations (sans précision sur les posologies utilisées.
Ciclosporine :
Elle reste un traitement suspensif dans l’UCS et non dénué d’effets secondaires. Pour certains auteurs elle serait efficace chez 70 à 80 % des patients après 6 semaines de traitement.
Méthotrexate :
Un essai randomisé multicentrique en cours dans le cadre d’un protocole hospitalier de recherche clinique (NCT01960283) devrait permettre de définir la place de cette molécule dans la prise en charge de l’UCS.
a. Les anticorps monoclonaux :
L’omalizumab :
Il s’agit d’un anticorps monoclonal humanisé, IgG1k se fixant aux immunoglobulines E (IgE) et empêchant leur fixation sur le récepteur de haute affinité de IgE (FceRI) exprimé à la surface des mastocytes. L’omalizumab a obtenu l’autorisation de mise sur le marché européen, en traitement additionnel dans l’UCS chez les adultes et adolescents (à partir de 12 ans) présentant une réponse insuffisante aux traitements antihistaminiques anti-H1. Plusieurs études randomisées contrôlées retrouvent une efficacité significative dans l’UCS chez 75 à 80 % des patients, avec un bon profil de tolérance.
Aucune étude randomisée de l’efficacité de l’omalizumab dans les UCI1 n’a été publié, par contre il existe des observations de l’efficacité et parfois de l’échec, de l’omalizumab dans les UCI. La conférence d’experts internationaux de 2014 positionne l’omalisumab en 3e ligne de traitement de l’UCS, au même niveau que la ciclosporine ou le montélukast. L’initiation de l’omalizumab dans l’UCS est hospitalière, annuelle et son renouvellement est réservé aux spécialistes en dermatologie, pédiatrie ou médecine interne ; la dose recommandée est de 300 mg en injection sous-cutanée toutes les 4 semaines, la demi-vie d’élimination est d’environ 26 jours. La durée de traitement n’est actuellement pas définie. Il reste au même titre que les antihistaminiques dans l’urticaire chronique spontanée un traitement symptomatique et non curatif.
Le rituximab :
Seuls des cas cliniques isolés sont publié, pour lesquels les résultats en termes d’efficacité sont contradictoires.
Les anti-TNFα :
Ils ont été utilisés dans des études ouvertes ou rétrospectives
1 UCI = Urticaire Chronique inductible chez un petit nombre de patients atteints d’UC ou d’urticaire retardée à la pression.
b. Autres traitements :
D’autres traitements ont été essayés dans le traitement de l’UC avec des résultats variables dans des études non contrôlées ; la colchicine, la dapsone, la salazopyrine, le plaquénil ou encore les anticoagulants ou la persantine.
Urticaires chronique et régimes d’éviction : Les données publiées concernant le rôle des aliments dits « riches en histamine » ou « histamino-libérateurs » ainsi que les régimes sans pseudo-allergènes au cours de l’UC ne sont pas convaincantes.
3. Urticaires chronique et qualité de vie :
L’UC a un réel impact sur la qualité de vie des patients, les poussant à rechercher la cause de la maladie, alors que cette maladie est multifactorielle. Différents scores d’activité de la maladie ont été développés pour évaluer l’impact de l’UC sur la qualité de vie : score Urticaria Activity Score sur les 7 derniers jours (UAS7), Chronic Urticaria Quality of Life Questionnaire (CU-Q2oL) ou l’urticaria Control Test (UCT). Certaines équipes proposent des séances d’éducation thérapeutique dans l’UC ; cette démarche, en apportant une information claire et détaillée au patient, et en évaluant son retentissement sur la vie quotidienne, peut améliorer l’adhésion thérapeutique et la prise en charge globale.
4. Traitement de l’urticaire chronique, en pratique :
a. Mesures générales :
En plus du contrôle des facteurs favorisants et de la prise en compte du retentissement de l’UC sur la vie quotidienne des patients, un traitement de fond adapté doit être institué. L’objectif du traitement n’est pas d’obtenir une disparition complète des lésions, mais une qualité de vie et des lésions acceptables du point de vue du patient. En effet, de nombreux patients sous-antihistaminiques ont encore des plaques d’UC mais peu ou non symptomatiques, non prurigineuses.
b. Traitement médicamenteux :
Les antihistaminiques anti-H1 de 2e génération prescrits initialement à posologie AMM (soit 1 comprimé par jour). Rapidement (dans un délai de 2 semaines), ce traitement peut être majoré (augmentation jusqu’à 4 comprimés par jour des antihistaminiques en cas de non-contrôle de l’UC.
En cas d’échec de ce traitement l’ajout d’un anti-leucotriène peut être essayé.
Ce traitement doit être systématiquement associé à des antihistaminiques à posologies augmentées et doit être prescrit en continu pour plusieurs semaines avant de parler d’échec. En troisième ligne de traitement, plusieurs options thérapeutiques sont possibles : l’omalizumab, le méthotrexate ou la ciclosporine.
Ces traitements doivent être associés aux antihistaminiques. L’omalizumab à 300g/semaines en injections sous-cutanées reste le seul qui ait l’AMM dans cette indication, il est d’initiation hospitalière.
Références :
Zuberbier T. Et al. The EAACI/GA(2)LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urtica- ria: the 2013 revision and update. Allergy. 2014 Jul;69(7):868-87.
Zuberbier T, Asero R, Bindslev-Jensen C, Walter Canonica G, Church MK, Giménez-Arnau AM, et al. EAACI/GA(2)LEN/EDF/WAO guide line: management of urticaria. Allergy 2009;64(10):1427–43.
Doutre M-S, Joly P. Management of chronic urticaria: consensus conference’s recommendations. Rev Med Interne 2003;24(10):637–9.
Nosbaum A, Augey F, Nicolas J-F, Bérard F. Pathophysiology of urticaria and therapeutic approaches. Rev Med Interne 2010;31(Suppl. 1):S18–22.
Augey F, Guillot-Pouget I, Gunera-Saad N, Berard F, Nicolas JF. Effet de l’arrêt des corticoides au cours de l’urticaire chronique (étude prospective de 17 malades). Ann Dermatol Venereol 2008; 135:21–5.
Bernstein JA, Lang DM, Khan DA, Craig T, Dreyfus D, Hsieh F, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol 2014;133(5):1270–7.
