F. MADACI,Service de Gynécologie-Obstétrique, Clinique Ibrahim Gharafa CHU Lamine Debaghine, Bab El Oued, Alger
Abstract : Genitourinary syndrome of menopause (GSM), previously known as atrophic vaginitis or vulvo-vaginal atrophy, affects more than half of postmenopausal women. Caused by low estrogen levels after menopause, it results in bothersome symptoms, including vaginal and urinary symptoms and signs. the GSM may have a profound negative impact of the quality of life of post menopausal women. Patients must be encouraged to talk about their difficulties and treated with an appropriate effective therapy. Several effective treatments exist, but low-dose vaginal estrogen therapy is the standard criterion. It is effective and safe for most patients.
Key-words : Genito-urinary syndrome of menopause, Vulvo-vaginal atrophy, menopause
Résumé : Le syndrome génito-urinaire de la ménopause (SGUM OU SGM) connu sous le vocable de l’atrophie vulvo-vaginale (AVV) moins spécifique, regroupe un ensemble de manifestations cliniques génitales et urinaires en rapport par le déclin des stéroïdes sexuelles en particulier les œstrogènes. Il affecte plus de la moitié des femmes ménopausées, survenant dès les premières années la ménopause. Il n’a aucune tendance à la régression nécessitant ainsi un traitement au long cours afin d’améliorer la qualité de vie des femmes, qui est profondément altérée sur le plan sexuel et émotionnel. Plusieurs traitements sont proposés mais le traitement ostrogénique local reste le traitement de référence car il est efficace et ne présente aucune conte indication.
Le syndrome génito-urinaire est la nouvelle terminologie de l’atrophie vulvo-vaginale (AVV) proposée et adoptée par NAMS (North American Menopause Society) et ISSWSH (International Society Study Women’s Sexual Health) lors du congres de Chicago tenu en 2014 (1,2).
Ce récent concept regroupe un ensemble de signes et de symptômes en rapport avec la diminution du taux d’œstrogènes et d’autres stéroïdes sexuels.
Le GSM dépasse le concept de la carence ostrogénique pour inclure les symptômes en rapport avec le vieillissement qui sont représentés par les symptômes vulvo-vaginaux (sécheresse, brûlures, irritation), les symptômes sexuels (dyspareunie) et les symptômes urinaires (urgenturie, infection urinaire à répétition, prolapsus…). Les symptômes liés à ce syndrome sont en augmentation depuis le déclin du traitement hormonal de la ménopause en rapport avec les conclusions de la WHI (Women’s Health Initiative) sur les risques cardiovasculaires et carcinologiques mis en exergue dans cette étude randomisée.
Il affecte plus de la moitié des femmes ménopausées. Survenant le plus souvent au moment de la ménopause ou dans l’année suivante, il est constant dans 60 % des cas (3,4). Si les troubles vasomoteurs ont tendance à diminuer spontanément, le SGUM ne se résout pas spontanément. Ce dernier nécessite une thérapeutique au long cours en raison des effets sévères sur l’équilibre émotionnel sexuel et la qualité de vie qui se trouve profondément altérée.
Les manifestations cliniques
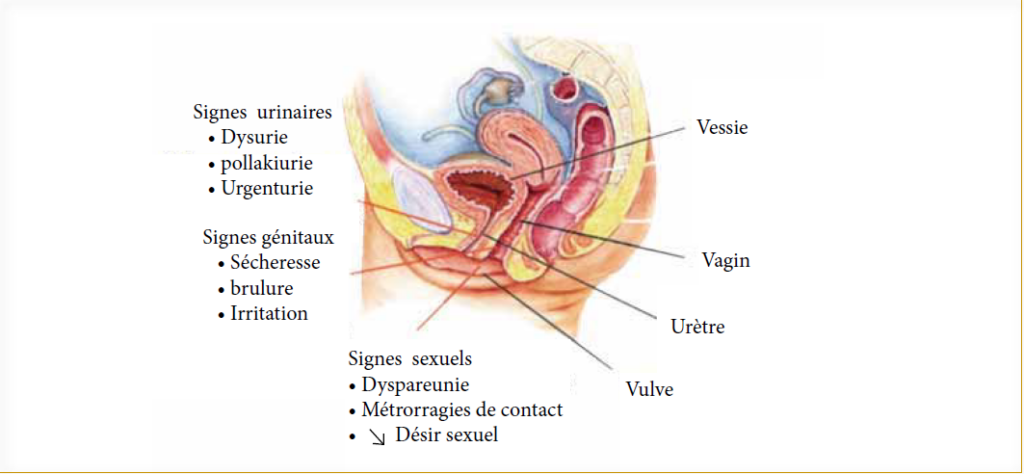
Les cliniciens jouent un rôle majeur dans la reconnaissance des signes du SGUM, la plupart des femmes n’en parlent pas par pudeur car c’est un sujet sensible (sexualité). Les manifestations cliniques se traduisent par des signes urinaires et génitaux (figure 1) qui peuvent être mis en évidence par l’interrogatoire et l’examen clinique.
Figure 1 : Sturdee DW, Panay N. Recommandations pour la prise en charge de l’atrophie vaginale post ménopausique. Climacterics. 2010 ; Early Online : 1-28
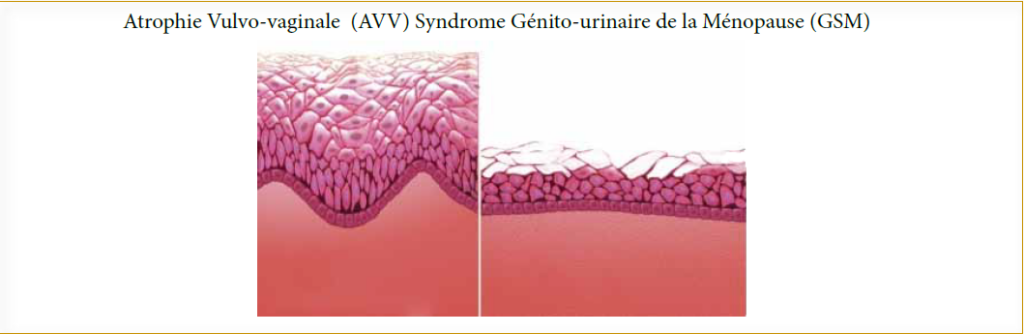
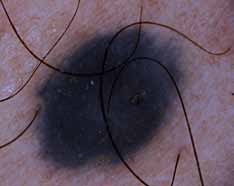
L’atrophie vaginale est observée 2 à 3 ans seulement après la ménopause. Les altérations atrophiques sont responsables du rétrécissement du hiatus génital ne permettant pas l’introduction de 2 doigts et une diminution de la profondeur du vagin favorisent l’apparition de blessures et d’infections, la vulve est pale et atrophiée (figure 2).
La vessie et l’urètre peuvent aussi être concernés, avec des symptômes tels que dysurie, l’incontinence d’effort (ou de stress) et les infections urinaires récidivantes.
Suite à l’atrophie vaginale, l’activité sexuelle est souvent perturbée.
La prévalence de la difficulté sexuelle augmente avec l’âge elle est de 22 % entre 40 et 44 ans, elle avoisine les 60 % après 60 ans. Les trois principaux symptômes sont la sécheresse vaginale (70 %), l’irritation vaginale (33 %) et la dyspareunie (29 %). Les patientes ayant une ménopause chirurgicale sont plus exposées (8,9).
Physiopathologie
Les œstrogènes ont un rôle majeur dans l’hydratation et la prolifération de l’épithélium vaginal. Ils ont un effet trophique direct sur le tissu conjonctif et les fibres élastiques. La baisse des œstrogènes est à l’origine de l’atrophie vaginale ; le vagin perd son élasticité et devient étroit et peu profond (5).
En raison d’une origine embryologique commune (sinus urogénital), cette atrophie va s’étendre à la vessie et à l’urètre ce qui explique la survenue de la symptomatologie urinaire.
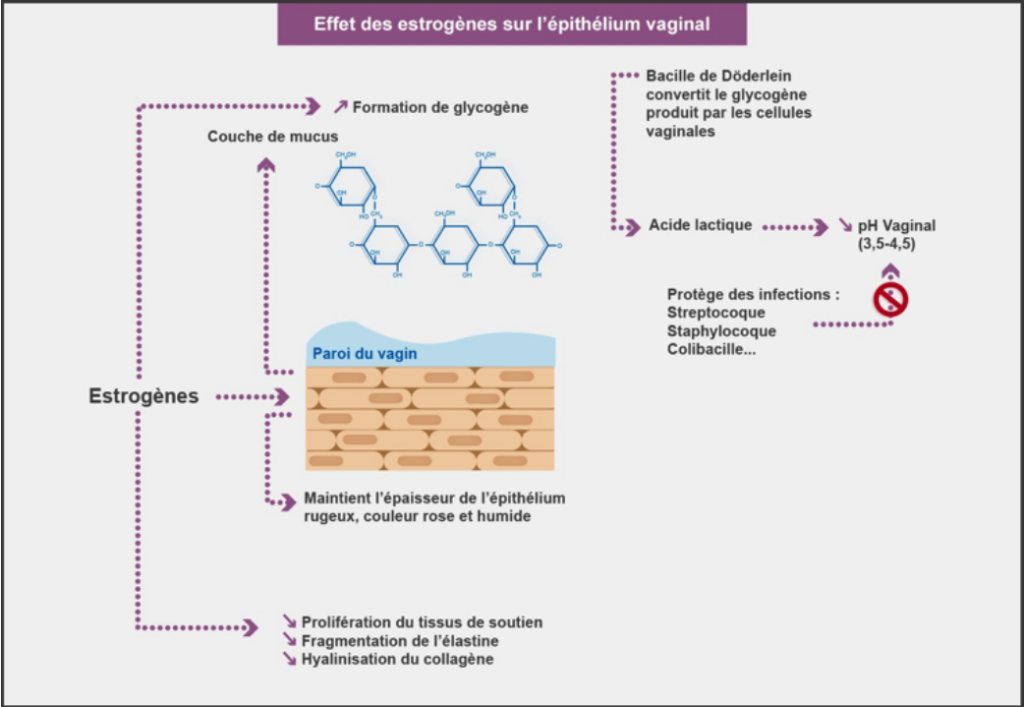
En pré ménopause la flore vaginale est dominée par le lactobacille (bacille de Doderline) qui fabrique de l’acide lactique à partir du glycogène libéré par les cellules épithéliales du vagin ce qui permet de maintenir une acidité vaginale (PH entre 3 et 5), qui empêche la prolifération d’autres colonies microbiennes évitant ainsi la survenue d’infections.
Avec la baisse des œstrogènes, le glycogène se raréfie avec le lactobacille, il s’ensuit une alcalinisation du PH vaginal (≥ 7), ce qui favorise la colonisation de germes pathogènes. Cette alcalinisation du PH est aussi responsable de mauvaises odeurs (figure 3).
Figure 3 : Sturdee DW, Panay N. « Recommandations pour la prise en charge de l’atrophie vaginale post-ménopausique. Climacterics. 2010 ; Early Online : 1-2 »
Les modifications subies par l’urètre sont (5) :
Diminution de l’épaisseur du muscle strié, ce qui est à l’origine du raccourcissement de l’urètre
Hypertrophie du muscle lisse responsable de la rigidité de ce dernier
Disparition des anastomoses artério-veineuses avec une diminution de la pression de clôture
Atrophie de la muqueuse.
La vessie
Le détrusor est peu à peu envahi par le collagène. L’activité sympathique diminue et l’activité parasympathique augmente ; tous ces phénomènes expliquent l’incontinence urinaire par hyperactivité vésicale ainsi que la survenue fréquente des infections urinaires.
Signes et symptômes urogénitaux liés aux déficits en estrogènes (extrait de Sturdee et al.)
• Vagin
Sécheresse et hydratation insuffisante
Diminution du flux sanguin
Dyspareunie
Prurit
Sensation de brûlures
Douleurs
Perte d’élasticité
Amincissement des tissus vaginaux et altération de la kératinisation
Anomalies muqueuses incluant des pétéchies, des microfissures, ulcération et inflammation
Raccourcissement, fibrose, fermeture de la cavité vaginale et/ou amincissement de l’entrée du vagin
Perte du relief du fornix et des plis vaginaux
Susceptibilité aux blessures mécanique
Difficultés à cicatriser
Index de la maturation vaginale anormal : diminution du pourcentage des cellules superficielles et des cellules parabasales
Diminution du contenu en glycogène des cellules de l’épithélium vaginal
Modification de la flore vaginale au profit des micro-organismes pathogènes
Augmentation du pH vaginal au-dessus de 5
Leucorrhées et/ou pertes louches
Infiltration des couches sous-muqueuses par des lymphocytes et des cellules plasmatiques
•Vessie et urètre
Augmentation du résidu vésical post-mictionnel
Diminution de la capacité de la vessie
Diminution de la pression maximale du détrusor pendant la miction
Diminution du seuil de sensibilité à l’extension de la vessie (seuil du sentiment de la nécessité d’uriner)
Diminution de la pression de fermeture urétrale
Diminution de la perfusion du plexus veineux périurétral
Diminution du flux d’urine au niveau de l’urètre
Diminution de l’index de maturation urétrale : diminution du pourcentage des cellules superficielles et augmentation des cellules parabasales
Incontinence urinaire
Infections urinaires récidivantes
Anomalies de la biosynthèse du collagène dans le tissu de soutien périurétral
Symptômes de dysurie, de nycturie et de mictions impérieuses
Traitement
L’objectif du traitement est de rétablir l’équilibre physiologique dans la zone génito-urinaire et de lutter ainsi contre les symptômes liés à l’atrophie avec un impact positif non négligeable sur la sexualité et la qualité de vie.
Le traitement doit être précoce avant que les complications de l’atrophie soient irréversibles. Il doit être poursuivi dans le temps pour maintenir les effets bénéfiques de ce dernier.
a. Traitement hormonaux
Œstrogènes locaux : ce sont les plus efficaces, le passage systémique est très faible, inférieur à 20pg/l, ils ne présentent aucune contre-indication. Une œstrogénothérapie locale est préférable pour traiter l’atrophie vaginale quand un traitement systémique n’est pas nécessaire pour d’autres raisons (9). Les œstrogènes locaux n’augmentent ni le risque de cancer du sein ni celui du risque cardiovasculaire (19). La protection de l’endomètre par un progestatif n’est pas indiquée. Plusieurs formes galéniques sont disponibles ovules, crèmes (promestriene, colpotrophine ) ; anneau vaginal (estring) non disponible en Algérie. Il n’existe aucune différence d’efficacité des différents produits selon une étude de la Cochrane 2016. La prescription est quotidienne pendant 2 à 3 semaines puis bi hebdomadaire.
Autre thérapeutiques hormonales
Tibolone (voie orale – Livial®) (16). Hormone synthétique qui a une activité analogue aux trois hormones sexuelles dans les différents tissus. Effets oestrogéniques favorables sur le vagin et la vessie, un effet progestatif anti prolifératif sur l’endomètre. La tibolone a une activité analogue à la testostérone qui peut jouer un rôle sur la libido. Mais ses effets secondaires cardiaques restreignent son emploi. Elle est contre indiquée en cas d’antécédent de cancer du sein.
Déhydroepiandrostérone (DHEA) (14). La transformation androgénique/ostrogénique varie selon les cellules elle n’a pas d’effet systémique, la DHEA améliore l’index de maturation vaginale, normalise le PH et stimule la
formation de collagène. Elle est classiquement prescrite sous forme d’ovules ; cependant en absence de cette forme galénique, une voie orale peut être prescrite à faible dose.
Ospemifene (SERM-Osphena°) (13). Pris par voie orale (60 mg/j), autorisé par la FDA aux USA depuis 2013 ; proposé comme traitement de la VVA modérée à sévère (en particulier dyspareunie) chez la patiente refusant une thérapeutique locale. Le médicament est contre indiqué en cas d’antécédent thromboembolique (ou d’antécédent de cancer hormono dépendant).
a. Traitements non hormonaux
Particulièrement indiqués chez les patientes présentant un cancer de l’endomètre ou du sein sous anti aromatases qui favorisent la survenue de SGUM ; chez lesquelles le traitement œstrogénique systémique est contre indiqué (17, 18,19).
Les lubrifiants sont utilisés principalement pour remédier à la sécheresse vaginale lors des rapports sexuels. Leur action ne repose sur aucun mécanisme physiologique particulier ; ils apportent un soulagement temporaire mais ne constituent pas une solution à long terme en cas de sécheresse vaginale.
Les produits hydratants. Ils sont constitués de polymères hydrophiles non solubles. Ces polymères bioadhésifs adhèrent aux cellules épithéliales de la paroi vaginale et lient des molécules d’eau. Les effets bénéfiques des hydratants sur les symptômes de l’atrophie vaginale sont surtout liés à leurs propriétés tampon qui entrainent une réduction du pH vaginal alcalin. Ils existent en gel à base d’acide lactique ou en ovule à base d’acide hyaluronique (Cicatrine®) .
Le laser CO2 fractionné (Monalisa°) ou le laser YAG (Intima). Nouveau concept de rejuvénation. Intéressant chez les patientes ayant une contre-indication à l’hormonothérapie (17). Le laser CO2 fractionné (3 séances toutes les 4 semaines), améliore la vascularisation, la production de glycogène, de collagène et de matrice extracellulaire. Il a une action clinique persistant jusqu’à 12 semaines après traitement, 85 % des patientes retrouvent une vie sexuelle normale à 12 semaines (18).
Activité sexuelle : Augmenter l’activité sexuelle des patientes avec ou sans partenaire (20), augmente l’élasticité, la lubrification du vagin et améliore la vascularisation génito-urinaire.
Conclusion
Environ la moitié des femmes ménopausées sont concernées par le SGUM qui altère leur qualité de vie en présentant des symptômes d’intensité très variable parmi lesquels, la sécheresse des muqueuses, le prurit et les douleurs lors des rapports sexuels, la baisse de la libido
et l’augmentation des infections vaginales et des cystites. En présence de symptômes légers, on peut recommander des produits hydratants. Si les symptômes sont d’intensité moyenne à sévère, il convient de recourir à un traitement œstrogénique local, car les symptômes ont plutôt tendance à augmenter au fil du temps. Plusieurs études ont confirmé l’efficacité d’un tel traitement contre le SGUM. Des données récentes confirment que le traitement vaginal topique par œstrogènes n’accroît pas le risque de cancer du sein ni le risque cardiovasculaire, ce qui est en faveur de sa sécurité. Il n’y a pas de durée prédéfinie du traitement. SGUM récidive cependant souvent à l’arrêt du traitement local.
Références :
Portman DJ et al, Maturitas, 2014, 79, 349-354
Gandhi J et al, Am J Obstet Gynecol, 2016, 1-8
Nappi RE, Palacios S, Panay N et al, Climateric, 2016, 19, 188-197
Rahn DD et al. Obstet Gynecol 2014 ;124:1147-1156Utian WH.
Biosynthesis and physiologic effects of estrogen and pathophysiologic effects 4 of estrogen deficiency: a review. American journal of obstetrics and gyneco- logy. 5 1989 ;161(6 Pt 2):1828-1831
Robinson D, Cardozo LD. The role of estrogens in female lower urinary tract dysfunction. Urology. 2003 ;62(4 Suppl 1):45-51
Nappi RE, Lachowsky M, Maturitas, 2009, 63, 138-141
Nappi RE, Palacios S, Panay N et al, Climateric, 2016, 19, 188-197
Palma F & al. Vaginal atrophy of women in postmenopause. Results from a multicentric observational study The AGATA study. Maturitas. 2015: 1-5
Sanchez-Borrego R et al, Maturitas, 2014, 78, 146-150 ; Blake J, Best Practice & Res Clin Obstet Gynaecol, 2006, 20, 799-839;
Gandhi J et al, Am J Obstet Gynecol, 2016, 1-8; Al-Baghdadi O et al, Clima- teric, 2009, 12, 91-105;
Edwards D et al, Climateric, 2016, 19, 151-161
Management of symptomatic vulvo-vaginal atrophy : 2013 position sta- tement of The 11 North American Menopause Society. Menopause. 2013 ;20(9):888-902; quiz 903-884
Labrie F, Archer DF, Koltun W, et al. Efficacy of intra vaginal dehydroepian- drosterone 24 (DHEA) on moderate to severe dyspareunia and vaginal dryness, symptoms of 25 vulvo-vaginal atrophy, and of the genito-urinary syndrome of menopause. Menopause. 26 2016 ;23(3):243-256
Mendoza N, Abad P, Baro F, et al. Spanish Menopause Society position sta- tement : use of 20 tibolone in postmenopausal women. Menopause (New York, NY). 2013.
Salvatore S, Nappi RE, Zerbinati N, et al. A 12-week treatment with frac- tional CO2 laser 6 for vulvo-vaginal atrophy : a pilot study. Climacteric. 2014 ;17(4) :363-369.
Salvatore S, Nappi RE, Parma M, et al. Sexual function after fractional micro- ablative 8 CO (2) laser in women with vulvo-vaginal atrophy. Climacteric. 2015 ;18(2) :219-225.
Crandall CJ et al. Breast cancer, endometrial cancer, and cardiovascular events in participants who used vaginal estrogen in the Women’s Health Initia- tive Observational Study. Menopause 2018 ; 25(1) 1
Gandhi J. Am J Obstet Gynecol 2016. – 2. Management of symptomatic vul- vo-vaginal atrophy: 2013 position statement of The NAMS. Menopause 2013.
S. BOUZID, M. DERGUINI, Hôpital Bachir Mentouri , Kouba, Alger,
Abstract : Menopausal transition or the menopause itself is not a disease, it is a natural process, however, it is accompanied by a deterioration of physical, psychological and social well-being in a variable way according to the individual, it is the climacteric syndrome. (With its multi-dimensionality and subjectivity) which has a repercussion on the quality of life of women. Management is based mainly on MHT, which is the most effective treatment for the climacteric system, and on the prevention of osteoporosis and its consequences, but which may include certain oncological and metabolic risks reported by the studies, as well as the benefit/risk balance is the most favorable for MHT initiated before age 60 or in the first 10 years after menopause, with a preference for percutaneous estrogen and natural progesterone if necessary, the treatment must be individualized for each patient. Hygiene of life remains the first prescription that is unavoidable. In women with contra-indications to MHT there are alternative treatments for vasomotor flush, vulvo-vaginal dryness and prevention of osteoporosis treatment.
Key-words : Menopause, hormonal treatment of menopause, climatere, quality of life.
Résumé : La transition ménopausique ou la ménopause elle-même ne constitue pas une maladie, c’est un processus naturel néanmoins il est accompagné d’une détérioration du bien-être physique, psychologique et social de façon variable selon l’individu, c’est le syndrome climatérique (avec sa multidimensionnalité et sa subjectivité) qui induit un retentissement sur la qualité de vie de la femme. La prise en charge repose essentiellement sur le traitement hormonal de la ménopause (THM), qui est le traitement le plus efficace sur le climatère, et sur la prévention de l’ostéoporose et ses conséquences, mais qui peut comporter certains risques carcinologiques et métaboliques relatés par les études. Ainsi la balance bénéfice/risque est la plus favorable pour le THM initié avant 60 ans ou dans les 10 premières années suivant la ménopause, en préférant un œstrogène percutané et progestérone naturelle si nécessaire, le traitement doit être individualisé pour chaque patiente. L’hygiène de vie reste la première prescription qui est incontournable. Chez les femmes ayant des contre-indications au THM, il existe des traitements alternatifs pour les BVM1, la sècheresse vulvo-vaginale ainsi que la prévention de le traitement de l’ostéoporose.
Mots-clés : Ménopause, traitement hormonal de la ménopause, climatère, qualité de vie.
Généralités – Introduction
La transition de la ménopause implique souvent des symptômes gênants, notamment des symptômes vasomoteurs, une sécheresse vaginale, une diminution de la libido, de l’insomnie, de la fatigue, des douleurs articulaires et les troubles cognitifs qui représentent les conséquences à court et moyen terme ; ensuite lorsque la ménopause est installée il faudra prévenir et prendre en charge les complications cardiovasculaires, thromboemboliques et les fractures ostéoporotiques.
Lorsque les femmes consultent pendant cette période pré-ménopausique, nous saisissons l’occasion pour aborder la santé de l’os, le sevrage tabagique, l’évaluation et la gestion des risques cardiovasculaires et le dépistage et la prévention du cancer.
La prise en charge est globale tant sur l’hygiène de vie, la diététique, l’activité physique, l’éducation pour la gestion des bouffées de chaleur, etc.
L’hormonothérapie ménopausique (MHT) est le traitement le plus efficace pour les symptômes vasomoteurs et autres symptômes du climatère. Les avantages peuvent dépasser les risques pour la majorité des femmes ménopausées symptomatiques qui ont moins de 60 ans ou moins de 10 ans de ménopause.
Il faudrait individualiser la thérapie en fonction des facteurs cliniques et des préférences de la patiente. Et dépister les femmes avant d’initier un THM pour le risque de maladie cardiovasculaire et de cancer du sein et recommander la thérapie la plus appropriée en fonction des risques/avantages. Les données actuelles ne justifient pas l’utilisation de THM pour prévenir les mala- dies coronariennes, le cancer du sein ou la démence.
D’autres options sont disponibles pour les femmes présentant des symptômes vasomoteurs, qui préfèrent ne pas utiliser THM ou qui ont des contre-indications. Les œstrogènes vaginaux à faible dose et l’ospémifène (SERM), constituent une thérapie efficace pour le syndrome génito-urinaire de la ménopause, et les hydratants vaginaux et les lubrifiants sont disponibles pour celles qui ne choisissent pas la thérapie hormonale.
Depuis dix ans, de nombreuses ré-analyses des anciennes études (telle que la WHI2 2002) et études plus récentes semblent modérer les conclusions sans doute trop hâtives sur le THM.
Diagnostic et symptômes de la ménopause :
Le diagnostic de la ménopause est clinique et rétrospectif. La ménopause est une étape biologique dans la vie d’une femme (50-51 ans) marquée par l’arrêt des menstrues pendant 12 mois consécutifs traduisant la fin de sa vie reproductive naturelle. Associant des changements qui surviennent lorsque les ovaires cessent d’ovuler et de sécréter des œstrogènes et de la progestérone.
La femmes ménopausées signifie les femmes en péri ménopause et en post ménopause.
La péri ménopause est le moment où une femme a des cycles irréguliers (ovulation et menstruation) menant à la ménopause et se poursuivant jusqu’à 12 mois après sa dernière période. Elle est également connue comme la transition ménopausique ou climatérique. La post ménopause est l’étape après la ménopause, le début de la ménopause étant la période de 12 mois consécutifs. La ménopause peut être influencée par certains facteurs, pouvant ainsi être iatrogène.
Dans la situation où la patiente a subi une hystérectomie avec conservation ovarienne, on peut se baser sur la présence de symptômes vasomoteurs (VMS) et sur les tests de laboratoires avec mesure répétées de FSH et E2 < 20 pg/ml ou encore utiliser le test aux progestatifs à raison de 10 j par mois pendant 3 mois consécutifs pour confirmer la ménopause [1].
Après la ménopause, si l’absence de progestérone n’induit aucun trouble avéré, la carence en œstrogènes a de nombreuses conséquences, désagréables ou pathologiques, associées à d’autres troubles, non hormono-dépendants.
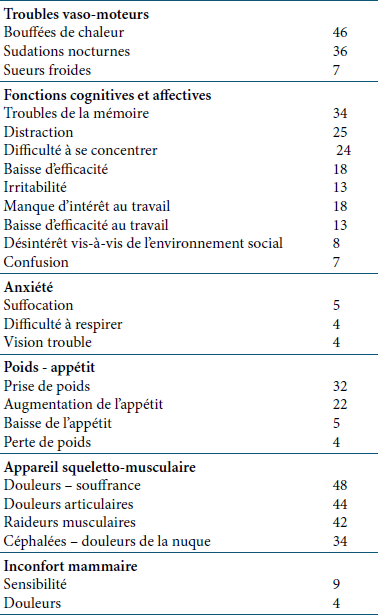
Symptômes (en %)
Étude PEPI (Postmenopausal Estrogen/Progestin Intervention, Greendale et al. 1998) menée en double aveugle pendant 3 années chez 875 femmes âgées de 45 à 64 ans aux USA
Prise en charge de la ménopause :
Impact de la symptomatologie climatérique sur la qualité de vie :
L’espérance de vie ne cesse d’augmenter et la qualité de vie devient un important paramètre de santé dans une popu lation vieillissante. « La qualité de vie comme mesure clinique est une construction compliquée et controversée nécessitant, pour pouvoir être évaluée, d’utiliser des échelles. Certaines sont spécifiques à la transition ménopausique, d’autres ont une vision plus large de la santé, incluant des questions sur la fonction physique, psychologique, sociale, et sont des échelles globales de qualité de vie.
On admet que la qualité de vie est proportionnelle au degré de satisfaction des besoins et à la réalisation des objectifs dans la vie de l’individu.
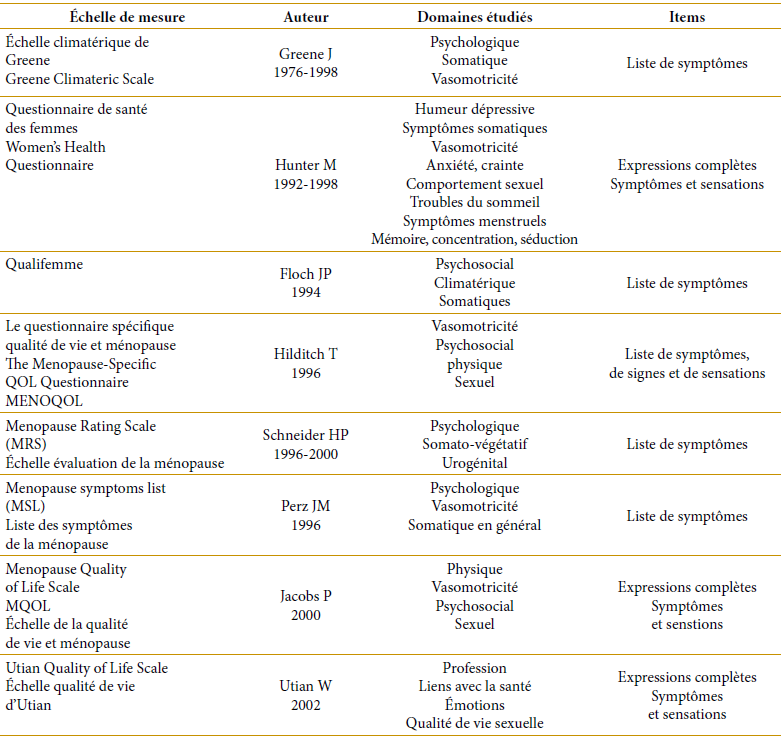
La qualité de vie constitue un paramètre essentiellement subjectif ne pouvant être évalué qu’à l’aide de questionnaires. Certains sont spécifiques pour la transition de ménopause. Ils mesurent l’intensité ou bien la gravité jugée par l’individu des symptômes (physique, mental et partiellement social ou syndrome climatérique). Les plus utilisés sont le Kuppermann, le Menopause Rating Scale, le Green Climacteric Scale» [22].
Échelle de mesure de la qualité de vie couramment utilisées spécifiquement pour la ménopause [4]
Echelle de mesure de la qualité de vie couramment utilisées spécifiquement pour la ménopause [4]
La sévérité des symptômes climatériques et le degré avec lequel ils interféreront dans leur activité et leur qualité de vie est particulièrement variable d’une femme à l’autre. Environ 20 % de ces femmes rapporteront des symptomatologies sévères ; 10 à 15 % seulement consulteront la recherche d’une prise en charge thérapeutique [5].
« En cela la plupart des études évaluant la qualité de vie associée à la symptomatologie climatérique ont mis en évidence l’intérêt du traitement hormonal substitutif ; mais malheureusement nous subissons encore dans cette ère post-WHI un bouleversement de la prise en charge de la symptomatologie climatérique en rapport avec les risques cardiovasculaires et carcinologiques mis en exergue dans cette étude randomisée.
Depuis, les prescriptions du traitement hormonal, même en début de ménopause, ont considérablement chuté (baisse de plus de 60 %) et les femmes se tournent désormais vers des stratégies différentes : thérapeutiques alternatives, phytothérapie, modifications d’hygiène de vie, etc. » [22].
La prise en charge de la ménopause est d’ordre global. Elle nécessite un plateau pluridisciplinaire, comportant le diététicien, le rhumatologue, le cardiologue et le gynécologue créant ainsi plusieurs volets thérapeutiques : les mesures hygiéno-diététiques, le traitement hormonal, les traitements non-hormonaux, les médecines alternatives telles que la phytothérapie, l’homéopathie, l’acupuncture, sans oublier la prise en charge des symptômes qui nécessitent des consultations et des prescriptions.
1. Hygiène de vie :
MHD générales : arrêt du tabac et pratiquer régulièrement une activité physique, adopter une alimentation équilibrée, perdre du poids avec une diététique alimentaire.
Bouffées de chaleur : éducation pour la gestion des bouffées de chaleur, habits adaptés, baisser le chauffage, éviter les boissons chaudes, serviette froide sur la nuque, apport phosphocalcique.
Sécheresse vaginale : utilisation d’un lubrifiant, de crème hydratante, ou œstrogéno-thérapie locale.Trouble du sommeil : se coucher à des heures stables, éviter la caféine.
Dépression : rester active, conserver son entourage social et si nécessaire traitement approprié (anxioly- tiques).
2. Traitement hormonal de la ménopause :
La symptomatologie climatérique peut être transitoire (un ou deux ans) mais peut durer bien plus longtemps chez certaines femmes [7]. Il est bien évident qu’une symptomatologie sévère est facilement repérée (sueurs nocturnes altérant le sommeil, fatigue altérant les facultés cognitives, les performances physiques et donc l’estime de soi et enfin la sexualité…) peut affecter profondément la vie personnelle, sociale et la qualité de vie de certaines femmes [8].
Malgré des décennies de recherche sur la ménopause et sur le THM, il est surprenant de voir que peu d’études apprécient les variations de la qualité de vie en rapport avec cette symptomatologie climatérique durant la transition ménopausique [9].
Le traitement hormonal de la ménopause (THM) joue un rôle important dans le maintien et l’amélioration de la qualité de vie (QDV) des femmes ménopausées.
« Si le THM a prouvé son efficacité sur la symptomatologie climatérique, notamment en début de ménopause, en dehors de cette symptomatologie climatérique il n’est pas possible aujourd’hui de conclure de l’impact du THM sur la qualité de vie globale. La balance bénéfices/risques du THM débutée en début de ménopause, notamment en cas de symptomatologie climatérique, apparaît tout à fait favorable et recommandée par la plupart des sociétés savantes de ménopause » [22].