S. HAMzAOUI, Y. ABI AYAD ,z. ELOSMANI, A. SERRADJ ; Service de Dermatologie, EHU 1er Novembre, Oran.
Abstract : Pemphigus is a rare acquired autoimmune bullous dermatosis. Objectives : This study of patients with pemphigus over a period of 9 years was aimed at identifying the epidemiological peculiarities of this pathology in the dermatology department of the Oran EHU. Materials and methods : Descriptive and analytical retrospective study based on patient files in the dermatology department of the Oran EHU conducted in 48 patients with pemphigus from January 2009 to August 2017. Data entry and analysis was done by EpiData Ver 3.1 and Microsoft Office Excel. Results : Forty-eight patients were included (28 women and 20 men) with a sex-ratio (M / F) of 0.6 and an average age of 57 years. Deep form was the most common (64.6 %) most often triggered in summer. Inaugural mucosal involvement was noted in 41.7 % of cases. Average consultation time was 158 days, and average duration of hospitalization was 42 days. Corticosteroids were used in 95.8 % of patients, Immunosuppressants were prescribed in 26 patients (54.6 %), sulfones in 15 patients (31.25 %) and rituximab in 6 patients (12.5 %). The evolution was good in 68.8% of cases. Infectious complications were the most frequent (44 %). Death was noted in 3 patients (6.3 %). Conclusion : Results of our study are consistent with literature data, however controlled studies are needed to confirm these results.
Résumé : Le pemphigus est une dermatose bulleuse auto-immune acquise rare. Objectifs : Cette étude de patients atteints de pemphigus réalisée sur une période de 9 ans a eu pour objectif de relever les particularités épidémiologiques de cette pathologie au niveau du service de dermatologie de l’EHU d’Oran. Matériels et méthodes : Étude rétrospective descriptive et analytique à partir de dossiers de malades au service de dermatologie de l’EHU d’Oran menée chez 48 patients atteints de pemphigus sur une période allant de Janvier 2009 à Août 2017. La saisie et l’analyse des données ont été effectuées par EpiData Ver 3.1 et Microsoft Office Excel. Résultats : Quarante huit patients ont été inclus (28 femmes et 20 hommes) ; avec un sex-ratio (H/F) de 0,6 et une moyenne d’âge de 57 ans. La forme profonde était la plus fréquente (64,6 %) déclenchée le plus souvent en été. L’atteinte muqueuse inaugurale a été notée dans 41,7 % des cas. Le délai moyen de consultation était de 158 jours. La durée moyenne d’hospitalisation était de 42 jours. Les corticoïdes ont été utilisés dans 95,8 % des patients. Les immunosuppresseurs prescrits chez 26 malades (54,6 %), les sulfones chez 15 patients (31,25 %) et le rituximab chez 6 patients (12,5 %). L’évolution était bonne dans 68,8 % des cas. Les complications infectieuses étaient les plus fréquentes (44 %). Le décès était noté chez 3 patients (6,3 %). Conclusion : Les résultats de notre étude concordent avec les données de la littérature, cependant, des études contrôlées sont nécessaires pour confirmer ces résultats.
Le pemphigus est une dermatose bulleuse auto immune intra épidermique acquise touchant la peau et les muqueuses caractérisée par la production d’auto-anticorps dirigés contre des protéines d’adhésion de la membrane cytoplasmique des kératinocytes (principalement les desmogléines). La fixation des auto-Ac sur ces protéines de jonction entraîne une perte de la cohésion interké-ratinocytaire (acantholyse) aboutissant à la formation d’une bulle intra-épidermique [1].
On distingue deux grandes formes de pemphigus auto-immuns selon le siège préférentiel du clivage intra-épidermique : les pemphigus profonds (pemphigus vulgaire et pemphigus végétant) (figure 1, 2, 3) ; les anticorps reconnaissent des protéines exprimées pré-férentiellement dans les couches basales de l’épiderme qui a pour résultat un clivage qui se situe au niveau de la couche suprabasale de l’épiderme, et les pemphigus superficiels (pemphigus érythémateux, pemphigus foliacé) (figure 4, 5) ; les antigènes cibles sont exprimés de façon prédominante dans les couches superficielles et le clivage se situe plus haut dans l’épiderme, sous la couche granuleuse [2] .
Depuis les années 1970, de nouvelles formes de pemphigus ont été décrites qui échappent à cette dichotomie : le pemphigus paranéoplasique (figure 6), pemphigus herpétiforme et pemphigus à IgA. Ainsi, si le diagnostic est suspecté cliniquement, son affirmation nécessite la réalisation d’une biopsie cutanée ou muqueuse montrant un clivage intra-épidermique par acantholyse ainsi que des examens complémentaires pour la recherche des auto-anticorps permettant d’affirmer le diagnostic.
L’évolution spontanément mortelle du pemphigus a été transformée par la corticothérapie générale et les immunosuppresseurs et récemment le rituximab, mais le pronostic reste réservé du fait des complications iatrogènes [3].
Étude rétrospective descriptive et analytique à partir de dossiers de malades au service de dermatologie de l’EHU d’Oran menée chez 48 patients atteints de pemphigus sur une période de 9 ans, allant de Janvier 2009 à Août 2017. La saisie et l’analyse des données ont été effectuées par EpiData Ver 3.1 et Microsoft Office Excel.
Résultats :
Quarante huit patients ont été inclus ( dont 28 femmes et 20 hommes) avec un sex-ratio (H/F) de 0,6 (graphe 1) et une moyenne d’âge de 57 ans.
Graphe 1 : Répartition des cas de pemphigus selon le sexe
Le pemphigus profond (vulgaire et végétant) était le plus fréquent (64,6 % des cas), le pemphigus superficiel (séborrhéique et foliacé) était retrouvé chez 27,1 % des cas (graphe 2). L’atteinte muqueuse inaugurale était notée chez 41,7 % des patients et le début était cutané dans 50 % des cas.
Graphe 2 : Répartition des cas selon le type du pemphigus
Le délai moyen de consultation était de 158 jours avec un pic de fréquence au mois de Juin (20,8 %). La durée moyenne d’hospitalisation était de 42 jours.
Les corticoïdes étaient utilisés chez 46 patients (95,8 %) à une dose d’attaque variant de 0,5-2 mg/kg/jour maintenue pendant une durée moyenne de 58 jours, la durée de la dégression était de 21 mois. Les immunosuppresseurs (Imurel® et Cellcept®) étaient utilisés chez 26 malades (54,6 %), les sulfones chez 15 patients (31,25 %) et le rituximab chez 6 patients (12,5 %) (graphe 3).
Graphe 3 : Répartition des cas selon les traitements utilisés
L’évolution du pemphigus était bonne dans 68,8 % des cas, mauvaise dans 4,3 % des cas, des rechutes étaient notées chez 26,9 % des patients liées à l’arrêt prématuré du traitement, diminution rapide du traitement et diminution au dessous de la dose minimale efficace (graphe 4).
Les complications infectieuses étaient les plus fréquentes (44 %) suivies par les complications métaboliques (18,75 %) et endocriniennes (14,5 %) (graphe 5). Le décès était noté chez 3 patients (6,3 %) (graphe6).