Composition du THM :
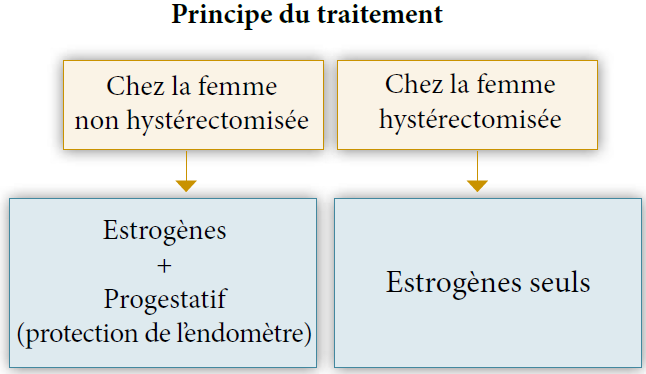
Le principe de ce traitement est d’associer un traitement par estrogène et un progestatif sans induire un effet prolifératif sur l’endomètre. En cas d’hystérectomie il ne sera donc pas utile d’associer le progestatif.
Plusieurs voies d’administration peuvent être utilisées pour le THM :
Le 17 beta estradiol est l’estrogène naturel de référence qui peut être utilisé par voie orale, vaginale ou percutanée, provoquant des impacts différents sur le risque métabolique, il existe un effet de premier passage hépatique, lors de l’utilisation de la voie orale induisant un effet métabolique délétère tel que la diminution de l’antithrombine III et l’augmentation de triglycérides et de l’angiotensinogène.
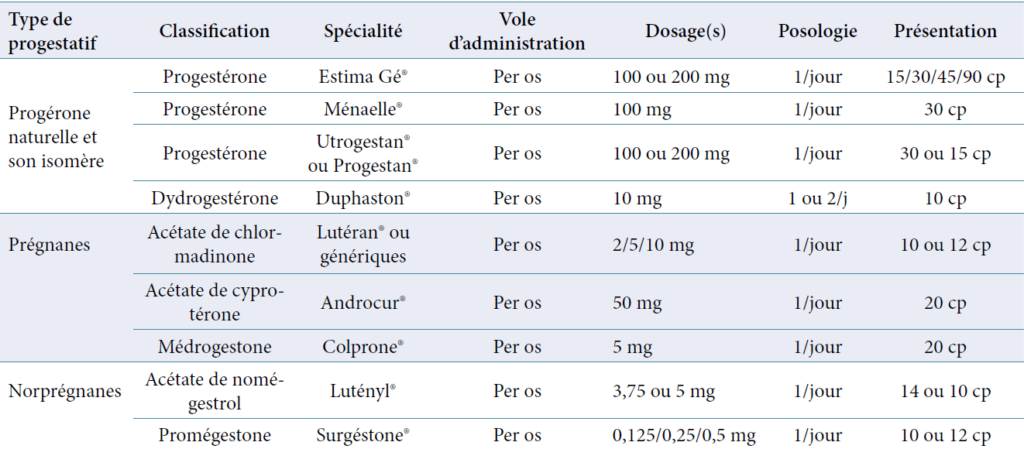
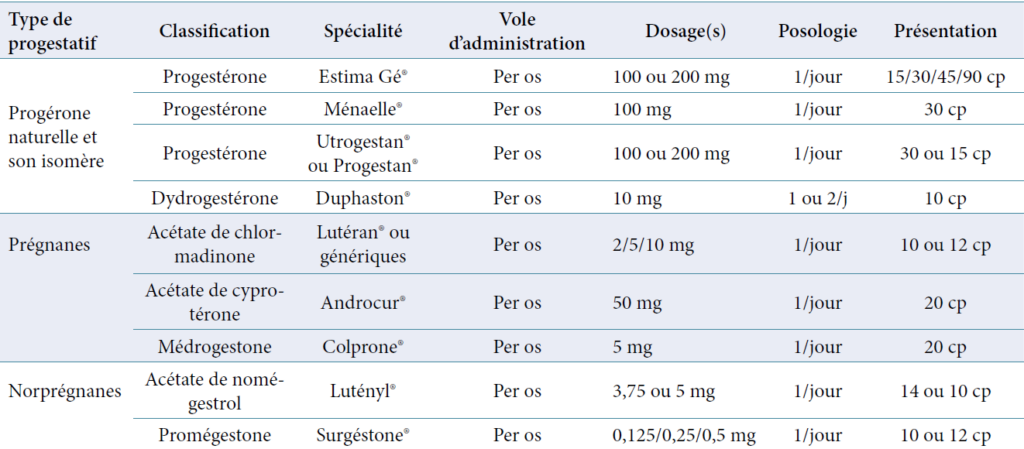
Les progestatifs utilisables sont repartis en plusieurs groupes :
Progestérone naturelle et son isomère la dydrogesterone
Les dérivés de la 17-OH progestérone (pregnanes)
Les dérivés de la 19-norprogesterone (norpregnanes)
Les dérives de la spironolactone.
Schémas :
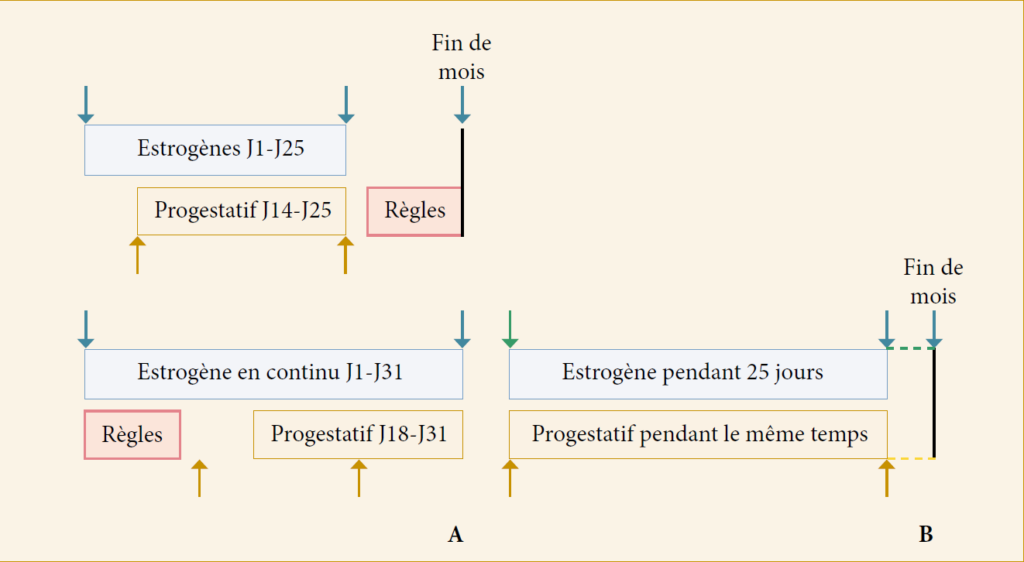
Schéma séquentiel
Permet de restaurer des hémorragies de privation lors de l’arrêt du progestatif.
Le schéma séquentiel discontinu
consiste à donner un œstrogène du 1j au 25j du cycle auquel on associe un progestatif pendant au moins 12j, du 14ème au 25ème j/mois.
Liste des progestatifs disponibles en France 2012 (Vidal 2012)Tableau 4 : Propriétés des progestatifs.
Le schéma séquentiel continu
consiste à poursuivre l’estrogène pendant tout le mois c’est à dire du 1j au 31j en association avec le progestatif pendant au moins 14j soit du 18ème au 31ème j.
Schéma combiné
La prise continue d’un œstrogène associé à un progestatif pour la même durée induit une atrophie endométriale supprimant l’hémorragie de privation.
Prise discontinue du j1 au j25 avec arrêt de 5jours ou en prise continue du j1 au j31 sans arrêt. Ce traitement est prescrit à une femme ménopausée depuis au moins 2 ans.
Indication et contre-indication :
L’ANSM [10], stipule qu’aucune durée limite d’utilisation du THM n’a été établie tant que durent les symptômes climatériques en commençant par les doses les plus faibles puis augmenter progressivement jusqu’à l’arrêt des symptômes. En informant clairement les risques inhérents à ce traitement. Certaines situations devraient faire évaluer la balance bénéfice /risque.
Surveillance :
Avant la prescription il convient de demander au préalable :
mammographie dépistage plus une échographie pelvienne à la recherche de pathologies intra-utérines. La surveillance comporte un examen clinique annuel (palpation mammaire plus examen gynécologique). Surveiller les signes d’hypo ou hyper œstrogène, échomammographie/2ans échographie endo-vaginale en cas de métrorragie si endomètre > à 6mm exploration par hystéroscopie plus ou moins biopsie.
Pratiquer une DMO pour bilan osseux initial (femmes ayant des facteurs de risque).
Bilan métabolique.
Évaluation du risque cardiovasculaire.
Effet du THM sur QDV :
Toutefois depuis la publication des résultats des études sur la WHI, MWS [11], l’hypothèse des effets néfastes du traitement hormonal a conduit à une perception radicalement différente expliquant la baisse de prescription mais aussi la baisse de la demande des femmes [12].
Trois études d’intervention randomisées en double insu ont pu aborder les relations entre THM et qualité de vie.
L’étude PEPI [13] sur trois ans conclut à une diminution significative des symptômes vasomoteurs d’autant plus prononcés que les symptômes étaient sévères au départ et à une amélioration des symptômes musculo-articulaires.
Par contre aucune différence sur les fonctions cognitives, l’affectivité ou l’anxiété. En résumé les effets du THM sont jugés positifs chez les femmes symptomatiques.
L’étude HERS [14] également randomisée en double insu sur trois ans, conclut à une amélioration de la qualité de vie surtout chez les femmes se plaignant de bouffées de chaleur et notamment sur les critères émotionnels. Par contre les scores de fonctions physiques et de fatigue déclinaient chez les femmes asymptomatiques au départ et sous THM par rapport au placebo, et reste sans effet sur la qualité de vie chez les femmes ne présentant pas de symptomatologie vasomotrice (moyenne de 67 ans). WHI conclut globalement à une absence d’amélioration de la QDV en sachant que dans cette étude la population était symptomatique et âgées entre 50 et 79 ans et que
20 % des cas étaient ménopausées depuis plus de 5 ans. D’autres études d’observation ont démontré les mêmes résultats.
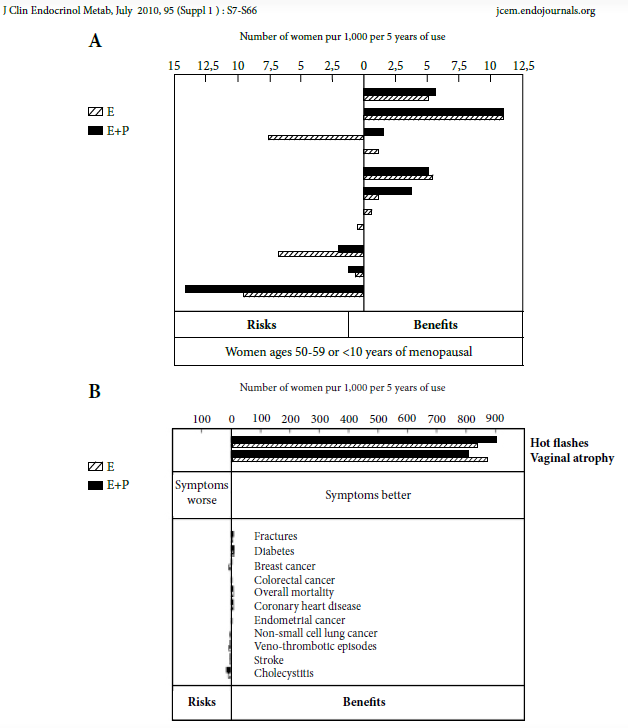
Selon les résultats tout récemment publiés (juillet 2010) de la société scientifique d’endocrinologie et en fonction des grades de recommandation [15]
Pour la QDV :
Le THM améliore en diminuant la symptomatologie (trouble du sommeil et de l‘humeur) (grade B) mais sur la qualité de vie globale il passe à un (grade C).
Bouffées vasomotrices :
La dose standard d’œstrogène (0,5 à 1mg/j d’E2 per os, 50μg/j transcutané et 0,5 mg /j percutané) diminue significativement la fréquence et la sévérité des BVM (grade A). Sans oublier la prévention de l’ostéoporose.
Sexualité :
300µg/j de testostérone augmente le nombre de rapports sexuels (grade A)
Pas d’effet sur troubles de l’humeur (grade C)
Pas d’effet sur le déclin cognitif (grade B)
Le THM débuté après 65 ans n’améliore pas les désordres cognitifs (grade C) :
Troubles génito-urinaires
Les E améliorent l’instabilité vésicale et diminuent les infections récurrentes (grade A)
Atrophie vaginale :
10µg d’EE par voie vaginale 2 fois /semaine ou 7,5 µg/j anneau vaginal restaure la trophicité vaginale grade A
Bénéfices/risques du THM débuté en début de ménopause (fenêtre d’intervention)
THM et os :
L’accélération de la déminéralisation osseuse expose à la fracture pathologique. Cette perte de l’os touche surtout le col du fémur le rachis et le poignet. Le THM a fait ses preuves dans la réduction de cette perte.
THM et cancer du sein :
La modulation du risque de cancer du sein sous THM, selon le temps d’instauration du THM / au début de la ménopause est rapportée par les études de la WHI, E3N et MWS. L’arrêt du sur-risque après l’arrêt du THM prouve l’effet promoteur et pas initiateur du cancer du sein. Les résultats montrent un sur-risque pour une prescription en début de ménopause en comparaison d’une instauration au bout de 3 à 5 ans [16] ; ce qui est en contradiction avec le risque cardiovasculaire.
La NAMS dans sa dernière recommandation estime que la balance bénéfices / risques concernant les moments d’instauration d’un THM penche en faveur de la femme jeune en début de ménopause.
THM et risque cardiovasculaire :
L’IMS dans ses dernières recommandation insiste sur les mesures primaires de prévention des accidents cardiovasculaires que sont le contrôle du poids, de la pression artérielle, la pratique d’un exercice physique, l’arrêt du tabac et le contrôle d’une dyslipidémie et d’un diabète [17].
THM et risque veineux thromboembolique : l’utilisation du THM est largement en faveur de l’association d’un estrogène percutané faiblement dosé et de la progestérone naturelle (Étude MEVE) [18].
THMetendomètre:il est donc recommandé d’associer un progestatif aux œstrogènes au moins 12j/mois chez les femmes non hystérectomisées.
THM, cancer de l’ovaire et cancer du côlon : Il est impossible de conclure de façon tranchée à un effet néfaste pour le cancer de l’ovaire, les études concluent à une diminution du risque colorectal EN3 [19].
1. Alternatives au THM :
• Traitement hormonal par voie orale :
La Tibolone, Livia® l 2.5mg/j appartient à la famille des 19-norsteroides et possède, selon les tissus cibles, des propriétaires ostrogéniques, progestatives et légèrement androgéniques ; c’est un stéroïde d’action sélective (sas), il est efficace sur les BVM de façon identique au THM, sur la trophicité vulvovaginale et stimule la libido, elle augmente légèrement la densité osseuse mais n’a pas d’action préventive sur les fractures ostéoporotiques, pas d’effet ostrogénique sur l’endomètre donc traitement sans hémorragie de privation, a les mêmes contre-indications que le THM. Certains auteurs ont testé la progestérone qui aurait une action sur les BVM [20].
• Traitements hormonaux locaux :
Plusieurs œstrogènes faibles à tropisme local agissant spécifiquement sur les muqueuses vulvovaginales.
Traitementssymptomatiquesdelaménopause: Différents traitements non hormonaux peuvent être prescrits afin d’améliorer les symptômes d’hypoestrogénie avec des résultats variables, ils sont intéressants chez les patientes présentant des contre-indications au THM. Peuvent réduire les bouffées vasomotrices jusqu’à 40% :
Certains antidépresseurs de la classe des inhibiteurs de la recapture de la sérotonine (IRS) tels que la paroxetine, fluoxetine, ou lexitopram et les inhibiteurs de la recapture de la sérotonine et la noradrénaline (IRSNA) comme venlafaxine sont efficaces sur les BVM [21] mais n’ont pas l’AMM dans cette indication.
Anti hypertenseur de la famille des agonistes des récepteurs alpha2 adrénergique, (la clonidine) peuvent être efficace sur les BVM.
Traitement anticonvulsivant a été utilisé.
Beta-alanine, acide aminé ou abufène qui possède l’AMM dans cette indication.
Phyto-œstrogène tels les flavones, les lignanes et les coumestans, ont une efficacité variable sur les BVM.
3. Traitement et prévention de l’ostéoporose ménopausique :
Les SERMs (modulateurs sélectifs des récepteurs aux œstrogènes) possèdent à la fois une action agoniste et antagoniste des estrogènes variable selon les tissus, raloxifene (Evista® 60mg) à une action préventive démontrée sur l’ostéoporose vertébrale uniquement (étude MORE).
Les biphosphonates ont une action anti-ostéoclastique et constitue, de ce fait, de puissant inhibiteurs de la ré- sorption osseuse. Ils ont prouvé leur action sur la réduction du risque fracturaire vertébral et périphérique.
La dernière recommandation de GEMVI sur ces différents traitements de l’ostéoporose post-ménopausique incluant le THM ne montrent pas de supériorité d’un traitement/à un autre, cela dépend de l’âge des antécédents.
Conclusion :
La ménopause est une période de transition marquant la vie d’une femme nécessitant une prise en charge adéquate et une surveillance qui permettent de soulager les désagréments occasionnés par le bouleversement hormonal lié à cette période et de prévenir éventuellement des complications notamment métaboliques et osseuses avec un accompagnement physique et médicamenteux lui permettant de garder une qualité de vie compatible avec l’espérance de cette même vie qui augmente, avec la dignité qu’exige l’époque actuelle.
Références :
Atkins D, Best D, Briss PA, et al. Grading quality of evidence and strength of recommendations BMJ 2004; 328:1490
J Bitzer, M. Lachowsky. Ménopause et qualité de vie. Réalités en gynécologie- obstétrique septembre 2007;123:1.
WHO. Constitution of the WHO Geneva A. Switzerland: Word Health Orga- nization, 1946.
Hilditch JR, Lewis J, Peter A et al. A menopause-specific quality of life ques- tionnaire: development and psychometric properties. Maturitas 1996; 24:161-75.
Guthrie J, Dennerstein L, Taffe J, Donnelly. V. health care-seeking for meno- pausal problems. Climacteric 2003; 6:112-7.
Gelfan MM, Moreau M, Ayotte NJ, Hilditch JR et al. Clinical assessment and quality of life of postmenopausal women treated with a new intermittent pro- gestogen combination hormone on replacement therapy : a placebo-controlled study. Menopause 2003;10: 29-36.
Politi MC, Schleinitz MD, col NF. Revisiting the duration of vasomotor symp- toms of menopause: a meta-analysis. J Gen Intern Med 2008; 23:1507-13.
Anderson E, Hambourger S, Liu JH, Rebar BW. Characteristics of menopau- sal women seeking assistance. Am J Obstet Gynecol 1987;156:428-33.
Avis NE, Colvin A, Bromberger JT et al. Change in health-related quality of life over the menopausal transition in a multiethnic cohort of middle-aged wo- men: study of Women’s Health Across the Nation. Ménopause 2009; 16:860-9.
Afssaps. Mise au point actualisée sur le traitement hormonal de la meno- pause (THM) 2006.
Ressouw JE, Anderson GL, Prentice RL et al. Risks and benefit of oestrogen plus progestin in healthy post-menopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321-33.
Genazzani AR, Schneider HP, Panay N et al. The European Menopause Sur- vey 2005: Women’s perceptions on the menopause and postmenopausal hor- mone therapy. Gynecol Endocrinol 2006; 22: 369-75.
Reeves GK, Beral V, Green J et al. Hormonal therapy for menopause and breast-cancer risk by histological type: a cohort study and meta-analysis. Lan- cet Oncol.2006; 910-8.
Hlatky MA, Boothroyd D, Vittinghoff E, Sharp P, Whooley MA for the Hers Research Group. Quality of life and depressive symptoms in postmenopausal women after receiving hormone replacement therap. JAMA 2002; 287:591-7.
Santen RJ et al. 2010. Postmenopausal Hormone Therapy: An Endocrine Society Scientific Statement. The journal of Clinical Endocrinology & Metabo- lism 95, Supplement 1:S1-S66.
Beral V, Reeves G, Bull D, Green J, and the Million Women Study Colla- borators. Breast cancer risk in relation to the interval between menopause and starting hormone therapy. J National Cancer Inst 2011; 103:296-305.
Strudee DW, Pines A, Archer DF, Barber RJ, Barlow D, Birkhauser MH, et al Updated IMS recommendations on postmenopausal hormone therapy and preventive strategies for Midlife health. Climacteric 2011;14:302-20.
Olié E, Plu- bureau G, Conard J, Horellou MH, Canonico M, Scarabin PY. Hormone therapy and recurrence of venous thromboembolism among post- menopausal women. Menopause 2011; 18:488-93.
Morois S, Fournier A, Clavel-Chapelon F, Mesrene S, Boutron-Ruault MC. Menopausal hormone therapy and risks of colorectal adenomas and cancer in the French E3N prospective cohort: true associations or bias. Eur J Epidemiol 2012; 27:439-52.
Hitchcock CL, Prior JC. Oral micronized progesterone for vasomotor symptoms: a placebo-controlled randomized trial in healthy postmenopausal women. Menopause 2012; 19:886-93.
Nelson HD, Vesco KK, Haney E, Fu R, Nedrow A, Miller J et J, et al. Non hormonal therapies for menopausal hot flushes. Systematic review and meta- analysis. JAMA 2006;295:2057-71.
B. LETOMBE CNGOF 2010. « Le syndrome climatérique et la qualité de vie : balance bénéfices/risques du THM et des traitements symptomatiques », Lille. Extrait des Mises à jour en gynécologie médicale, Volume 2010, publié le 10 Décembre 2010. http://www.cngof.asso.fr/d_livres/2010_GM_513_letombe.pdf
Baffet H, Robin G, Letombe B. Ménopause. EMC – gynécologie 2015.
N. KESRI, Service d’Endocrinologie, Centre Pierre et Marie Curie, Alger.
Abstract : The premature ovarian failure (POF) commonly called early menopause, observed in 1-2% of female population, defines the appearance of amenorrhea before the age of 40 years associated with high levels of FSH checked twice in one month apart. It is a pathological situation within multiple pathogenic mechanisms of autoimmune or genetic origin responsible of depletion or dysfunction follicular causing an early alteration of the endocrine function and fertility. Clinical symptomatology translated early, acute or gradual installation of hormonal deficiency mainly estrogenic with significant psychosomatic component related to the reduction of the natural reproductive capacity. In the absence of early diagnosis, the main complications are bone and cardiovascular. In evolutionary terms, phases of reversibility of unpredictable duration are reported in beginner forms. The etiological investigation remains unsuccessful in nearly two thirds of the cases, and management is based on immediate and and indisputable institution of estrogen and progestogen hormonal substitution which the only formal contraindication is currently the breast cancer. Patterns are personalized, adapted to the diagnosis stage and need to focus on the natural hormones and the transdermal route. In iatrogenic POF fertility prognosis is improved by the use of prior cryopreservation of the ovarian cortex while only infertility curative therapy is currently based on egg donation, which is forbidden by Muslim religion. New cell therapy techniques are currently being tested such as the maturing of primordial follicles or isolation of ovarian stem cells providing to these young women in future may be close opportunities for natural conception from their own eggs.
Résumé : L’insuffisance ovarienne prématurée (IOP) communément appelée ménopause précoce, observée chez 1-2% de la population féminine, définit l’apparition d’une aménorrhée avant l’âge de 40 ans associée à des taux élevés de FSH vérifiés à deux reprises à un mois d’intervalle. Il s’agit d’une situation pathologique relevant de multiples mécanismes pathogéniques d’origine génétique ou auto immune responsables de déplétion ou de dysfonction folliculaire entrainant une altération précoce de la fonction endocrine et de la fertilité. La symptomatologie clinique traduit la carence hormonale précoce, essentiellement estrogénique, d’installation aigue ou progressive avec une importante composante psychosomatique liée à la réduction des capacités de procréation naturelle. En absence d’un diagnostic précoce, les principales complications sont d’ordre osseux et cardio-vasculaire. Au plan évolutif, des phases de réversibilité de durée imprévisible, sont rapportées notamment dans les formes débutantes. La prise en charge comprend une enquête étiologique qui reste infructueuse dans près de deux tiers des cas, et repose sur l’institution immédiate et indiscutable d’une substitution hormonale œstro-progestative dont la seule contre-indication formelle reste actuellement le cancer mammaire. Les schémas sont personnalisés, adaptés au stade diagnostique et doivent privilégier les hormones naturelles et la voie transdermique. Dans les IOP iatrogènes le pronostic de fertilité est amélioré par le recours à la cryoconservation préalable du cortex ovarien tandis que la seule thérapeutique curatrice de l’infertilité des IOP spontanées repose actuellement sur le don d’ovules, interdit par la religion musulmane. De nouvelles techniques de thérapie cellulaire sont actuellement en cours d’expérimentation telles que la maturation de follicules primordiaux résiduels ou l’isolement de cellules souches ovariennes offrant à ces jeunes femmes dans un avenir peut être proche, des possibilités de conception naturelle à partir de leurs propres ovules.
Mots-clés : Ménopause précoce, anomalies de la folliculogénèse, syndrome climatérique précoce, traitement hormonal substitutif, hypofertilité iatrogène.
Introduction :
La ménopause est une étape physiologique et inéluctable de la vie génitale féminine marquée par la cessation définitive des menstruations et de la fonction de reproduction relevant d’un mécanisme unique de déplétion en follicules primordiaux potentiellement fonctionnels (1). Elle est en général diagnostiquée rétrospectivement après 12 mois d’aménorrhée et son âge de survenue est stable autour de 50 ans en moyenne (46-54 ans pour 95 % femmes, et vers 40-45 ans pour moins de 5 % d’entre elles) (1).
Une situation similaire d’aménorrhée avec son cortège climatérique classique peut être observée avant l’âge de 40 ans chez environ 1-2 % des femmes de la population générale, naturellement dénommée « ménopause précoce » (2).
De nombreux auteurs considèrent cette appellation inadéquate car en plus de son âge de survenue, cette entité s’oppose à la ménopause naturelle par plusieurs aspects :
Les mécanismes pathogéniques nombreux, de mieux en mieux cernés grâce aux progrès de génétique moléculaire réalisés au cours de ces vingt dernières années.
Les modalités évolutives fluctuantes et imprévisibles de la fonction endocrine et même exocrine responsables d’une grande variabilité clinique et biologique.
Le caractère indiscutable et prolongé du traitement hormonal de substitution (THS) dans cette situation, auquel ne sauraient s’appliquer les conclusions des études WHI.
Ainsi, les dénominations de « dysfonction ovarienne prématurée » ou « d’insuffisance ovarienne prématurée », sont actuellement privilégiées (2) dont l’avantage principal est d’éviter la connotation psychologique péjorative, inéluctablement associée à l’annonce d’un diagnostic aussi dévastateur dans cette tranche d’âge que celui de « ménopause précoce ».
Quelques données épidémiologiques :
Une estimation précise de la prévalence des IOP manque encore, il faut distinguer les :
IOP spontanées dont la prévalence serait de 1 % avant l’âge de 40 ans, de 1/1.000 avant 30 ans et de 1/10.000 avant 20 ans. Leur prévalence est stable d’après les dernières études épidémiologiques.
IOP iatrogènes dont la prévalence est en nette progression, du fait de l’amélioration de la survie liée à l’efficacité indéniable des traitements carcinologiques mais au prix d’effets gonadotoxiques inévitables.
Certaines études épidémiologiques rapportent une association avec le tabagisme et certains facteurs ethniques vu la prévalence moindre chez les femmes chinoises ou japonaises comparées aux femmes caucasiennes ou africaines. Il n’a pas montré de lien entre la survenue d’une IOP et l’âge des premières règles, le niveau d’éducation ou la prise de contraception orale.
Aspects cliniques :
Diverses circonstances peuvent révéler une IOP, essentiellement des troubles du cycle menstruel, un bilan d’infertilité, un syndrome génétique ou une maladie auto–immune ainsi que les suites de traitements gonadotoxiques ou d’une ovariectomie bilatérale.
Le plus souvent l’IOP spontanée s’installe après une puberté normale suivie de cycles réguliers de durée variable. Quelques fois les cycles peuvent s’arrêter brusquement : absence de retour des cycles après une grossesse ou à l’arrêt d’un contraceptif oral mais le plus souvent l’aménorrhée est précédée par des prodromes sur un terrain de stress tels qu’une oligo-spanioménorrhée, polyménorrhée, ou métrorragies.
La symptomatologie est inconstante, corrélée au degré du déficit oestrogénique, variable du fait de la possibilité d’une réversibilité transitoire dans les formes débutantes, mais aussi selon l’ancienneté du déficit et de l’étiologie : en général minime dans les causes génétiques, et intense si les causes sont chirurgicales. Les conséquences des insuffisances ovariennes liées aux carences hormonales précoces sont mal connues ; elles sont décrites à partir d’observations sur des séries d’IOP secondaires essentiellement chirurgicales. Initialement ce sont des signes de déficits oestrogénique et progestatif qui apparaissent à court ou moyen terme : manifestations vasomotrices à type de bouffées de chaleur, crises sudorales surtout nocturnes, sécheresse vaginale par diminution des sécrétions vaginales source de dyspareunie gênante.
Des signes de retentissement sur l’appareil urinaire se surajoutent avec risque d’incontinence d’effort, d’infection urinaire, de dysurie et de miction impérieuse.
A long terme, le tableau se complète par l’installation de troubles osseux et cardiovasculaires.
Le tableau peut comporter également un syndrome de déficit androgénique non spécifique suspecté en absence d’amélioration sous substitution œstro-progestative, baisse de vitalité, asthénie croissante et fatigabilité, modifications corporelles comme la perte de la forticilisation des poils pubiens, et l’atrophie génitale. De même que la baisse de la masse et du tonus musculaires, des anomalies de la répartition des graisses et l’installation de troubles sexuels comme la perte du désir, des fantasmes, des rêves érotiques, et de la capacité orgasmique.
L’examen physique est capital, permet d’apprécier, en plus de la symptomatologie fonctionnelle, le degré du développement pubertaire (absent, incomplet ou normal) et de retrouver des signes d’orientation étiologique d’une IOP syndromique génétique ou auto-immune ou conclure à une IOP isolée spontanée.
Profil hormonal des IOP :
Le profil est variable fonction du stade au diagnostic, de la cause et de l’ancienneté de l’affection ; l’IOP spontanée correspond à un phénomène de vieillissement ovarien précoce évoluant en deux phases :
La première, celle de la baisse de la réserve folliculaire caractérisée par des ovulations moins fréquentes : la FSH est élevée, la 17βestradiol(E2) reste élevée pendant quelques années puis la phase folliculaire et l’intervalle intercycle se raccourcissent.
La seconde est un stade d’épuisement folliculaire où les taux d’E2 baissent à moins de 50 pg/ml, les taux de FSH s’élèvent à plus de 30µU/ml ; la LH s’élève mais à des valeurs moindres ce qui différencie l’IOP d’un dosage effectué lors d’un pic ovulatoire. Par ailleurs, diminution des sécrétions androgéniques de Delta 4 androstènedione, Testostérone, DHEA et de la sécrétion des peptides ovariens (inhibines, AMH…).
Critères diagnostiques des IOP :
L’absence de standardisation des critères diagnostiques est encore cause de retard diagnostique des IOP, les critères actuellement retenus sont d’ordre clinique et biologique.
Auplanclinique,il n’y a pas encore de réel consensus sur la durée de l’aménorrhée : l’aménorrhée de plus de 4 mois chez une femme de moins de 40 ans est actuellement retenue.
Au plan biologique : les dosages d’E2, FSH et d’AMH sont proposés.
L’intérêt du dosage d’E2 est encore discuté : du fait de sa variabilité selon l’ancienneté de l’IOP, il n’est actuellement pas retenu comme critère diagnostique.
Le niveau d’élévation de la FSH n’est pas encore consensuel, le seuil de 30-40 µU/ml est admis, à condition de le vérifier à deux reprises, à au moins un mois d’intervalle. Certains proposent un seuil inferieur de 20-40 µU/ml en gardant présent à l’esprit que l’élévation de FSH en dessous de ce seuil, n’est pas toujours liée à une IOP (3).
Certaines équipes proposent d’introduire parmi les paramètres diagnostiques d’IOP le dosage de l’hormone anti Mullerienne (AMH), qui reflète le nombre des follicules primitifs et l’âge ovarien. En plus du fait que l’AMH peut être dosée à tout moment durant le cycle menstruel, ce dosage semble être dans l’IOP plus utile que celui de la FSH car ses modifications apparaissent avant celles de la FSH.
Dans ces études, la précision diagnostique de l’AMH est plus élevée que celle de la FSH et à des différences significatives (4).
La dernière réunion du Groupe d’Étude des IOP a recommandé en 2016 l’utilisation des critères diagnos- tiques suivants (5) :
Survenue d’une oligo-aménorrhée depuis au moins 4 mois ou la présence de signes de déficit E2.
Avant l’âge de 40 ans.
Des taux élevés de FSH supérieurs à 25UI/ml contrôlés deux fois à un mois d’intervalle.
Aspects étiopathogéniques :
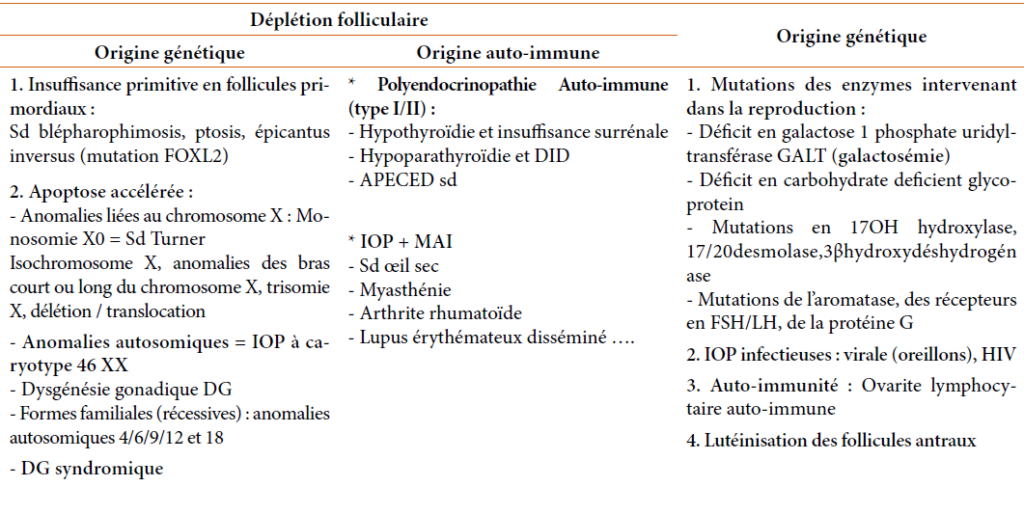
A l’opposé de la ménopause physiologique qui résulte d’un épuisement folliculaire, plusieurs mécanismes sont incriminés dans la survenue d’une IOP, il s’agit :
Soit d’une déplétion folliculaire de cause génétique ou auto-immune : il n’y a pas de follicules primordiaux dans l’ovaire par échec primitif de constitution ou du maintien d’un pool suffisant de follicules primordiaux, ou atrésie des follicules, secondaire à une apoptose accélérée ou une destruction auto immune ou toxique des follicules.