Graphe 4 : Evolution du pemphigusGraphe 5 : Répartition des complications secondaires aux traitementsGraphe 6 : Evolution des patientsTableau 1 : Tableau comparatif des paramètres épidémiologiques du pemphigus dans certaines régions du monde (PS : Pemphigus superficiel ; PP : Pemphigus profond ; PV : Pemphigus vulgaire)
Discussion :
Dans notre série, le pemphigus atteint les adultes d’âge moyen avec une prédominance féminine rejoignant ainsi les données de la littérature et de la plupart des études (tableau 1).
Les formes profondes sont plus fréquentes concordant avec les résultats de l’étude Tunisienne [4], Française [5], et Indienne [6].
Le début cutané a été noté dans la moitié des cas alors que l’atteinte muqueuse inaugurale a été retrouvée dans 41,7 % des cas se rapprochant de l’étude Indienne [6] et Oranaise [7].
Le délai moyen de consultation était plus long que celui de l’étude Française [8], relevant les difficultés et les errances qui accompagnent le diagnostic du pemphigus.
Les poussées coïncident avec la saison estivale soulignant probablement le caractère déclenchant du soleil, ce dernier a été bien incriminé, à l’origine du pemphigus dans diverses études [9,10,11].
L’approche thérapeutique principale était la corticothérapie générale ; néanmoins, il est à souligner une efficacité et bonne tolérance du rituximab à travers notre petit échantillon traité par ce dernier.
Les complications infectieuses sont les plus fréquentes ce qui est en accord avec l’étude Marocaine (71,5%) [12], Tunisienne (55%) [4] et Iranienne (60,6 %) [13], suivies par les complications métaboliques et endocriniennes ; ces effets secondaires doivent être pris en compte et toute prescription doit prendre en considération le rapport bénéfice/risque pour le patient.
Le taux de mortalité estimé à 6,25% est plus élevé que celui rapporté par les Marocains [3]; moins élevé que chez les Tunisiens [2] et les Français [4], ce taux est revu à la baisse ces dernières années, lié probablement à une prise en charge plus adéquate.
Conclusion :
Les résultats de notre étude concordent avec les données de la littérature, cependant, d’autres études plus contrôlées sont nécessaires pour confirmer ces résultats.
Références :
Centre de référence des maladies bulleuses auto-immunes / HAS. Mai 2018.
A. Serradj. Thèse : Pemphigus à Oran : Etude épidemiologique, clinique, immunologique et thérapeutique. 2010.
A. Khaled, S. Ben Taazayet, A. Souissi, M. Kharfi, F. Zeglaoui, B. Fazaa. Service de dermatologie, hôpital Charles-Nicolle, Tunis, Tunisie. Étude pronostique de 47 cas de pemphigus. doi: 10.1016/j. ann der.2011.10.032
L. Jelti et All Incidence et mortalité du pemphigus en France.http:// dx.doi.org/10.1016/j.annder.2016.09.147
Joyeeta Chowdhury et All. A Clinicopathological Study of Pemphigus in Eastern India with Special Reference to Direct Immunofluorescence. 2016 May-Jun; 61(3): 288–294.
A. Serradj, H. Saleh, Y. Abiayada, N. Mahmoudib, Y. Zemmour, N. Midoun. Le pemphigus à Oran. doi:10.1016/j. ann der. 2011.10.036
Almugairen ET All. Délai diagnostique des différentes formes de pemphigus. Ann Dermatol Vénérol. Vol 136 n 12 S2009
Kaplan RP et al. Physically induced pemphigus after cosmetic procedures. Int J Dermatolo 1993;32:100-3
Langan S M et al. Bullous pemphigoid and pemphigus vulgaris indice and mortality in the UK: population based cohort study. BMJ 2008 ;337 :a 180 :1-7
B. DAHMANI,O. BOUDGHENE STAMBOULI, Service de Dermatologie, CHU Tidjani Damerdji, Tlemcen
Abstract : Skin diseases are a frequent reason for consultation in Algeria and thus represent a public health issue that has long been underestimated. Little data are available in general on epidemiological criteria for these dermatoses. Psoriasis, a chronic disease with a major impact on quality of life is one of the major skin diseases listed in order of frequency after eczema and acne with a rate that varies between 3 and 5% according to the publications. It is characterized by its high frequency, its impact on quality of life, increased frequency of comorbidities, the oncological risk related to the proposed treatment, treatment cost of the disease is increasing with marketing of various products from new technology and increased mortality due to above mentioned factors. This skin disease is common in dermatology department consultation of Tlemcen CHU, representing 5.8% (consultation register). Psoriasis affects mainly the quality of life to the same extent as other chronic diseases, especially visible diseases. Smoking and alcohol can contribute as triggers of psoriasis. The existence of an association between psoriasis and addictive disorders is suspected for several years. Accurate information on the cost of the disease are needed to make decisions and the development of a health policy based on real data.
Key-words : Psoriasis, comorbidities, cost, public health.
Résumé : Les dermatoses représentent un motif fréquent de consultation en Algérie, et de ce fait représentent un enjeu de santé publique longtemps sous-estimé. Peu de données sont disponibles en général concernant les critères épidémiologiques de ces dermatoses. Le psoriasis, une maladie chronique avec un impact important sur la qualité de la vie est une des principales dermatoses recensées par ordre de fréquence après les eczémas et l’acné, avec un taux qui est variable entre 3 et 5 % selon les publications. Elle est caractérisée par sa fréquence élevée, son impact important sur la qualité de vie, la fréquence accrue de comorbidités associées, le risque carcinologique rapporté avec les traitements proposés, le coût de la prise en charge de la maladie qui ne cesse d’augmenter avec la commercialisation de divers produits issus de la nouvelle technologie, et l’augmentation de la mortalité du fait des facteurs sus cités. Cette dermatose est fréquente en consultation dermatologique au service de dermatologie du CHU Tidjani Damerdji de Tlemcen, (représente 5,8%, registre de consultations). Le psoriasis touche essentiellement la qualité de la vie dans la même mesure que d’autres maladies chroniques surtout affichantes. Le tabagisme et l’alcool peuvent contribuer en tant que facteurs déclenchant du psoriasis. L’existence d’une association entre le psoriasis et les troubles addictifs est suspectée depuis plusieurs années. Des informations précises sur le coût de la maladie sont nécessaires pour pouvoir prendre des décisions et l’élaboration d’une politique de santé basée sur des données réelles.
Les maladies dermatologiques représentent un motif fréquent de consultation en Algérie et de ce fait, un enjeu de santé publique longtemps sous-estimé. Peu de données sont disponibles en général concernant les critères épidémiologiques de ces dermatoses.
Le psoriasis, une maladie chronique avec un impact important sur la qualité de vie, est une des principales dermatoses recensées par ordre de fréquence après les eczémas et l’acné avec un taux qui varie entre 3 et 5 % selon les publications. Elle est caractérisée par sa fréquence élevée, son impact important sur la qualité de vie, la fréquence accrue des comorbidités associées, le risque carcinologique rapporté avec certains traitements proposés, et le coût de la prise en charge de la maladie qui ne cesse d’augmenter avec la commercialisation de divers produits issus de la nouvelle technologie notamment les traitements biologiques [1].
Fréquence :
Cette dermatose est fréquente en consultation dermatologique au service de Dermatologie du CHU Tidjani Damerdji de Tlemcen, où elle représente 5,8 % des consultations (registres des consultations). Elle atteint les deux sexes avec une prédominance masculine et l’atteinte infantile est retrouvée dans 22,6 % ; les formes compliquées représentent 5,7 % de l’ensemble des psoriasis [2].
Ce profil épidémiologique se rapproche des études du centre de l’Algérie (3 à 6 %) [3]. Par contre en Tunisie, le psoriasis représente 3 % des consultations [4]. Au Maghreb ; dans une étude récente multicentrique PSOMAG (réalisée dans les 03 pays du Maghreb) ; dont l’objectif principal était de déterminer la prévalence du psoriasis en consultation quotidienne, qui avait inclut plus de 221 médecins généralistes et 60 dermatologues dans différentes régions : 12,08/1.000 IC 95 % (10,96/1.000 ; 13,19/1.000) [5].
Dans notre pays, le peu d’études réalisées sur les petits échantillons de sujets atteints de psoriasis ne reflète pas la prévalence réelle de cette maladie, d’ou l’intérêt d’accorder encore plus d’importance aux études épidémiologiques.
En Algérie, il est difficile de donner des chiffres exacts sur l’incidence du psoriasis pour plusieurs raisons : ab- sence d’une base de données nécessaire pour effectuer ce genre d’étude, les hôpitaux et les services hospitalo- universitaires ne sont pas dotés de moyens logistiques humains et matériels pour réaliser ce genre de travail et un manque de collaboration avec les médecins.
En se référant aux registres de consultation quotidienne des services hospitaliers et de quelques dermatologues libéraux, les chiffres ont révélé une fréquence variant entre 2 à 3 % de la population générale [1].
Impact sur la qualité de vie :
La qualité de vie est définie par le World Heath Organisation (WHO) comme un état de bien être complet mental, physique et social.
Le psoriasis touche essentiellement la qualité de vie au même titre que d’autres maladies chroniques, surtout affichantes. Le vécu de la maladie est en effet très différent d’un malade à un autre. Les échelles de la qualité de vie proposées évaluent l’impact de la maladie sur l’état psychologique du patient, sur ses activités quotidiennes et sur ses relations sociales.
Selon la gravité de la maladie et le degré de la surface cutanée atteinte, les psoriasiques peuvent souffrir d’un certain inconfort physique allant jusqu’au handicap fonctionnel dans les formes palmo-plantaires ou encore en cas d’atteinte articulaire associée.
Les personnes touchées peuvent être gênées par cette affection affichante et avoir une mauvaise estime de soi induite par la crainte d’être socialement rejeté ou par des difficultés d’ordre psychologique et sexuel.
On observe souvent chez elles, une détresse psychologique due en particulier à la stigmatisation dont elles font l’objet et qui peut être à l’origine d’une discrimination dans l’emploi et d’un isolement social.
De nombreuses études ont fait état d’un taux de dépression et d’anxiété plus élevé chez les personnes atteintes de psoriasis que dans la population générale.
Dans une publication récente en 2015, la comorbidité psychiatrique chez les psoriasiques est estimée approximativement à 30 %. Les troubles observés étaient essentiellement la dépression, l’anxiété et les idées suicidaires. La prévalence de la dépression est estimée à un taux alarmant allant jusqu’à 62% [6].
Un cas de suicide a été rapporté chez un patient recevant une biothérapie ; l’auteur explique que le sujet était déjà en état de dépression avant la mise en route du traitement et conclut de ce fait, que la cause était le désespoir du malade arrivant à la dernière ligne des thérapeutiques de cette maladie sans efficacité notable, et on soulève ainsi le problème de l’accompagnement psychologique primordial [7].
Le tabagisme et la consommation d’alcool :
Le tabagisme et l’alcool peuvent contribuer en tant que facteurs déclenchants du psoriasis [8]. L’existence d’une association entre le psoriasis et les troubles addictifs est suspectée depuis plusieurs années. Ce lien a fait l’objet de plusieurs évaluations épidémiologiques transversales, dont les résultats sont parfois controversés, plusieurs études ont conclu que le tabagisme est un facteur de risque pour le développement du psoriasis [8].
Le tabac chez nos malades psoriasiques était un facteur associé qui peut influencer l’aggravation de cette maladie et surtout la survenue de comorbidités d’une façon générale. Notre étude ne peut pas être comparée avec les grands échantillons qui se sont intéressé au sujet du fait d’abord de la taille relativement réduite de notre échantillon. Ainsi, vu le contexte socioculturel de notre population où le tabagisme est un tabou, tout particulièrement pour le sexe féminin, ce qui constitue un point faible pour notre étude car elle n’a pas permis d’obtenir des résultats concluants et fiable [1].
Ceci serait encore plus compliqué pour l’alcool ; car pour certains, si ce n’est pour la plupart, soit ils ne donnent aucune information exacte, soit ils nient simplement, pour les mêmes raisons que précédemment [1].
Une analyse systématique de la littérature faite en 2012 sur la consommation d’alcool et le psoriasis a retenu 5 études dont 4/5 trouvent l’alcool comme facteur de risque du psoriasis : trois études cas-témoins dont les OR varient de 2,5 à 3,4 et une étude cohorte : le risque relatif est de 1,7 avec IC 95 % (1,2-2,7), tandis que la cinquième étude ne trouve pas de lien [9].
Le risque carcinologique et le psoriasis :
Le psoriasis est une affection chronique nécessitant des traitements locaux et systémiques qui peuvent engendrer des effets secondaires en particulier carcinologique surtout chez les sujets de la race blanche aux antécédents d’exposition solaire prolongée ou de traitement physique par les UVA. Une étude faite par Margolis et collaborateurs, qui a comparé le taux du cancer chez les psoriasiques par rapport à un groupe de témoins ; a conclu que les sujets atteints de psoriasis ont plus de risque de développer les carcinomes en particulier [10].
Un risque élevé de développer des carcinomes épidermoïdes sous ciclosporine chez les sujets ayant reçu
plus de 200 séances de puvathérapie ; (RR de 7.3 à 9). L’étude PUVA follow-up study a montré un risque de carcinomes épidermoïdes 03 fois plus élevé sous ciclosporine.
Dans une étude observationnelle sur 1.252 patients psoriasiques, la fréquence des cancers était 6 fois plus élevée sous ciclosporine. Pour le méthotrexate, il n’y a pas de risque de développement de cancer en se référant à l’étude PUVA follow-up study, les résultats sont contro- versés pour cette molécule même à forte dose [10].