Soit d’un mécanisme de dysfonction folliculaire par blocage de la croissance folliculaire ; les follicules restent dans l’ovaire mais un processus pathologique empêche leur fonctionnement normal.
Les principales étiologies des IOP sont résumées dans le tableau ci-dessous :
Bilan étiologique recommandé par l’ESHRE en 2016 :
Le bilan doit comporter au minimum un bilan génétique et un bilan d’auto-immunité (5) :
Un caryotype doit être réalisé chez toute patiente présentant une IOP non-iatrogénique : une gonadectomie devrait être recommandée chez toute femme avec matériel chromosomique Y détectable complétée par la recherche systématique de la premutation de l’X fragile.
– La recherche anomalies autosomiques n’est indiquée que s’il existe des signes cliniques suggérant une mutation spécifique (ex blépharophimosis-ptosis–épicanthus inversus syndrome).
Le dépistage des anticorps (AC) anti thyroïdiens et d’AC anti 21OHhydroxylase ou anti corticosurrénale doit être considéré chez toutes les femmes atteintes d’IOP de cause idiopathique ou si elle est associée à un désordre auto immun. En cas de positivité de ces derniers, les patientes devront être référées en endocrinologie pour évaluer la fonction surrénalienne et éliminer une maladie d’Addison.
Il n’y a pas assez de preuves pour recommander le dépistage systématique d’un diabète sucré. et d’infections ovariennes chez les femmes porteuse d’IOP.
Cependant malgré tous les progrès réalisés, aucune cause ne peut être identifiée au terme de l’enquête étiologique chez un nombre significatif de femmes, il s’agit d’IOP inexpliquée ou idiopathique.
Risques liés à l’IOP non traitée :
ils sont la traduction de la souffrance des principaux tissus cibles secondaire à la carence hormonale :
La première conséquence est une grande souffrance psychologique, prévisible dans une tranche d’âge aussi vulnérable qui devra nécessiter une assistance psychologique régulière et prolongée.
Elle se traduit par divers troubles de l’humeur : irritabilité, susceptibilité, hostilité, mélancolie, état anxio-dépressif menant à un épuisement psychologique, tendance à l’isolement, repli sur soi, céphalées, insomnies aggravées par les troubles vaso moteurs et réveils de plus en plus précoces aggravant l’état d’asthénie avec perte de l’estime de soi et sentiment d’infériorité (6).
Altération de la sexualité : La fonction sexuelle chez la femme est modulée par les œstrogènes intervenant dans la congestion vaginale et la lubrification et par la testostérone dans la promotion du désir et la libido par effet central sur la dopamine. L’IOP non traitée est à l’origine de troubles sexuels tels que perte du désir, des fantasmes et des rêves érotiques, de la capacité orgasmique, et de l’activité sexuelle ainsi que de signes de carence oestrogénique, cause d’amincissement vaginal,dessèchement, perte d’élasticité et dyspareunie.
Risque cardio-vasculaire : une augmentation significative du risque cardiovasculaire a été démontrée chez les femmes atteintes d’IOP, et de la mortalité cardiovasculaire dans 80 % par ischémie cardiaque (7,8), soit un risque plus précoce de 10 ans par rapport à une ménopause naturelle. Différents facteurs de risque ont été incriminés, essentiellement la dysfonction endothéliale, réversible sous THS (9), la baisse de l’insulino sensibilité (10), ainsi que la présence de syndrome métabolique (11) avec profil lipidique délétère (12).
Risque osseux : Compte tenu du fait que l’IOP s’installe souvent avant l’acquisition du pic de la masse osseuse, de nombreuses études ont démontré que l’IOP est associée à une densité minérale osseuse (DMO) réduite significativement par rapport à des femmes de même âge (13) : deux tiers des IOP ont une DMO basse un an et demi après le diagnostic. La DMO des IOP idiopathiques ou malignes est beaucoup plus basse que la DMO des IOP bénignes.
Pour le groupe de l’ESHRE (5), Il est important d’évaluer la santé osseuse lors du diagnostic initial (14), par mesure de la DMO pour toutes les femmes, surtout lorsqu’il existe des facteurs de risque supplémentaires.
Impact de l’IOP sur les fonctions cognitives : Des études d’observation ont suggéré que la carence prolongée d’E2 ovarien doublait le risque de démence et multipliait par cinq la mortalité par affections neurologiques, mais il reste encore des controverses. D’autres études ciblant les IOP ont montré un risque accru de troubles neurologiques concernant la mémoire verbale et les fonctions cognitives (15,16). Dans ces études, il est démontré que l’IOP quelle que soit sa cause, est associée au long cours avec des effets délétères sur la fonction cognitive qui ne sont pas entièrement réversibles sous THS.
PriseenchargedesIOP:elle a un double objectif.
D’une part la correction du déficit oestrogénique permettant de soulager et traiter les symptômes de l’IOP et surtout d’en prévenir les effets délétères et d’autre part d’envisager les possibilités d’un traitement curatif des troubles de la fertilité. Cette prise en charge doit tenir compte de plusieurs paramètres : l’âge au diagnostic de l’IOP, son caractère induit ou spontané, l’ancienneté, l’étiologie et le terrain comportant d’éventuelles comorbidités.
Les intérêts du THS sont multiples : de nombreuses
études ont démontré un effet de prévention primaire de la détérioration osseuse (5) : en effet, les ostrogènes naturels contenus dans le THS ont un effet protecteur indéniable sur l’os ; pris au moins 3 ans le THS diminue significativement le risque de fractures ultérieures (17). Par ailleurs, ces ostrogènes naturels auraient un effet cardio-protecteur : malgré le manque de données du devenir au long cours, l’introduction rapide du THS dès le début de l’IOP est fortement recommandée pour contrôler le risque ultérieur de maladies cardiovasculaires : il est conseillé de poursuivre le traitement au moins jusqu’à l’âge de la ménopause naturelle. Les données concernant l’effet neuroprotecteur sont encore discordantes surtout après IOP chirurgicale, cependant le THS apparait plus bénéfique s’il est initié dès la ménopause.
Le THS est actuellement fortement recommandé par l’ensemble des sociétés savantes s’occupant de pathologie gonadique féminine (6) et ce au moins jusqu’à l’âge de la ménopause naturelle, voire au-delà si les symptômes persistent. La correction hormonale est accompagnée de mesures hygiénodiététiques permettant le contrôle des facteurs de risque cardiovasculaires (poids, TA), et de préservation du capital osseux. Ces mesures hygiéno-diététiques reposent d’une part sur des apports vitamino-calciques adéquats pour préserver le capital osseux, soit par respect des apports alimentaires recommandés pour l’âge, sinon par compléments alimentaires. Comme il n’y a pas de données spécifiques à ces jeunes femmes, il est possible de s’inspirer des recommandations de la société nord-américaine de ménopause : 1200-1500 mg/j de calcium élément et de vitamine D, sous sa forme active D3 : 800-1000 U/j, à maintenir dans les normes recommandées (25 OHD > 30 mg/ml). D’autre part sur l’exercice physique régulier : là aussi, il n’y a pas de données spécifiques à ces femmes pour la fréquence et l’intensité, les exercices conseillés sont des séances de musculation, jogging, footing, monter les escaliers à pied, et éviter les ascenseurs.
L’arrêt du tabac et de l’alcool sont fortement recommandés chez toutes les femmes à risque d’IOP. Certaines équipes proposent le recours à l’acupuncture qui aurait un certain effet sur l’amélioration des menstruations et du syndrome climatérique, probablement lié à l’augmentation des taux d’œstrogènes (18).
Prescription du THS : L’estrogène de choix est le 17βestradiol dont les effets sont supérieurs à ceux de l’éthynil-œstradiol ou aux œstrogènes conjugués, prescrit préférentiellement par voie transdermique, percutanée du fait de ses moindres effets sur les facteurs hémostatiques, ou orale en cas de réticence. Des études contrôlées ont montré que cette voie a moins d’accidents veineux thromboemboliques que les œstrogènes oraux (19).
La dose d’initiation
doit être supérieure à la dose des œstrogènes contenus dans le THS des ménopauses naturelles pour un meilleur contrôle des symptômes (surtout vasomoteurs), elle permettrait d’atteindre une concentration plasmatique moyenne de la mi-phase folliculaire d’un cycle d’une femme normalement réglée soit environ 300-400 pg/ml assurant la protectrice osseuse. Cette dose est autour de 100 µg/j par voie transdermique (75-200 µg/j) et de 0,06 % x 2 / j de gel par voie percutanée.
Les progestagènes restaurent correctement un endomètre sécrétoire et protègent contre le cancer de l’endomètre, ils sont prescrits par voie orale durant 12-14 j par mois. Il s’agit d’isomères de la progestérone à la dose de 10 mg / j ou de la progestérone micronisée orale à la dose de 200 mg / j.
Il existe d’autres voies d’administration : transdermiques / gel : 12 j / mois ou la voie utérine par dispositif intra-utérin à libération continue de progestérone.
Les schémas de prescription mimant parfaitement la sécrétion physiologique des E2/P n’existent pas et il y a nécessité d’associer la patiente dans le choix du schéma et de la voie d’administration pour une meilleure compliance au long cours, parfois même de considérer les produits contraceptifs (20).
Les schémas proposés sont :
Le schéma séquentiel avec règles qui permet l’obtention d’hémorragie de privation à l’arrêt du progestatif, a un impact psychologique extrêmement bénéfique par prescription d’estradiol du J1 au J25 associé à la progestérone du J14 au J25.
Le schéma combiné sans règles, comportant la prescription continue de comprimés contenant de l’E2 et de la progestérone du J1 au J25, généralement utilisé après 1 an de THS séquentiel.
Si l’IOP comporte une activité résiduelle, le progestatif prescrit seul du J14 au J25 est suffisant pour l’obtention de règles.
Sous THS, la dose d’E2 doit être modulée selon la réponse clinique et la DMO : il n’y a pas d’intérêt à répéter les dosages d’E2 ni de FSH laquelle peut ne pas se normaliser.
L’androgénothérapie peut être envisagée chez des femmes ovariectomisées, en cas d’absence d’amélioration sous THS bien conduit. Cependant il y a lieu d’informer ces patientes que les données concernant ce type de traitement sont encore limitées et que ses effets au long cours ne sont pas encore clairs : ils doivent être évalués après 3–6 mois et devraient être limités à 24 mois (6).
Indications du THS :
A l’opposé des recommandations actuelles concernant les ménopauses naturelles de ne traiter par THS que les femmes avec symptômes modérés à sévères, de carence oestrogénique, toutes les IOP spontanées quel que soit l’âge au diagnostic sont une indication formelle au THS sauf contre- indication absolue. Les femmes atteintes d’IOP doivent être informées qu’il n’a pas été démontré de sur-risque de cancer du sein sous THS avant l’âge de la ménopause, (21). La prévention du cancer de l’endomètre passe par la précaution d’associer un progestatif avec l’estradiol pour protéger l’endomètre des femmes avec utérus intact (22).
IOP et situations particulières :
IOP induites : le THS reste une contre-indication formelle après un cancer du sein (23), chez les femmes porteuses de mutation du gène BRCA, cependant certaines équipes ont montré que le THS pouvait constituer une option thérapeutique bénéfique pour les femmes porteuses des mutations BRCA1/2 sans antécédent personnel de cancer du sein, institué après salpingo-ovariectomie bilatérale prophylactique (24).
IOP et gynécologie : Dans l’endométriose, si une ovariectomie est réalisée, le THS peut être indiqué pour le traitement du syndrome climatérique et pourrait prévenir la réactivation de la maladie (25). Le fibrome ne représente pas une contre-indication au THS (26).
IOP et migraine : la migraine ne doit pas être considérée comme une contre-indication au THS. Une éventuelle aggravation peut être réversible par le réajustement de la dose, de la voie, ou du schéma de prescription du THS. La voie transdermique présente le risque le plus faible d’une migraine avec aura (27).
IOP et situations à haut risque cardiovasculaire : plusieurs études ont montré que ces dernières ne constituent pas de contre-indications au THS, telles que des antécédents de thrombo-embolie veineuse ou de troubles thrombophiliques, d’HTA, obésité, excès de poids ; dans ces cas la voie transdermique est fortement recommandée (28).
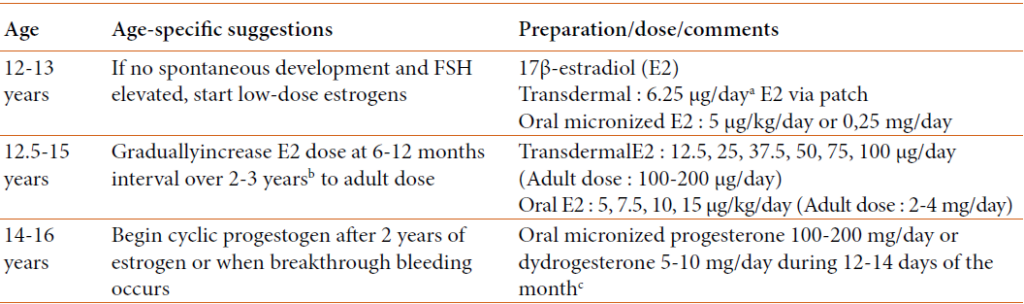
Cas particulier de l’IOP à adolescence : Les modalités d’induction pubertaire proposées par le groupe d’étude du syndrome de Turner en 2007 sont résumées dans le tableau ci-dessous.
Tableau : Traitement de substitution aux œstrogènes à l’adolescence (adapté de “Bondy Turner Syndrome Study Group,2007”)
AlternativesauTHS:Les preuves sur l’efficacité et l’innocuité de traitements alternatifs ou complémentaires dans les IOP iatrogènes sont très insuffisantes ou absentes (29).
Bilan de suivi des IOP recommande par l’ESHRE (5) :
La surveillance d’une IOP doit comporter un examen clinique annuel visant à évaluer la compliance des patientes atteintes. L’absence de signes d’appel ne justifie pas d’examen complémentaire particulier.
Les modalités de suivi de la santé osseuse se basent sur la mesure de la DMO : Si la DMO est normale et un THS adéquat est introduit, l’intérêt de la répétition de la mesure est faible. Si une ostéoporose est diagnostiquée et le remplacement d’œstrogène ou d’un autre traitement osseux est initié, la mesure de DMO doit être renouvelée dans les 5 ans : une diminution de la DMO devrait réajuster la dose d’œstrogénothérapie et rechercher d’autres facteurs potentiels. Il y aura lieu alors de confier la patiente en milieu spécialisé.
Les modalités de suivi des facteurs de risque cardiovasculaire comportent la surveillance annuelle du poids, de la pression artérielle, du tabagisme et le dépistage de tous autres facteurs de risque. Chez les femmes atteintes du syndrome de Turner, les facteurs de risque cardiovasculaires devraient être recherché au moment du diagnostic et réévalué chaque année (au moins PA, tabagisme, poids, profil lipidique, glycémie à jeun et l’ HbA1c)
Les patientes avec AC TPO positifs doivent bénéficier d’une surveillance annuelle de la TSH (30,31).
Fertilité et IOP : des possibilités thérapeutiques ?
La prise en charge de l’infertilité résultant d’une IOP reste un grand challenge (32) : d’importants progrès ont été réalisés essentiellement dans les IOP iatrogènes grâce au développement des techniques de procréation médicalement assistée ayant permis chez certaines femmes une conception naturelle avec leurs propres ovules. Le préalable est l’abaissement des taux de FSH à des taux physiologiques avant d’entamer le traitement : l’éthynil œstradiol est classiquement utilisé, il permet de stimuler la glaire cervicale et le développement endométrial et n’interfère pas dans le dosage de l’estradiol. Des gonadotrophines exogènes à faibles doses sont parfois nécessaires pour booster la maturation associées à la progestérone prescrite en phase lutéale (33).
Des techniques préventives et/ou curatrices de l’infertilité des IOP ont été développées par fertilisation in vitro utilisant le don d’ovocytes avec 50 % de succès selon l’âge du donneur, ayant permis 40-50 % de naissances vivantes par embryon transféré. Cette technique reste interdite par la religion dans les pays musulmans.
Pour les patientes devant subir un traitement antitumoral, des techniques de cryoconservation sont proposées (34,35) utilisant soit des embryons, des ovocytes ou des fragments du cortex ovarien, suivi par la transplantation orthotopique et la maturation in vitro des oocytes. Cependant, bien que des résultats prometteurs aient été obtenus, les taux de grossesse restent très bas. Des techniques de thérapies cellulaires sont actuellement en cours d’expérimentation ciblant l’activation des follicules ovariens résiduels de la folliculogénèse ainsi que l’isolement de cellules souches ovariennes (36).
Références :
H. Rozenbaum Ménopause -EMC d’endocrinologie nutrition [10- 035-A-10]
A. Graff, S. Christin-Maitre : Insuffisance ovarienne prématurée EMC gynécologie [147-A-40]
Cohen J, and al. Diminished ovarian reserve, premature ovarian fai- lure, poor ovarian responder – a plea for universal definitions. J Assist Reprod Genet. 2015
Alipour F : Comparison of Specificity and Sensitivity of AMH and FSH in Diagnosis of Premature Ovarian Failure. Dis Markers. 2015 ;
L. Webber and all. European Society of Human Reproduction and Embryology ESHRE 2016: « Management of women with premature ovarian insufficiency †: The ESHRE Guideline Group on POI, in Hu- man Reproduction, 2016.