La survenue des cancers cutanés non mélanique sous puvathérapie est la plus admise surtout chez les psoriasis de race blanche ayant reçu plus de 200 séances de Puvathérapie. L’incidence des carcinomes épidermoïdes est 14 fois plus élevée chez les psoriasis avec une exposition légère (moins de 100 séances). Tandis que pour les carcinomes baso-cellulaires et les mélanomes malins, le risque est moindre. En 2001 un nouveau rapport de l’étude follow-up study a montré que le risque du mélanome malin est lié à la dose reçue et la durée d’exposition.
Pour les traitements biologiques dans le psoriasis, il est admis qu’il y a un risque légèrement accru de développer des cancers non mélaniques [10]. Dans une étude récente, le risque potentiel de malignité a été évalué au cours des traitements systémiques du psoriasis. L’exposition a long terme des essais pivots pour l’infliximab, l’adalimumab et l’etanercept ont généralement montré un taux faible et stable comparable aux taux de base des données [11,12].
Les comorbidités associées :
La découverte des comorbidités liées au psoriasis est ancienne, puisqu’en 1978, Mc Donald et coll. constataient que le risque de maladie occlusive vasculaire était augmenté chez les patients atteints de psoriasis [13]. Depuis, les travaux sur ce sujet se sont multiplié. En 1995, dans une première étude cas-témoins, Henseler comparait
40.000 patients psoriasiques et témoins appariés selon le sexe et l’âge et montrait un risque relatif augmenté d’obésité, d’hypertension artérielle, d’insuffisance cardiaque et de diabète sucré dans le groupe psoriasis [14].
Le syndrome métabolique (SM) est estimé à un taux alarmant de 24 % des adultes aux États-Unis, avec une prévalence plus élevée chez les sujets 60 à 69 ans (44 %) [15].
En Europe, sa prévalence était respectivement de 41% et 26 % ; pour les hommes et les femmes de 55 ans et plus [16]. Dans notre série la prévalence des psoriasiques qui ont
présenté un SM était de 34,8 % des cas, légèrement supérieur par rapport aux témoins qui était de 22,6 % des cas, et un OR de 1,76 IC à 95% (1,33-2,31) ; p très significatif < 0,000 [1].
Ainsi en Tunisie, dans une étude cas-témoins sur 164 patients psoriasiques et 216 témoins, la prévalence du syndrome métabolique était plus élevée chez les cas, que chez les témoins ; mais sans différence statistiquement significative ; légèrement supérieure par rapport à nos résultats (35,5 % vs 30,8 %, odds ratio (OR) : 1,39 IC: 0,88 à 2,18 ; p=0,095) [17].
En Égypte les résultats publiés étaient très superposables aux nôtres. Ainsi, une étude de cas réalisée en milieu hospitalier sur 210 patients adultes atteints de psoriasis en plaques et 389 patients atteints d’autres dermatoses non-inflammatoires, l’OR était de 1,94 IC de (1,24-3,02) [18].
Une étude japonaise a également montré que la préva- lence du syndrome métabolique était augmentée chez les patients psoriasiques [OR = 1,72 (IC 95 % 0,98 à 3,01] [19]. En Allemagne, les patients hospitalisés pour un psoriasis comparés aux patients atteints de mélanome hospitalisés ; ont présenté une prévalence du syndrome métabolique très augmentée (OR 5,92, IC 95%: (2,78 à 12,8)) après ajustement pour l’âge et le sexe [20].
Comme ces risques cardiovasculaires sont des complications connues du syndrome métabolique, sa présence accrue dans le psoriasis peut contribuer à l’augmentation du risque de ces complications observées chez les personnes atteintes de psoriasis [21].
L’impact économique de la prise en charge du psoriasis :
Des informations précises sur le coût de la maladie sont nécessaires pour pouvoir prendre des décisions et l’élaboration d’une politique de santé basée sur des données réelles. Peu de données sont disponibles dans la littérature quant à l’impact économique des médicaments
utilisés pour le traitement du psoriasis.
Depuis la première mise sur le marché des traitements biologiques, la stratégie thérapeutique du psoriasis a totalement changé. L’introduction de ces produits a fourni des traitements supplémentaires pour les formes modérées à sévères du psoriasis, mais avec une facture plus élevée retentissant sur le budget global de la santé.
Dans une étude Allemande faite entre 2003 et 2004 sur un total de 180 malades suivis sur une période de 12 mois, les coûts moyens étaient variables entre 6.709 euros et 8.831 Euros pour les psoriasis modérés à sévères en fonction du PASI [22].
Le coût moyen de la prise en charge d’un psoriasis varie en fonction de la gravité de l’affection et des traitements reçus en particulier les thérapeutiques biologiques ; il englobe deux volets :
Les coûts directs qui comportent les consultations spécialisées, la consommation des médicaments: dans les conditions normales de la mise en route d’un traitement biologique à titre d’exemple en suivant les schémas proposés par les experts dans le domaine et les prix tels qu’ils sont mentionnés dans le Vidal 2012 [23]; le coût annuel du traitement se situe entre 11.280 euros (1.588.860 DA) et 70.444 euros (9.566.295 DA) en fonction de la molécule utilisée ( tableau 1), les hospitalisations du jour ou avec un séjour à l’hôpital, les bilans pratiqués et les traitements anti-infectieux en particulier antituberculeux.
Les coûts indirects en particulier l’absentéisme suite aux hospitalisations répétées avec une perte de productivité.
Tableau 1 : Évaluation du coût des traitements biologiques.
Conclusion :
Le psoriasis est une dermatose chronique sous-estimée, de par sa prévalence, sa fréquence ainsi que ses associations et ses complications. Les études épidémiologiques sont nécessaires pour planifier la stratégie de la prise en charge de cette affection chronique, considérée comme affichante voire invalidante. Les médicaments utilisés actuellement visent à améliorer la qualité de vie au dépend de certains effets secondaires parfois graves avec une facture plus élevée, tout cela doit être pris en compte. Si la santé n’a pas de prix, elle a par contre un coût.
Photos : Pr B. DAHMANI
Références :
Dahmani B. Thèse de doctorat en sciences médicale. Mai 2013 ; faculté de médecine. Université Aboubakr Belkaid Tlemcen ; Étude de la prévalence des facteurs de risque du syndrome métabolique au cours du psoriasis à Tlemcen.
Boudghène Stambouli. O. Thèse de doctorat en sciences médicale. 1999; faculté de médecine. Université Aboubakr Belkaid Tlemcen ; profil épidémiologique des affections dermatologiques dans la wilaya de Tlemcen 1981-1995 ; page 121.
Ammar khodja, A Bachar, Ysmail Dahloug M, Benkaidali Ismail, et coll. Aspects du psoriasis ; étude rétrospective portant sur 410 cas. IIIème congrès maghrébin de dermatologie. Alger 29-30 mai 1996. livre des resumés, page 38.
Zartal, Fazaa B, Kamoun M R. Profil épidémiologique du psoriasis en Tunisie : IIIème congrès maghrébin de dermatologie, .Alger 29-30 mai 1996.livre des résumés, page 2.
Ammar Khodja. A, Benkaidali .I, Denguezli M,N. Doss, Serradj A, Bouadjar, A. Titi, Sekkat .A; Eude Psomag: prévalence des cas de psoriasis au Maghreb ; Ann. Dermatol. Venereol. B162-JDP 2012.
Chamoun A, Goudetsidis L, Poot F, Bourdeaud’hui F, Titeca G. Psoriasis and depression. Rev Med Brux. 2015 Jan-Feb; 36(1):23-8
Ellard R1, Ahmed A, Shah R, Bewley A. Suicide and depression in a patient with psoriasis receiving adalimumab: the role of the dermatologist. Clin Exp Dermatol. 2014 Jul; 39(5):624-7.
Poikolainen K, Karvonen J, Pukkala E. Excess mortality related to alcohol and smoking among hospital-treated patients with psoriasis. Arch. Dermatol. 135(12), 1490-1493 (1999).
Bernault et al. L’alcool serait un facteur de risque mais pas un facteur de sévérité du psoriasis: analyse systématique de la littérature .COO32 JDP 2012 Ann. Dermatol. Venereol. ;139(hors série) Décembre 2012.
Margolis D, Bilker W, Hennessy S, Vittorio C, Santanna J. The risk of malignancy associated with psoriasis. Arch Dermatol 2001 ; 137: 778-783
Fiorentino D, Ho V, et coll. Risque de malignité avec les traitements systémiques du psoriasis dans un registre d’évaluation longitudinale. J Acad Dermatol .2017.845-854.
Dreyers L et al. Incidences des cancers globaux et spécifiques chez les patients traités par un inhibiteur du TNF alpha atteints de polyarthrite rhumatoïde. Une étude de suivi du registre DANBIO. Ann Rheum Dis 2013 ;72 :79-82.
Kremers HM, Mc Evoy MT, Dann FJ, Gabriel SE, Heart diseases in psoriasis; J Am Acad Dermato 2007;57:347-57.
Azfar RS, Gelfand JM. Psoriasis and metabolic disease : epidemiology and pathophysiology. Opin Rheumatol 2008 ;20 (4) 416- 42218525354
Bebis S .Psoriasis sévère, au-delà de la peau, quel est le risque à ne pas traiter? Ann. Dermatol. Venereol. (2008) 135, supplément 5, S285-S289.
Mebazaa et al. Psoriasis and metabolic syndrome in tunisian psoriatic patients : prevalence and determinants; J Eur Acad Dermatol Venereol. 25(6):705-9 ;2011.
El-Shahat Farag Ahmed, Mohammed Khaled Seliem. Prevalence of metabolic syndrome in Egyptian patients with psoriasis ; Egypt. J. Derm & Androl. Vol. 29. No (2) December 2009
Takahashi, H ; Takahashi, Honma M et al. La prévalence de troubles métaboliques syndromes chez les patients atteints de psoriasis japonais. Sci J Dermatol 2010 ; 57: 143-144.
Sommer DM, Jenisch S, Suchan M, et al. Increased prevalence of the metabolic syndrome in patients with moderate to severe psoriasis. Arch Dermato ! Res. 2006 ;298 :321-328.
Thorvardur Jon Love, MD, MMSc; Abrar A. Qureshi, MD, MPH; Elizabeth Wood Karlson, Prevalence of the Metabolic Syndrome in Psoriasis Results From the National Health and Nutrition Examination Survey, 2003-2006 FREE Arch Dermatol. 011;147(4):419-424.
Cost of Moderate to Severe Plaque Psoriasis in Germany:A (Multicenter Cost-of-Illness StudyS. Sohn a O. Schoeffskia J. Prin- zb K. Reichc E. Schubertd K. WaldorfeM. Augustin Dermatology 2006;212:137–144)
Le dictionnaire Vidal – 2012. EAN13 : 9782850912023.
N. NEBTI, Service d’Endocrinologie et de Diabétologie, CHU Isaad Hassani, Béni Messous, Alger , Alger
Abstract : Primary hyperparathyroidism (HPT) has long been considered a rare disease, responsible for pain, pathological fractures, deformities and recurrent kidney stones that may be complicated by end-stage renal failure. Primary hyperparathyroidism is now a common disease representing the second endocrinology consultation motif after thyroidopathia: its prevalence reaches 1 in 1,000 (1). The condition predominates largely among women population after the age of menopause, which is explained by the revealing effect of eostrogen deficiency on osteoblastic activity. In this population in particular, the majority of the forms observed are asymptomatic, accidentally discovered by the calcemia measurement, and are not very evolutionary. Therapeutic management is mainly surgical in the symptomatic forms and in the young subjects. On the other hand, the whole discussion is about asymptomatic forms where several international consensuses have succeeded one another to help the therapeutic decision.
Résumé : L’hyperparathyroïdie primaire (HPT) a longtemps été considérée comme une maladie rare, responsable de douleurs, de fractures pathologiques, de déformations et de calculs rénaux récidivants pouvant se compliquer d’insuffisance rénale terminale. L’hyperparathyroïdie primaire est actuellement une maladie fréquente représentant le 2ème motif de consultation en endocrinologie après les thyroïdopathies : sa prévalence atteint 1 sujet sur 1.000 (1). Avec une nette prédominance féminine après l’âge de la ménopause, expliqué par l’effet révélateur de la carence œstrogénique sur l’activité ostéoblastique. La majorité des formes observées est asymptomatique, fortuitement découverte par la mesure de la calcémie, et s’avère peu évolutive. La prise en charge thérapeutique est principalement chirurgicale dans les formes symptomatiques et chez les sujets jeunes. Toute la discussion est en revanche entamée dans les formes asymptomatiques où plusieurs consensus internationaux se sont succédés pour aider à la décision thérapeutique.
L’hyperparathyroïdie primaire (HPTP), asymptomatique dans la majorité des cas, est caractérisée par une hypercalcémie causée par une hypersécrétion autonome de parathormone, le plus souvent par un adénome parathyroïdien (85 %). Après avoir exclu une cause d’élévation secondaire de la parathormone, une recherche de complications rénales et osseuses fait partie du bilan d’HPT1. La présentation classique associant une ostéite fibrokystique, une chondrocalcinose, une néphrocalcinose, des coliques néphrétiques, et les signes cliniques digestifs et neuropsychiques de l’hypercalcémie. Il faut différencier l’hyperparathyroïdie primaire (HPTP) qui est une hypersécrétion de parathormone (PTH) non freinable par l’hypercalcémie, de l’hyperparathyroïdie (HPT) secondaire (réactionnelle à une hypocalcémie, p. ex. : dans l’insuffisance rénale, et le déficit en vit D) ; et tertiaire (c’est une HPT secondaire qui s’autonomise malgré la correction de l’hypocalcémie Très rarement, dans environ 2 % des cas, l’HPT1 s’intègre dans le cadre de l’une des trois formes d’HPTP familiales.