Risque psychol Shmidt JCEM 2011)
Jacobsen B Kand al. Age at natural menopause and total mortality and mortality from ischemic heart disease: the Adventist Health Study in J Clin Epidemiol. 1999
Roeters van Lennep JE, and all collaborators of the Dutch Multi- disciplinary Guideline Development Group on Cardiovascular Risk Management after Reproductive Disorders : Cardiovascular disease risk in women with premature ovarian insufficiency: A systematic review and meta-analysis in Eur J Prev Cardiol. 2016.
Kalantaridou SN, and all : Impaired endothelial function in young women with premature ovarian failure : normalization with hormone therapy J Clin Endocrinol Metab. 2004
Corrigan EC, and all : Effects of ovarian failure and X-chromo- some deletion on body composition and insulin sensitivity in young In Menopause. 2006
Eshtiaghi R : Menopause is an independent predictor of metabolic syndrome in Iranian women In Maturitas. 2010.
Knauff EA, and all Lipid profile of women with premature ovarian failure. Menopause. 2008
Popat VB and alla : Bone mineral density in estrogen-deficient young women. J Clin Endocrinol Metab. 2009
BakhshH and all Gynecol Endocrinol. 2015 Premature ovarianinsufficiency in young girls: repercussions on uterine volume and bone mineral density. in Gynecol Endocrinol. 2015.
Scott EL and al. Premature menopause and risk of neurological disease: basic mechanisms and clinical implications. in Mol Cell En- docrinol. 2014.
Ryan J and all : Impact of a premature menopause on cognitive function in later life. In BJOG. 2014
Van der Klift M and al Risk factors for incident vertebral fractures in men and women: the Rotterdam Study. In J Bone Miner Res. 2004.
Zhongguo Zhen Jiu. 2014. [Acupuncture for premature ovarian failure : a prospective cohort study].
Langrish JP and all : Cardiovascular effects of physiological and standard sex steroid replacement regimens in premature ovarian fai- lure. Hypertension 2009.
Sassarini J and al : Sex hormone replacement in ovarian failure – new treatment concepts. Best Pract Res Clin Endocrinol Metab. 2015 Jan.
Soares PM, and all Breast density in women with premature ova- rian failure or post menopausal women using hormone therapy: ana- lytical cross-sectional study. Sao Paulo in Med J 2010.
Furness S and al. Hormone therapy in postmenopausal women and risk of endometrial hyperplasia. Cochrane Database Syst Rev 2012.
Antoine C, and all : Safety of hormone therapy after breast cancer : a qualitative systematic review.in Hum Reprod 2007.
Rebbeck TR et al. Effect of short-term hormone replacement therapy on breast cancer risk reduction after bilateral prophylactic oophorectomy in BRCA1 and BRCA2 mutation carriers : the PROSE Study Group. J Clin Oncol 2005.
Dunselman GA et al. ESHRE guideline : management of women with endometriosis in Hum Reprod 2014.
Nappi RE and all : Course of primary headaches during hormone replacement therapy in Maturitas 2001 ;
Ciarmela P, Ciavattini A, Giannubilo SR, Lamanna P, Fiorini R, Tranquilli AL, Christman GM, Castellucci M. Management of leio- myomas in perimenopausal women in Maturitas 2014
Langrish JP and all : Cardiovascular effects of physiological and standard sex steroid replacement regimens in premature ovarian fai- lure. In Hypertension 2009
Rada and al. : Non-hormonal interventions for hot flushes in women with a history of breast cancer. in Cochrane Database Syst Rev 2010:
Kim TJ, and all. Routine endocrine screening for patients with ka- ryotypically normal spontaneous premature ovarian failure in. Obstet Gynecol 1997.
Goswami R, and all. Prevalence of thyroid autoimmunity in spo- radic idiopathic hypoparathyroidism in comparison to type 1 diabetes and premature ovarian failure. J. Clin Endocrinol Metab 2006 ;
Gougeon A and all : [Present and future strategies for women at risk, or suffering from premature ovarian failure (POF)] in Gynecol Obstet Fertil. 2012.
Blumenfeld Z and al : Gonadotropin-Releasing Hormone Agonist Cotreatment During Chemotherapy May Increase Pregnancy Rate in Survivors in Oncologist. 2015.
Ravel C and al : Fertility preservation in pre-pubertal girls with cancer: the role of ovarian tissue cryopreservation in Gynecol Obstet Fertil. 2016
Silvestris [Ovarian failure : New treatments in perspective ?].J Ovarian Res. 2015.
N. MAHMOUDI (1), S. SAHNOUNE (1),I. BELHADJI (1), A. KHELIL (1),A. CHIALI (1), A. SERRADJ (2) Service de Dermatologie, CHU Benaouda Benzerdjeb, Oran, Service de Dermatologie, EHU 1er Novembre, Oran.
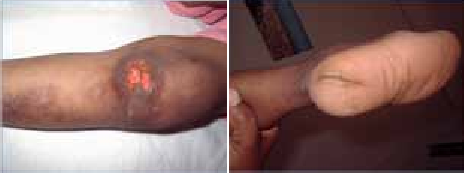
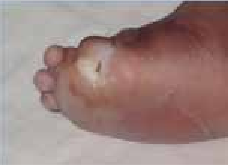
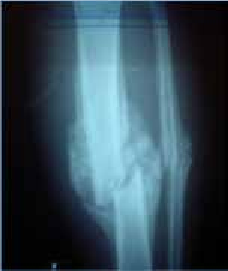
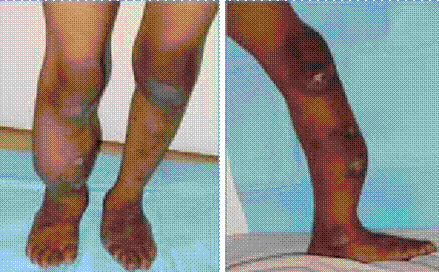
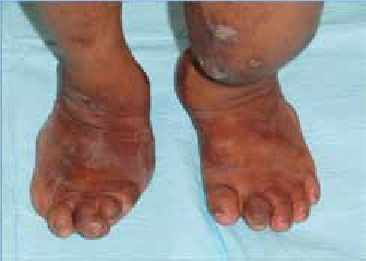
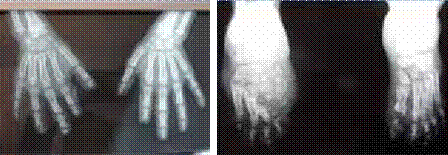
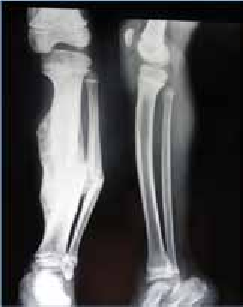
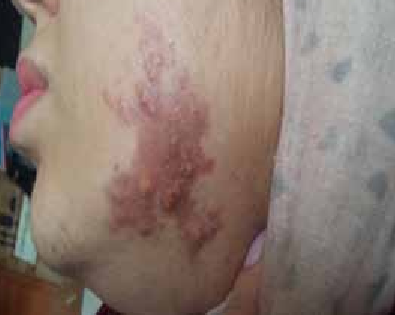
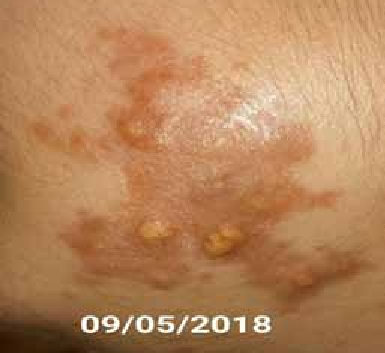
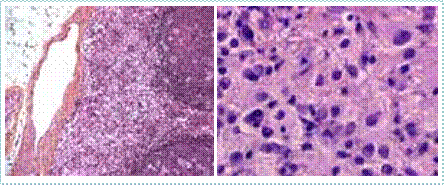
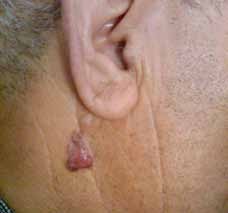
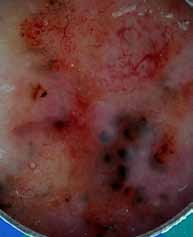
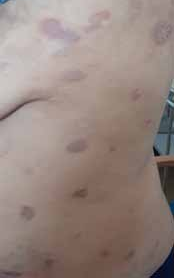
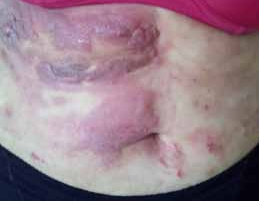
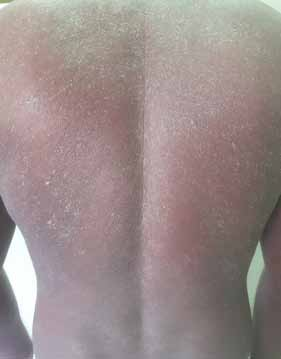
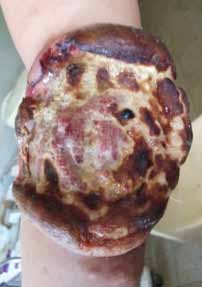
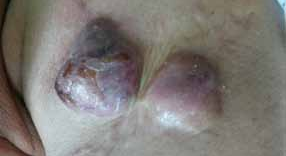
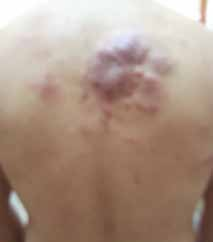
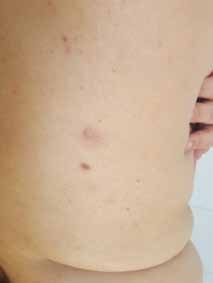
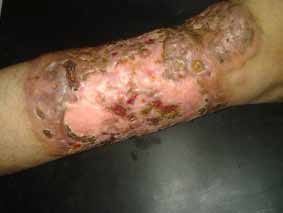
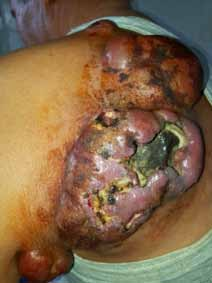
Abstract : Congenital insensitivity to pain (CIP) is a very rare genetic syndrome, characterized in its most severe form by an absence of pain sensation of congenital origin. There are several types integrating in the context of autonomous hereditary sensory neuropathies (HSAN). ICD with anhidrosis (HSAN IV) remains the rarest and most radical form. Autosomal recessive inheritance is manifested by painful and thermal numbness, self-injury, unexplained episodes of major hyperthermia, anhidrosis, bone and oral manifestations, and mental retardation. It is thought to be related to a sensory involvement of the fine-sized fibers in relation to a mutation of the TRKA gene coding for the high affinity receptor for NGF (Nerve Growth Factor) playing a survival role for nociceptors and sympathetic fibers. We report the case of a 5-year- old girl presenting a typical CIP.
Key-words : Congenital insensitivity, pain, anhidrosis, CIP, HSAN type IV.
Résumé : L’insensibilité congénitale à la douleur (ICD) est un syndrome génétique très rare, qui se caractérise dans sa forme la plus sévère par une absence de sensation douloureuse d’origine congénitale. Il en existe plusieurs types s’intégrant dans le cadre de neuropathies sensitives héréditaires autonomes (HSAN). L’ICD avec anhidrose (HSAN type IV) reste la forme la plus rare et la plus radicale. De transmission autosomique récessive, elle se manifeste par une insensibilité douloureuse et thermique, des automutilations, des épisodes inexpliqués d’hyperthermie majeures, une anhidrose, des manifestations osseuses, buccodentaires ainsi qu’un retard mental. Elle serait liée à une atteinte sensitive des fibres de fin calibre en rapport avec une mutation du gène TRKA codant pour le récepteur de haute affinité pour le NGF (Nerve Growth Factor) jouant un rôle de survie pour les nocicepteurs et les fibres sympathiques. Nous rapportons le cas d’une petite fille de 5 ans qui présente une ICD avec anhidrose typique avec différentes atteintes et complications définissant ce syndrome.
Mots-clés : Insensibilité congénitale, douleur , anhidrose, CIPA, HSAN type IV
Introduction :
L’insensibilité congénitale à la douleur (ICD) est un syndrome génétique très rare, qui se caractérise dans sa forme la plus sévère par une absence de sensation douloureuse d’origine congénitale. Il en existe plusieurs types s’intégrant dans le cadre de neuropathies sensitives héréditaires autonomes (HSAN). L’ICD avec anhidrose (HSAN type IV) reste la forme la plus rare et la plus radicale. Nous rapportons le cas d’une petite fille atteinte d’une ICD avec anhidrose présentant automutilations, troubles trophiques et diverses manifestations osseuses.
Observation :
Petite fille de 05 ans, du sud algérien (Adrar), issue d’un mariage consanguin du 1er degré, dernière d’une fratrie de 7, née hypotrophe par accouchement eutocique d’une grossesse à terme qui consulte pour des troubles trophiques à type d’ulcérations multiples indolentes des coudes et genoux (figure 1), une hyperkératose fissuraire des mains et des pieds (figure 2) avec amputation des phalanges distales des mains, index droit et gauche ; (figure 3, 4 et 5). Il n’existe pas de cas similaires dans la famille.
Figure 1 : Ulcération indolente du genou gauche Figure 2 : Hyperkératose fissuraire du pieds droitFigure 3 : Hyperkératose fissuraire de la main droite, amputation de l’index Figure 4 : Amputation de la phalange distale (Index gauche)Figure 5 : Amputation des phalanges distales des mains (index droit et gauche)
Le début du tableau semble remonter à la période post-natale, par la survenue de plusieurs épisodes fébriles sans source d’infection notable. Au cours de la première année de vie, les parents constataient des actes d’automutilation de la langue et des doigts depuis l’éruption dentaire sans qu’aucune douleur ne soit ressentie. (Amputations auto infligées). Au cour de l’évolution, apparaissaient fréquemment des troubles trophiques, à type d’ulcérations indolentes suite à des chutes et brûlures, difficiles à faire cicatriser.
Une chute de plusieurs dents permanentes spontanément et par auto extraction (figure 6) ainsi que des diarrhées d’allure motrice, complètent le tableau. Les parents rapportent l’absence de sudation. À partir de l’âge de 2 ans, sur le pied droit, la patiente commençait à faire des amputations spontanées du petit orteil, puis plusieurs mois après, celle du gros orteil (figure 7) laissant place à une ulcération (figure 8).
Figure 6 : Chute des dents permanentes spontanément et par auto extraction Figure 7 : Amputation spontanée des petit et gros orteils (Pieddroit)Figure 8 : Ulcération après amputation du gros orteil droit
Deux mois après, la patiente se présente pour une tuméfaction de ce même pied, avec écoulement de pus au site d’amputation du gros orteil, radiographie faite objective une ostéite du 1er, 2ème et 3ème métatarsiens avec de multiples séquestres osseux et de nombreuses modifications architecturales (figure 9) bénéficiant d’un traitement chirurgical (ablation des séquestres).
Figure 8 : Radiographie du pied droit : ostéite du 1er, 2éme et 3éme métatarsiens, multiples séquestres osseux
Dans les suites d’une chute, la patiente présente une fracture diaphysaire non déplacée des deux os de la jambe gauche bénéficiant d’une immobilisation plâtrée.
La radiographie de contrôle (1 mois) montre la formation d’un cal hypertrophique pathologique sur une fracture qui s’est déplacée (figure 10).
Figure 10 : Radiographie de la jambe gauche :Cal hypertrophique pathologique sur une fracture diaphysaire déplacée
La peau d’aspect épaissi est particulièrement sèche et chaude à la palpation. La patiente est d’intelligibilité jugée normale, cependant hyperactive et agitée. Un retard staturo-pondéral est présent estimé à (–) 2 DS. L’examen neurologique retrouve une anesthésie thermoalgique, un tonus musculaire diminué surtout au moment des pics fébriles et des réflexes ostéo-tendineux diminués. Le test à la sueur confirme l’anhidrose. La biopsie cutanée retrouve une réduction de la densité des fibres nerveuses dermiques. L’E.M.G confirme la neuropathie sensitive. La TDM cérébrale pratiquée s’est révélée sans particularités. La biopsie nerveuse a été refusée par les parents.
Sur le plan évolutif, la patiente a été revue 04 mois après avec un aspect angulé de la jambe gauche, des ulcérations persistantes (figure 11) et des pieds plus déformés que précédemment (figure 12 et 13).
Figure 11 : Évolution à 4 mois: aspect angulé de la jambe gauche, ulcérations persistantes, pieds plus déformés Figure 12 : Évolution à 4 mois : aspect angulé de la jambe gauche, ulcérations persistantesFigure 13 : Évolution à 4 mois :Pieds droit et gauche plus déformés
Au contrôle radiologique, on notait les foyers de fracture au niveau de la diaphyse du tibia et péroné gauche avec angulation et un cal de consolidation hypertrophique (figure 16) ainsi que des acrolyses et fontes osseuses intéressant les mains et les pieds (figure 14, 15).
Figure 14 : Radiographie des mains à 4 mois : acrolyses et fontes osseuses Figure 15 : Radiographie des pieds à 4 mois : acrolyses et fontes osseusesFigure 16 : Radiographie de contrôle : Foyers de fracture au niveau de la diaphyse du tibia et péroné gauches avec angulation et cal de consolidation hypertrophique
Discussion :
L’ICD est un syndrome génétique très rare, caractérisée par l’absence de sensation douloureuse d’origine congénitale. Lorsqu’elle s’intègre dans le cadre de neuropathies sensitives héréditaires (HSAN) de type IV, elle définit l’ICD avec anhidrose ou CIPA (Congenital Insensitivity to Pain with Anhidrosis) qui a été décrit pour la première fois en 1951.