Les néoplasies endocriniennes multiples NEM.
HPT familiale avec tumeur osseuse de la mâchoire.
HPT familiale isolée : celle-ci est souvent en rapport avec une hyperplasie thyroïdienne multifocale.
Épidémiologie :
L’HPTP s’observe à tout âge de la petite enfance jusqu’aux âges les plus avancés.
Cependant l’incidence s’accroit avec l’âge. L’âge moyen au moment du diagnostic est de 58 ans. L’incidence est de 27 nouveaux cas/ 100.000 habitants dans une étude New Yorkaise (2).
En Europe, la prévalence est de 1 sujet/1.000 (1/500 pour les femmes de plus de 45 ans).
Aucune évaluation dans notre pays n’a été faite jusqu’à maintenant.
La prédominance féminine est nette : de 2 à 3/1, avec un sex-ratio proche de 1 chez les moins de 40 ans, qui s’élève à 5 femmes pour 1 homme au-delà de 75 ans.
Physiopathologie et étiopathogénie: L’hormone parathyroïdienne PTH est une hormone hypercalcémiante et ce :
En augmentant le remodelage osseux, principalement l’ostéolyse.
En favorisant la réabsorption tubulaire rénale du calcium.
En majorant l’absorption intestinale du calcium : cet effet est indirect en activant la 1α hydroxylase rénale qui produit la 1,25 (OH)2 cholécalciférol .
La PTH est également hypophosphorémiante (effet phosphodiurétique) et réduit l’élimination rénale des ions H+, d’où le profil biologique de l’HPTP : hypercalcémie, hypoposphorémie, hyperphosphaturie, tendance à l’acidose hyperchlorémique, et à l’alcalinisation des urines. L’HPTP est due :
À un adénome parathyroïdien unique : dans 80 à 85% des cas, et dans 10 % des cas, les adénomes sont situés dans des parathyroïdes ectopiques : intra thyroïdien, l’espace rétro-œsophagien, médiastin,
Dans moins de 5 % des cas les adénomes sont multiples.
Hyperplasie des glandes parathyroïdes : 15-20 %, développées à partir des cellules principales.
Cancer des parathyroïdes : 1 % des cas.
L’HPTP est familiale dans 5 à 10 % des cas, en rapport avec des mutations génétiques bien identifiées : gène de la Ménine (pour les néoplasies endocriniennes multiples (NEM1), gène RET (NEM2), gènes HRPT1 et 2 (HPTP
familiale isolée) et le gène codant pour le récepteur sensible au calcium (syndrome de Marx et Aurbach).
La recherche des ces formes génétiques est justifiée en pratique clinique devant les situations suivantes : HPTP chez un sujet jeune de moins de 40 ans, hyperplasie des 4 glandes parathyroïdiennes, atteinte pluriglandulaire, adénome kystique, carcinome parathyroïdien, HPT persistante ou récidivante après chirurgie.
NEM type 1 ou syndrome de Wermer : associant : hyper-parathyroïdie primaire (retrouvée dans presque 100% des cas, sous forme d’hyperplasie des 4 glandes) + tumeur entéropancréatique (Insulinome, glucagonome, gastrinome) + Adénome hypophysaire (le plus souvent un prolactinome) + tumeur surrénalienne + tumeur bronchique.
NEM type 2a ou syndrome de Sipple : associant cancer médullaire de la thyroïde +/-
Phéochromocytome +/- hyperparathyroïdie (souvent sous forme d’adénome).
HPTP familiale isolée : évoquée devant l’absence de caractéristiques suggestives de NEM type 1 et 2. Dans cette situation, on note la fréquence des adénomes kystiques, carcinomes parathyroïdiens, et l’association possible à des tumeurs mandibulaires (Jaw Tumor) ou rénales.
Syndrome de Marx et Aurbach : C’est l’hypercalcémie hypocalciurique familiale. Certains auteurs inscrivent cette entité dans les HPTP bien que la présentation clinique soit différente : mutation du récepteur sensible au calcium dans 90 % des cas. L’intervention chirurgicale n’est pas justifiée en principe dans ces situations.
Présentation Clinique :
Différentes manifestations cliniques sont susceptibles de révéler une HPT1
Atteinte rénale : Parmi les formes symptomatiques, la forme rénale est la plus fréquente, retrouvée dans 60 à 70 % des cas. Les signes rencontrés sont : un syndrome polyuro-polydipsique, des lithiases rénales, récidivantes, bilatérales, spontanément radio opaques à l’ASP (abdomen sans préparation), responsables de crises de coliques néphrétiques, néphrocalcinose et insuffisance rénale représentant le risque évolutif majeur de l’hyperparathyroïdie.
Atteinte osseuse : Douleurs osseuses, fractures spontanées, déformations, tuméfactions. À la radiographie : déminéralisation diffuse, géodes, calcification des tissus mous, érosion sous périostée des os longs surtout, mais également des os plats, lésions lytiques, kystes ou tumeurs brunes.
Manifestations digestives : anorexie, nausées, vomissements, constipation. Plus évocatrice, la survenue d’une pancréatite aiguë ou chronique ou d’ulcères récidivants de l’estomac.
C. Manifestations neuropsychiques et musculaires :
asthénie, céphalées, irritabilité, dépression.
Manifestations cardio-vasculaires : HTA, troubles du rythme, diminution de l’espace QT à l’ECG.
La crise aiguë hypercalcémique : C’est une urgence métabolique. Il existe le plus souvent un facteur déclenchant survenant sur un fond d’hypercalcémie chronique modérée : déshydratation, diurétiques thiazidiques etc. La calcémie est ≥ 150 mg/l.
Sur le plan clinique : état d’agitation, délire, confusion, jusqu’au coma, vomissements, douleurs abdominales pseudo-chirurgicales, déshydratation sévère, fièvre, insuffisance rénale aigüe.
Confirmation diagnostique :
A. Diagnostic biologique :
Le tableau biologique typique d’HPTP : Hypercalcémie, hypophosphorémie, hypercalciurie, hyperphosphaturie, tendance à l’acidose hyperchlorémique, PTH élevée (parfois normale mais inappropriée à une hypercalcémie). A noter qu’à côté de ce tableau biologique typique, existent plusieurs formes, sources de pièges diagnostiques de l’HPTP :
Formes sans hypercalcémie : en cas de pancréatite aigüe, d’insuffisance rénale, carence en vitamine D, prise de diurétiques, HPTP normocalcémique par résistance tissulaire à l’action de la PTH.
Formes sans hypercalciurie : rencontrées en cas de déficit en vitamine D associé, insuffisance rénale, diurétiques thiazidiques, hypercalcémie hypocalciurique familiale.
Formes sans élévation de la PTH : en rapport peut être avec la présence d’Ac anti PTH ou d’anticorps hétérophiles interférant lors des dosages.
B. Diagnostic topographique :
Fréquemment négatif dans les NEM 1 où existe une hyperplasie multiglandulaire.
L’échographie cervicale : Elle fournit une image anatomique précise mais ne peut
visualiser les adénomes ectopiques.
La scintigraphie au Sestamibi marqué au technétium Tc99 m (MIBI) : reste l’examen le plus performant, permet de localiser avec précision les foyers en particuliers les parathyroïdes ectopiques.
Le scanner cervico-médiastinal triphasique : Il permet une localisation précise des adénomes parathyroïdiens. La mesure de densité permet de distinguer ces adénomes, des ganglions lymphatiques. Il est indiqué en 2ème intention quand les examens de 1ère intention (échographie et scintigraphie) ne sont pas contributifs.
Intervention chirurgicale : à la fois exploratrice et curatrice.
Diagnostic différentiel :
Il se pose avec les autres causes d’HPT :
HPT secondaire : réactionnelle à l’hypocalcémie en cas de carence en vit D, d’insuffisance rénale, ou de résistance à l’action de la PTH (forme génétique de la pseu-dohypoparathyroïdie).
HPT tertiaire : c’est une HPT secondaire qui s’autonomise malgré la correction de l’hypocalcémie nécessitant à ce stade une prise en charge spécifique de la maladie parathyroïdienne.
En cas d’hypercalcémie, le diagnostic différentiel se pose avec :
L’hypercalcémie d’origine néoplasique : métastases osseuses ostéolytiques, localisations osseuses des hémopathies (myélome), hypercalcémie humorale maligne : production d’un facteur hypercalcémiant (PTHrp, interleukine, prostaglandine, exceptionnellement PTH)
L’exérèse totale en cas d’adénome unique est indiquée et ablation de trois parathyroïdes et d’une partie de la quatrième en cas d’hyperplasie des 4 glandes.
En cas de cancer : exérèse large avec curage ganglionnaire. Les suites opératoires sont marquées par les risques suivants :
Hypoparathyroïdie post opératoire : Avec les signes d’hypocalcémie : crise de tétanie, jugulée par un traitement associant Vit D et calcium. Cette hypoparathyroïdie peut être transitoire ou définitive.
Récidive, ou geste chirurgical incomplet.
Risques de la chirurgie thyroïdienne : infection, hématome, atteinte du nerf récurent.
b. Traitement médical:
La crise hypercalcémique : Le traitement médical est urgent précédant la chirurgie, et conduit dans une unité de soins intensifs : réhydratation, Furosémide, Calcitonine : efficacité transitoire. Biphosphonates : Ils sont principalement indiqués dans les hypercalcémies malignes.
Dans les cas les plus extrêmes on peut recourir à l’hémodialyse.Dans l’hypercalcémie modérée :
Régime désodé ou les diurétiques de l’anse ou épargneurs de K+ sont à éviter car ils aggravent l’hypercalcémie en augmentant la réabsorption rénale du Ca++,
Assurer un apport normal en Ca++ (1g en moyenne) car un apport restreint aggrave encore l’hypersécrétion de PTH,
Substituer un déficit en vitamine D associé qui majore les risques de L’HPTP : 100.000, une ampoule buvable à répéter mensuellement tous les 2 mois avec surveillance de la calcémie et de la calciurie.
Les agents calcimimétiques (Cinacalcet) ou agonistes du récepteur sensible au calcium entraînent une diminution de la sécrétion de la PTH
B. Indications :
Le traitement chirurgical est impératif, précédé ou non par une préparation médicale selon l’intensité de l’hypercalcémie, dans tous les cas d’HPTP survenant chez les patients jeunes de moins de 50 ans et dans les formes symptomatiques.
Les indications opératoires dans les formes asymptomatiques ainsi que le protocole de surveillance en cas de non opérabilité ont fait l’objet de recommandations de sociétés savantes Française (SFE) et Américaine (NIH). Les nouvelles recommandations SFE 2014 sont résumées sur le tableau 2.
Un seul critère suffit à indiquer la prise en charge chirurgicale.
La chirurgie peut aussi être proposée chez les patients ne souhaitant pas ou ne pouvant pas être surveillés régulièrement, s’il n’existe pas de contre-indication à une chirurgie.
Concernant la mesure de la DMO, l’utilisation du T-score est recommandée, mais chez un homme jeune (< 50 ans) ou une femme avant la ménopause, l’utilisation du Z-score est préférable.
Le tableau 3 résume les modalités de surveillance pour les patients ayant une HPTP asymptomatique non opérés. Il est aussi rappelé que les techniques d’imagerie (échographie, scintigraphie au 99mTc-sestaMIBI, scanner,IRM) sont des techniques de localisation préopératoire et n’ont donc pas d’indication chez les patients chez lesquels la chirurgie n’est pas envisagée.
(IRM : imagerie par résonnance magnétique, VFA : vertebral fracture assessement, DFGe : débit de filtration glomérulaire estimé, ASP : radiographie de l’abdomen sans préparation, TDM : tomodensitométrie) Tableau 2 : Indication de prise en charge chirurgicale devant une hyperparathyroïdie primaire asymptomatique : recommandations en 2014, 2009 et de la SFE EN 2005.(SFE : Société française d’endocrinologie, DFG : débit de filtration glomérulaire, VFA : vertebral fracture assessment, TDM : tomodensitométrie) Tableau 3 : Surveillance des patients ayant une HPTP asymptomatique non opérée.
Conclusion
En définitive, le diagnostic et la prise en charge de l’hyperparathyroïdie primaire suivent un protocole bien précis, afin de limiter les explorations abusives et de réserver la chirurgie aux patients qui doivent en bénéficier.
Points à retenir
L’HPT primaire est affirmée biologiquement en présence de valeurs élevées de la calcémie, inappropriées à des valeurs élevées ou simplement normales de PTH.
L’os et le rein constituent les organes cible de l’HPT primaire. La réduction de l’apport calcique, les médications hypocalcémiantes, les états de carence en vitamine D majorent les risques des HPT primaires.
L’enquête morphologique (échographie, scintigraphie) ne doit s’envisager que pour les patients de moins de 50 ans, ou symptomatiques, relevant de l’intervention
chirurgicale. Elle a toutes chances d’être négative en cas d’HPT primaire liée aux NEM 1, qui sont responsables d’hyperplasies multiglandulaires.
Références :
Houillier P. Hyperparathyroïdie primitive. Enc Méd Chir Endocrinologie-Nutrition,10-012-B-10, 2002.
Calzada-Nocaudie M. Prise en charge de l’hyperparathyroïdie pri- maire asymptomatique, consensus d’experts de la SFE. Ann Endocri- nol (Paris). 2006 ;677-12.