De type autosomique récessif, elle se manifeste habituellement par une insensibilité douloureuse et thermique, des automutilations, des épisodes inexpliqués d’hyperthermie majeures, une anhidrose, des manifestations osseuses (ostéite, ostéomyélite, cals hypertrophiques), des manifestations buccodentaires (perte des dents spontanément ou par auto-extraction, morsures de la langue et ulcérations des lèvres) et un retard mental souvent présents. Elle serait liée à une atteinte sensitive des fibres de fin calibre en rapport avec une mutation du gène TRKA codant pour le récepteur de haute affinité pour le NGF (Nerve Growth Factor) jouant un rôle de survie pour de les nocicepteurs et les fibres sympathiques.
La présentation clinique chez cette petite fille est typique d’une CIPA et rend compte de nombreuses atteintes et de complications définissant ce syndrome. Sur le plan dermatologique, les troubles trophiques étaient de plus en plus difficiles à faire cicatriser. Les pics hyperthermiques ont pu être jugulés par refroidissement externe. Sur le plan orthopédique, le pronostic fonctionnel reste réservé en rapport avec les déformations osseuses et les amputations distales qu’elle présente. La chute de la majeure partie des dents posent un problème de mastication. Il est important de préciser que cette patiente vit dans une ville du sud algérien où la température excède les 50°C durant plus de la moitié de l’année, et que la surchauffe corporelle met la patiente dans un état de léthargie cédant après refroidissement corporel.
Cependant, il n’a pas été noté de retard mental important, comme dans la plupart des observations décrites dans la littérature.
Conclusion :
Le diagnostic d’ICD doit être évoqué le plus rapidement possible devant un enfant ayant un comportement d’automutilation afin de mettre en œuvre les mesures adéquates en collaboration étroite avec les parents. Un diagnostic précoce, des soins médicaux appropriés, une éducation de ces patients peuvent diminuer de la fréquence et de la gravité des plaies infectées et des ostéomyélites. Le conseil génétique est indispensable compte tenu de la gravité de la maladie.
Références :
John R. Person, MD ; Roy S. Rogers III, MD ; K. Hable Rhodes, MD, Congenital Sensory Neuropathy Report of an Atypical Case Arch Dermatol. 1977;113(7):954-957.
Okuno, A Inoue and S Izumo, Congenital insensitivity to pain with anhidro- sis. A case report, J Bone Joint Surg Am. 1990 ;72:279-282.
Rosemberg S, Marie SKN, Kliemann S. Congenital insensitivity to pain with anhidrosis (hereditary sensory and autonomic neuropathy type IV). Pediatr Neurol 1994 ;11:50-6
Sztriha László, PhD , Gilles G. Lestringant, Hertecant Jozef M, Philippe M. Fros- sard, I. Masouyé MD, Insensibilité congénitale à la douleur avec anhidrose , Neuro- logie pédiatrique Volume 25 Numéro 1, Juillet 2001, Pages 63-66
Sunil Karande, Nitin Satam, Hereditary Sensory and Autonomic Neuropathy Type IV, Images In Clinical Practice Indian Pediatrics ; VOLUME 42 JUNE 17, 2005
Redouani L., Léauté-Labréze C., Ramirez de Villar S., Taieb A., Sarlangue J., Difficulté de prise en charge d’une insensibilité congénitale à la douleur. Arch Pediatr 2002; 7: 701-4
Y. ABI AYAD, A. SERRADj,Z. ELOSMANI, S. HAMZAOUI, Service de Dermatologie, EHU 1er Novembre, Oran.
Abstract : Rosai-Dorfman’s disease (RDF), is a benign histiocytic disorder characterized by painless cervical lymphadenopathy with fever. The extra nodal location is frequent, including the skin; However, forms localized only to the skin are very rarely described in the literature. We report a case of cutaneous RDF disease revealed by solitary plaque of the face. Observation : 25-year-old woman consulted for erythematous infiltrated plaque of the face with reddish yellow papules evolving since 1 year followed by the occurrence of subcutaneous nodule 6 months later at the axillary site, without other abnormalities on clinical, biological and radiological examination. An initial skin biopsy showed a granulomatous polymorphic inflammatory process. Various diagnoses were discussed in the patient, including acne, sarcoidosis and cutaneous tuberculosis. A review of the histological lesion revealed histiocytic infiltration with image of emperipolesis allowing to retain the diagnosis of the cutaneous Rosai Dorfman disease. Because of the aesthetic prejudice the patient was treated by steroid associated with methotrexate. The evolution was marked by a slight improvement after 3 months of treatment. Discussion : RDF is a rare disease, more common in children and young adults with a slight male predominance. Extranodal localizations occur in 43% of cases , the most common site is the skin, (16%), which can reveal the disease, but The purely cutaneous form without lymph node involvement is very rare and have different demographic characteristics, with a median onset age of 46.7 years and female predominance. Cutaneous lesions are non-specific and varied causing confusion with other diagnostics. Diagnosis orientation by clinicians allows to search evocative histological signs represented by an important histiocytic infiltration and images of emperipolesis. Therapeutic management is still not codified, numerous treatment have been attempted; however, with often poor response. Our case illustrates the diagnostic difficulties encountered in this pathology, with often delayed diagnosis, on the other hand, it raises the difficulties of therapeutic management even if in most cases the evolution is benign with spontaneous regression in variable delays.
Résumé : La maladie de Rosai-Dorfman (RDF), est un trouble histiocytaire bénin caractérisé par des Poly adénopathies diffuses, surtout cervicales associées à une fièvre. Les localisations extra ganglionnaires sont fréquentes, parmi lesquelles la peau constitue un site fréquent; toutefois les formes strictement localisées à la peau sont très rarement décrites dans la littérature. Nous rapportons une observation de la maladie de RDF cutanée révélée par une plaque cutanée du visage. Observation : Une patiente de 25 ans consultait pour une plaque infiltrée érythémateuse du visage avec des lésions papuleuses rouges et jaunâtres évoluant depuis 1 année, suivies par l’apparition de nodule sous cutané 6 mois après au niveau axillaire, sans autres anomalies à l’examen clinique, biologique ou radiologique. Une biopsie cutanée mettait en évidence initialement un processus inflammatoire polymorphe granulomateux évoquant une tuberculose ou sarcoïdose. Différents diagnostics ont été évoqués chez la patiente notamment acné, sarcoïdose et tuberculose cutanée. Une relecture des lames histologiques a mis en évidence une infiltration histiocytaire avec image d’empéripolese permettant de retenir le diagnostic de la maladie de (RDF). En raison du préjudice esthétique, la patiente a été traitée par corticothérapie générale associée secondairement au méthotrexate entrainant une discrète amélioration après 3 mois de traitement. Discussion : La maladie de RDF est une affection rare, plus fréquente chez les enfants et les sujets jeunes avec une légère prédominance masculine. Les atteintes extra ganglionnaires peuvent se voir dans 43 % des cas, au premier rang desquelles une atteinte cutanée (16 %), qui peut être révélatrice. Les formes strictement cutanées sans atteinte ganglionnaire sont plus rarement rapportées et présentent des caractéristiques démographiques différentes avec un âge moyen de 46,7 ans et plus souvent de sexe féminin. Les lésions cutanées sont non spécifiques et variées entraînant une confusion avec d’autres diagnostics. L’orientation diagnostique par les cliniciens permet de rechercher des signes histologiques évocateurs représentés par une importante infiltration histiocytaire et des images d’empéripolèse. La prise en charge thérapeutique n’est toujours pas codifiée, de nombreux traitements ont été tentés ; avec une réponse souvent médiocre. Notre cas illustre les difficultés diagnostiques rencontrées dans cette pathologie, avec un diagnostic souvent retardé, d’autre part, il soulève les difficultés de prise en charge thérapeutique même si dans la plupart des cas l’évolution est bénigne avec régression spontanée dans des délais variables.
La maladie de Rosai-Dorfman (MRD), également connue sous le nom d’histiocytose sinusale avec lymphadénopathie massive, est une histiocytose non langerhansienne décrite pour la première fois en 1965 par Destombes [1] et reconnue par Rosai et Dorfman en 1969 comme une entité anatomo-clinique bien distincte [2]. Elle est caractérisée par des polyadénopathies diffuses, surtout cervicales associées à une fièvre, un syndrome inflammatoire, une hyper gammaglobulinémie et une hyper leucocytose [3]. Les atteintes extra ganglionnaires sont fréquentes avec une prédilection pour la peau ; cependant, les formes exclusivement cutanées ne sont que très rarement décrites [4,5].
Nous rapportons une observation de la maladie de RDF cutanée, révélée par une plaque cutanée initiale isolée du visage.
Observation :
Une patiente de 25 ans ayant pour seuls antécédents un ulcère gastrique traité consultait pour une plaque érythémateuse infiltrée du visage évoluant depuis plus d‘un an ayant débuté par des papules de couleur variable, suivies 6 mois plus tard par un nodule sous cutané au niveau axillaire dans un contexte général conservé.
L’examen clinique objectivait une plaque érythémato violacée infiltrée légèrement, prurigineuse, localisée au niveau du maxillaire gauche de 4 cm du grand axe aux contours irréguliers et surmontée de papules de couleur allant du jaune au rouge brun (figure 1).
Figure 1 : Plaque érythémato-violacée infiltrée au niveau du maxillaire gauche aux contours irréguliers et surmontée de papules de couleur allant du jaune au rouge brun.
Le reste de l’examen somatique notamment lymphogan-glionnaire ORL et ophtalmologique était sans anomalies.
La biopsie exérèse du nodule sous cutané mettait en évidence initialement un processus inflammatoire polymorphe granulomateux évoquant une tuberculose ou une sarcoïdose.
Les examens para cliniques ne révélaient aucune anomalie ni à L’hémogramme ni à l’électrophorèse des protéines plasmatiques, avec absence de syndrome inflammatoire.
La recherche d’anticorps antinucléaires, le dosage du quantiferon et IDR à la tuberculine étaient négatifs, la tomodensitométrie thoraco-abdomino-pelvienne était normale.
Ce tableau clinique a suscité plusieurs consultations au cours desquelles différents diagnostics ont été évoqués chez la patiente notamment acné, sarcoïdose et tuberculose cutanée.
Une relecture des lames histologiques complétée par l’immunohistochimie préconisée devant la discordance du tableau clinique et para clinique a permis finalement de retenir le diagnostic de la maladie de Rosai-Dorfman cutané.
Un traitement était préconisé en raison du préjudice esthétique par corticothérapie : 1 mg/kg/j, associée secondairement au méthotrexate à 20 mg/semaine. L’évolution était marquée au bout de 3 mois, par une discrète amélioration clinique (figure 2).
Figure 2 : Amélioration clinique, palissement de la lésion et affaissement des papules après 3 mois de traitement.
Discussion :
La maladie de Rosai-Dorfman (RDF) est une forme bégnine d’histiocytose non langerhansienne.C’est une entité rare dont la cause demeure inconnue. Les caractéristiques immuno-histochimiques des his- tiocytes sont en faveur d’une pathologie macrophagique et jusqu’à maintenant aucune cause infectieuse n’a été formellement identifiée [6].
Dans sa forme classique, la maladie survient habituellement chez les enfants et les sujets jeunes avec une légère prédominance masculine, elle se manifeste typiquement par des adénopathies cervicales bilatérales, de consistance dure, non douloureuses, survenant dans un contexte fébrile. D’autres sites peuvent être impliqués notamment axillaires, inguinaux et médiastinaux [3].
Les atteintes extra-ganglionnaires sont rapportées dans 43 % des cas, soit comme une manifestation initiale isolée de la maladie, ou en association à l’atteinte ganglionnaire.
Tous les organes peuvent être touchés en particulier la peau, suivie par les atteintes ORL, osseuses, orbitaires et neurologiques [4].
Environ 16 % des patients atteints de la maladie de RDF présentent des lésions cutanées, et dans 3 % des cas, la maladie se manifeste exclusivement au niveau la peau [6,7].
Elle se distingue de la forme systémique de la maladie par les caractéristiques démographiques.
Cette variante exclusivement cutanée de la maladie a été rapportée pour la première fois par Thawerani et al en 1978. Depuis, un certain nombre de cas ont été rapportés notamment chez les individus asiatiques et caucasiens. Elle affecte plutôt les femmes, tel qu’observé chez notre patiente, cependant avec un âge d’apparition plus tardif (âge médian : 43,5 ans) [8,9].
L’aspect cutané est varié et non spécifique à l’origine de difficultés diagnostiques. Les lésions siègent préférentiellement au niveau de la région cervicocéphalique et la partie haute du tronc. Elles se manifestent habituellement par des papules, nodules ou des plaques infiltrées de couleur rouge-brun parfois jaune et en nombre variable. Ces lésions sont habituellement asymptomatiques [9]. Ont également été rapportés, plus rarement, des lésions acnéiformes, xanthomateuses, psoriasiformes et des lésions de panniculites [10,11].
Chez notre patiente, il s’agissait initialement de lésions papuleuses du menton étiquetées « acné » réalisant secondairement une plaque infiltrée au niveau du visage.
Le diagnostic repose sur l’analyse histologique, l’orientation diagnostique par les cliniciens permet la recherche des signes histologiques caractéristiques en objectivant au niveau cutané un infiltrat dermique polymorphe, renfermant des lymphocytes, des plasmocytes, des polynucléaires et surtout de volumineuses cellules histiocytaires qui sont constamment CD68 (+), CD1a (−) ; et le plus souvent PS100 (+) en immunohistochimie ; associé à des images d’empéripolèse qui sont très suggestives de la maladie et correspondent à la pénétration active d’un lymphocyte ou d’un plasmocyte dans la cellule histiocytaire plutôt que sa phagocytose (figure 3).
Figure 3 : Plages, amas, ou cellules isolées d’histiocytes polygonaux avec empéripolèse et infiltrat inflammatoire floride avec des plasmocytes en plages ou bande. (Kong YY, Kong JC, Shi DR, Lu HF, Zhu XZ, Wang J et al. Cutaneous Rosai-Dorfman Disease : A Clinical and Histopathologic Study of 25 Cases in China. Am J Surg Pathol 2007 ; 31(3):341-35).
D’autres signes peuvent être décrits comme la présence de granulomes épithélioïdes [12,13,14]. C’est l’ensemble de ces critères qui permet de poser le diagnostic.
Le diagnostic différentiel de la maladie de Rosai-Dorfman cutanée peut se poser avec d’autres pathologies parmi lesquelles l’acné en raison de la non spécificité du tableau clinique, d’autres diagnostics peuvent être évoqués comme la sarcoïdose et la tuberculose d’autant plus que l’histologie peut comporter des granulomes tuberculoïdes à l’origine de difficultés et retard diagnostic qui peut aller de quelques mois à plusieurs années [15].
Un retard diagnostic de plusieurs mois était noté chez notre patiente chez laquelle la présentation initiale a fait évoquer le diagnostic d’acné, par la suite celui de sarcoïdose, puis de tuberculose cutanée devant l’aspect anatomoclinique. La relecture des lames histologiques a permis de confirmer le diagnostic.
Les anomalies biologiques fréquemment observées au cours de la forme systémique telles que le syndrome inflammatoire, hypergammaglobulinémie poly clonale, et hyper leucocytose à polynucléaires neutrophiles et une anémie ne sont pas retrouvés dans la forme cutanée pure de la maladie. Un bilan exhaustif initial est nécessaire car la maladie peut toucher tous les organes, d’où l’intérêt du TEP scanner au 18 FDG qui permet de réaliser un bilan lésionnel initial complet. Notre patiente n’avait aucune anomalie biologique ni radiologique rejoignant les données de la littérature [6]. L’évolution est dans la majorité des cas bénigne, la résolution spontanée en quelques mois ou années est la règle, parfois entrecoupée de poussées suivies de rémission [15,16].
Le traitement de la maladie n’est pas codifié en raison de l’hétérogénéité des modalités thérapeutiques publiées. L’abstention thérapeutique est souvent justifiée.
Le traitement peut être nécessaire en cas de gêne esthétique ou psychologique. Les atteintes cutanées sont particulièrement résistantes aux traitements avec une réponse dans 29 % des cas uniquement [6].
Plusieurs options ont été utilisées : la chirurgie, la cryothérapie, la corticothérapie locale ou générale, le laser, la radiothérapie, la dapsone, thalidomide, l’isotrétinoine et l’imatinib [17,18,19].
L’utilisation du méthotrexate seul ou en combinaison avec d’autres agents a été rapportée dans 9 cas de Rosai- Dorfman systémique, entrainant une réponse complète à partielle dans la plupart des cas. En revanche, dans les formes purement cutanées le traitement par méthotrexate a été plus rarement utilisé avec une efficacité moindre [20,21,22,23].
Dans notre cas, nous avions choisi l’association corticoïde-méthotrexate en raison du préjudice esthétique. Ce traitement a entrainé une légère amélioration au bout de 3 mois avec une bonne tolérance du traitement. Notre observation illustre d’une part les difficultés diagnostiques, avec un diagnostic souvent retardé, et souligne les difficultés de prise en charge rencontrées au cours de cette variante cutanée exclusive de la maladie de RDF.
Conclusion :
La maladie de Rosai–Dorfman est une entité rare, souvent méconnue et dont la présentation clinique peut être variable, source de difficultés et de retard diagnostic en particulier en cas de forme exclusivement cutanée de la maladie. L’analyse histologique permet de confirmer le diagnostic. Une meilleure compréhension de la pathogénie pourrait conduire à des stratégies thérapeutiques bien codifiées et consensuelles.
Références :
Destombes P: Adenitis with lipid excess, in children or young adults, seen in the Antilles and in Mali (4 cases). Bull Soc Pathol Exot Filiales 58: 1169 1175, 1965 (In French).
Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
Brenn T, Calonje E, Granter SR, Leonard N, Grayson W, Fletcher CD, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermato- pathol 2002;24:385-91
Wang KH, Chen WY, Liu HN, Huang CC, Lee WR, Hu CH. Cutaneous Ro- sai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277–86
Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphade- nopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73 [6]F. Cohen Aubart et al. La maladie de Destombes-Rosai-Dorfman : évolution du concept, classification et prise en charge / La Revue de médecine interne (2017) [7]Chu P, Le Boit PE. Histologic features of cutaneous sinus histiocytosis (Rosai- Dorfman disease): study of cases both with and without systemic involvement.J Cutan Pathol. 1992;19:201-206
Kong Y, Kong J, Shi D, Lu H, Zhu X, Wang J, et al. Cutaneous Rosai-Dorfman disease a clinical and histopathologic study of 25 cases in China. Am J Surg Pathol. 2007;31:341-50
Lu CI, Kuo TT, Wong WR and Hong HS: Clinical and histo¬pathologic spec- trum of cutaneous Rosai Dorfman disease in Taiwan. J Am Acad Dermatol 51: 931 939, 2004
Puppin D Jr, Chavaz P and Harms M: Histiocytic lympho¬phagocytic pannicu- litis (Rosai Dorfman disease): a case report. Dermatology 184: 317 320, 1992.
Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman disease: tumor biology, clinical features, pathology, and treatment.Cancer Control. 2014;21:322-327
Chu P, Leboit P. Histologic features of cutaneous sinus histiocytosis (Rosai- Dorfman disease): study of cases both with and without systemic involvement. J Cut Pathol 1992;19:201-6.
Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016;127:2672-81
Gameiro A, Gouveia M, Cardoso JC, Tellechea O. Histological variability and the Importance of clinicopathological correlation in cutaneous Rosai-Dorf- man disease. An Bras Dermatol 2016;91:6347.
Vuong V, Moulonguet I, Cordoliani F, Crickx B, Bezier M, Vignon-Pennamen MD, et al. Cutaneous revelation of Rosai-Dorfman disease: 7 cases. Ann Dermatol Venereol 2013;140:83-90
Chang LY, Kuo TT, Chan HL. Extranodal Rosai-Dorfman disease with cutaneous, ophthalmic and laryngeal involvement: report of a case treated with isotretinoin. Int J Dermatol. 2002;41:888-891
Chan CC and Chu CY: Dapsone as a potential treatment for cutaneous Rosai Dorfman disease with neutrophilic predom¬inance. Arch Dermatol 142: 428 430, 2006
Tjiu J-W, Hsiao C-H, Tsai T-F. Cutaneous Rosai– Dorfman disease: remis- sion with thalidomide treatment. Br J Dermatol 2003;148(5):1060–1 [20]Gebhardt C, Averbeck M, Paasch U, Ugurel S, Kurzen H, Stumpp P, et al. A case of cutaneous Rosai-Dorfman disease refractory to imatinib therapy. Arch Dermatol 2009; 145: 571–574.
Nasseri E, Belisle A, Funaro D. Rosai–Dorfman disease treated with metho- trexate and low- dose prednisone: case report and review of the literature. J Cutan Med Surg 2012;16(4):281–5
Sun NZ, Galvin J, Cooper KD. Cutaneous Rosai-Dorfman disease success- fully treated with low-dose methotrexate. JAMA Dermatol 2014; 150: 787–788 [23]Marion Nadal1, Thibault Kervarrec1 et all Cutaneous Rosai-Dorfman Di- sease Located on the Breast: Rapid Effectiveness of Methotrexate After Failure of Topical Corticosteroids, Acitretin and Thalidomid,Acta Derm Venereol 2015; 95: 758–759
S. ZOBIRI (1),H. HAMMADI (2). CHU Mustapha Bacha, Alger ; Hôpital Central de l’Armée, Ain Naâdja, Alger,
Abstract : The sun is known for its beneficial effects on the morale and the synthesis of vitamin D, however harmful effects of the UV are more and more debated. Sun is incriminated in skin aging, in DNA damage and in cutaneous carcinogenesis. Today, photoprotection has an important place and photo-protective products raise doubts about their real efficiency and their potential dangers. A good photo-protector is effective in the prevention of skin cancers, it must cover both UVA and UVB and must contain filters that are not toxic for humans and the ecosystem.
Key-words : Sun, Ultraviolet (UV), solar protection products, skin cancer.
Résumé : Le soleil est connu pour ses effets bénéfiques sur le moral et la synthèse de vitamine D, cependant ; les effets néfastes des UV sont de plus en plus débattus. Le soleil est incriminé dans le vieillissement cutané, dans les dommages de l’ADN et ainsi dans la carcinogenèse cutanée. La photoprotection occupe de nos jours une place importante et les produits photoprotecteurs suscitent des doutes sur leur réelle efficacité et sur leurs dangers potentiels. Un bon photoprotecteur est efficace dans la prévention des cancers cutanés, il doit couvrir aussi bien les UVA que les UVB et doit contenir des filtres non toxiques pour l’homme et l’écosystème.
Le soleil, étoile de notre système solaire, occupe une place importante depuis les civilisations égyptiennes et grecques jusqu’à nos jours. Autrefois, avoir une peau blanche était un signe de noblesse et on veillait dans les classes aisées à protéger sa peau du soleil pour ne pas être assimilés à des roturiers obligés de travailler et d’avoir ainsi un teint halé. Vers la fin du 19ème siècle, de par la place du travail dans la société et la pratique d’activités en plein air, le teint bronzé est devenu un effet de mode. Si on connait les bienfaits du soleil, ses effets néfastes sont découverts notamment sur la carcinogenèse cutanée [1].
Rappel sur le rayonnement solaire [2] :
Le rayonnement solaire est réparti de la manière suivante (figure 1) :
Les rayons infra rouges (800-5.000 nm) producteurs de chaleur ;
La lumière visible (400-800 nm), partie visible pour l’œil humain, qui se compose des couleurs de l’arc en ciel ;
Les ultraviolets sont non visibles à l’œil nu, on en distingue trois :
Les UVA (320-200nm) : représentent 95 % du rayonnement solaire qui atteint la terre. Ils pénètrent profondément la peau jusqu’au tissu conjonctif et peuvent ainsi induire le vieillissement cutané, ils stimulent également la synthèse de mélanine et sont responsables du bronzage et favorisent la production de radicaux libres qui sont cancérigènes.
À noter que les UVA ne sont pas arrêtés par le verre.
Les UVB (290-320 nm) représentent 2 % des rayons qui arrivent à la surface de la terre, ils atteignent le derme et sont arrêtés par le verre. Ils sont responsables de l’érythème solaire (coup de soleil) et augmentent le risque de cancer cutané lors des expositions prolongées.
Les UVC (190-290 nm) sont arrêtés par la couche d’ozone.
Le phototype permet de classer les individus selon la réaction de leur peau lors de l’exposition solaire,
6 phototypes ont été identifiés, correspondant à 6 types de peau et couleur de cheveux (tableau 1). Plus le phototype est faible, plus le sujet a besoin de se protéger du soleil [1].
Tableau 1 : Classification des phototypes
a. Notion d’indice UV :
Cet indice exprime l’intensité du rayonnement UV qui atteint la surface de la terre, plus l’indice est élevé, plus le risque de lésions cutanées et oculaires est important.
b. Notion de capital soleil :
C’est une notion abstraite définie par la quantité d’UV que la peau peut recevoir sans trop de dommages, elle est variable d’un individu à l’autre et dépend du phototype et des habitudes solaires.
Avantages et inconvenients du soleil :
a. Avantages :
Effet bénéfique sur le moral et le bien être, action anti-dépressive.
Action anti rachitique par stimulation de la synthèse de la vitamine D, une exposition quotidienne de quelques minutes en plein air est suffisante.
Le soleil a un effet bénéfique sur certaines pathologies comme le psoriasis.
a. Inconvénients :
Les effets néfastes du soleil sont représentés par le vieillissement cutané, les cancers cutanés et l’induction ou l’aggravation de photodermatoses [2].
Le vieillissement cutané :
Les UV entrainent une diminution de l’épaisseur de la peau qui devient fine et fragile, une diminution du nombre de mélanocytes et de cellules immunitaires. Le vieillissement photo- induit est appelé héliodermie donnant un aspect de peau fine, ridée et télangiectasique sur le visage et le dos des mains, des taches actiniques et des lentigos solaires (petites taches brunes sur les parties exposées au soleil) [4].
b2. Cancers cutanés :
Les cancers les plus fréquents sont les carcinomes cutanés divisés principalement en carcinomes basocellulaires et carcinomes épidermoïdes [5].
Lecarcinomebasocellulaire:
Le plus fréquent (80 % des cancers), d’évolution lente, locale, sans jamais de métastases. Ce cancer est lié aux expositions solaires intermittentes, aigues [6].
Lecarcinome épidermoïde
Touche le sujet âgé et se développe dans la majorité des cas en zone photo-exposée à partir d’une lésion pré-cancéreuse appelée kératose actinique (petites plaques érythémateuses, rugueuses) [7].
Les preuves de la responsabilité du soleil dans ce carcinome sont sa prévalence plus importante chez les phototypes clairs, sur zones photo-exposées. C’est un carcinome agressif, pouvant donner des métastases.
L’apparition du carcinome épidermoïde est liée à la dose totale cumulée d’UV, ce qui explique sa prévalence élevée chez le sujet âgé.
Lemélanome:
C’est une tumeur maligne développée aux dépens des mélanocytes, le facteur de risque majeur est représenté par l’exposition solaire, le phototype clair et la présence d’un grand nombre de naevi sont également des facteurs de risque [8].
b.3.Photodermatoses:
Les photodermatoses sont caractérisées par une sensibilité exagérée à la lumière, ainsi le soleil peut être à l’origine de dermatoses comme les lucites, il peut également aggraver ou déclencher une poussée de lupus ou de dermatomyosite [9].
Des effets néfastes sont également rapportés sur les yeux à type de photokératite et de conjonctivite [10].
Protection solaire (photo protection) :
Il s’agit de l’ensemble des moyens naturels ou artificiels permettant de se protéger des effets néfastes du soleil. Ce chapitre vise à répondre à 3 questions principales :
b. Qui protéger du soleil ?
Nous ne sommes pas tous égaux face au soleil, il faut identifier les sujets à risque représentés par les photo-types clairs (I, II et III), les enfants car leur peau est plus fine et ils possèdent moins de mélanine pour se protéger, les sujets possédant des antécédents personnels ou familiaux de cancers cutanés, les patients suivis pour une photodermatose comme le lupus, la dermatomyosite ou l’albinisme à titre d’exemple [9].
Certaines professions à l’air libre comme les marins ou les agriculteurs sont également des sujets à risque qu’il faut protéger.
c. Comment se protéger ? :
Les moyens de photoprotection sont naturels et artificiels.
Protection naturelle :
Les cheveux et la barrière cutanée assurent une protection contre les UV, ainsi les sujets chauves présentent des lésions précancéreuses précoces sur le cuir chevelu. La couleur de peau foncée permet une meilleure protection de par les volumineux mélanosomes répartis sur toute la hauteur de l’épiderme à l’opposé des peaux claires [11].
De plus, il existe au niveau de la peau des systèmes de réparation de l’ADN permettant d’éviter les dommages photo-induits [12].
Eviter de s’exposer entre 11h et 16h même si on reste à l’ombre sous un parasol, les UV sont réfléchis à 17% par le sable et 3 % par l’herbe. Les 2/3 des UV arrivent à la surface de la terre entre 11h et 16 heures.
Il faut se protéger dans certaines situations, par exemple, la neige réfléchit 90 % des UV en montagne car l’intensité des UV augmente avec l’altitude et même sous l’eau car à 50 cm de profondeur, 60 % des UV B et 85 % des UV A arrivent.
Protection vestimentaire :
Le chapeau et les lunettes permettent de protéger le visage et les yeux, il faudra choisir un chapeau à bords larges et des lunettes avec un pouvoir filtrant les UV, la teinte foncée des lunettes ne signifie pas une meilleure protection, elle permet juste d’éviter l’éblouissement (il ne faut pas confondre teinte et filtre).
Les vêtements assurent une bonne protection solaire en fonction de la nature du textile et de sa couleur, ainsi le jean est très efficace et les couleurs foncées ont un effet photoprotecteur plus important (à titre d’exemple, le facteur de protection d’une chemise en coton blanc est de 7 alors que celui d’un velours vert foncé est de 50) [13].
Les tissus secs et amples ont une meilleure protection que les tissus mouillés et moulants [14].
Les produits de protection solaire :
Un produit de photoprotection (écran solaire) est un produit appliqué sur la peau pour bloquer les UV, il est composé de filtres chimiques et/ou minéraux [2].
Un écran organique est constitué de molécules organiques qui absorbent les UV comme le benzophenone 3, le benzophenone 4.
Un écran chimique agit par un effet barrière qui réfléchit les UV, il est composé de dioxyde de titane (Ti O2) ou de monoxyde de zinc (Zn O).
Comment choisir un produit de protection solaire ?
Un bon photoprotecteur doit protéger aussi bien contre les UV A que les UV B. La protection contre les UV B est évaluée par le FPS (facteur de protection solaire ou sunburn factor protection).
Le FPS est mesuré au laboratoire par la capacité de protection contre les UV B donc contre les coups de soleil, il doit être ≥ 30. Par exemple, pour un phototype II, un coup de soleil sans photoprotection se voit en 10 minutes, si le SPF du photoprotecteur est à 2 le coup de soleil apparaitra après 20 minutes (2 du SPFx10 minutes), un SPF à 30 permettra de retarder le coup de soleil à 300 minutes.
La protection contre les UVA est plus difficile à apprécier car les méthodes de calcul sont moins claires, il faudra vérifier sur le tube de l’écran solaire le logo UV A entouré d’un cercle signifiant que le produit protège contre ces rayons [15].
Attention, appliquer un photoprotecteur ne permet pas une exposition prolongée, car le coup de soleil constitue notre système d’alarme nous incitant à nous mettre à l’ombre, cependant, privés de ce dernier par le FPS ≥30, on a tendance à s’exposer plus longtemps.
Comment appliquer un écran solaire ?
Il faut l’appliquer généreusement, 30 minutes avant l’exposition solaire et renouveler l’application après un bain ou une sudation intense. Il faut respecter la quantité préconisée qui est de 2 mg/cm2 car en réalité on applique une quantité moindre qui diminue ainsi l’efficacité. Cette quantité équivaut à 5 pressions pour un visage et à 3-4 pressions pour l’avant bras, il est préconisé de conserver le produit à l’abri de la chaleur. La forme galénique du produit dépend du type de peau, les crèmes sont préconisées pour les peaux sèches et les textures légères pour les peaux grasses, le spray ne doit pas être appliqué sur le visage car il possède une toxicité pulmonaire en cas d’inhalation (la pulvérisation ne doit pas se faire dans un endroit fermé).
Efficacité/innocuité des photoprotecteurs :
L’application habituelle d’un photoprotecteur permet la prévention des kératoses actiniques et du carcinome épidermoïde ainsi que le vieillissement cutané [16].
Les risques d’un écran solaire sont représentés par :
Des effets locaux assez rares (dermites allergiques de contact, dermites irritatives) [17].
Un impact écologique car un grand nombre de filtres anti UV est retrouvé dans les eaux naturelles avec une toxicité pour le corail et les micro algues.
Le benzophenone 3 (filtre chimique) a montré sur modèle animal et par voie orale des effets endocriniens, par voie cutanée, les études n’ont pas montré de passage systémique, cependant, la concentration du benzophenone 3 ne doit jamais dépasser 6 % et il ne doit pas être appliqué chez l’enfant. Les filtres minéraux sont plus sûrs (pas de pénétration cutanée en peau saine) et n’entraînent pas de photosensibilisation, cependant, ils ont un aspect inesthétique donnant à la peau un aspect blanchâtre.
En résumé, pour choisir un produit photoprotecteur, il faut :
Une protection contre les UV A et les UV B
Un bon SPF ≥ 30
Des filtres sûrs (Mexoryl® et Tinosorb®).
Photoprotection chez l’enfant :
Les recommandations américaines sont rapportées dans le tableau 2 [18]
Tableau 2 : Recommandations américaines de photoprotection chez l’enfant.
Photoprotection et vitamine D :
Un déficit en vitamine D pourrait théoriquement être évoqué devant l’utilisation d’un produit photoprotecteur au long cours, cependant, les études réalisées n’ont pas montré de carence en vitamine D lors de l’utilisation de ces produits [19].
Conclusion :
Le soleil reste un ami à condition de ne pas en abuser car c’est la surexposition qui est néfaste. Les personnes à risque doivent obligatoirement bénéficier d’une photoprotection, les bébés et les enfants ne doivent pas être exposés au soleil. Le produit de photoprotection doit être bien choisi en décryptant son emballage afin d’évaluer son efficacité et son innocuité.
Références :
H. Hammadi et col, L’apport de la dermatoscopie dans le diagnostic du carcinome épidermoïde in situ sur un terrain de kératose actinique ; thèse de DSM. Bibliothèque Virtuelle de l’université d’Alger
L. Meunier, Photoprotection (interne et externe), Encycl Med Chir 2008 ;98-944-A-10
Grange F. Épidémiologie des cancers cutanés en France. In : Guillot B, Dépistage et cancers cutanés ; 2008. p. 9-20.
E Grosshans, Carcinomes basocellulaires, Encycl Méd Chir, Derma- tologie, 98-620-A-10, 1999, 8 p.
Bonerandi, J.-J, Carcinome épidermoïde (spinocellulaire et ses pré- curseurs, Encycl Med Chirur Dermatol, 2011. Volume 6, Issue 1.
C. Gaudy-Marqueste, mélanome, Encycl Med Chir Dermatol, 98- 595-A-10
Amblard P. Photodermatoses. Photoprotection. Encycl Méd Chir. Pédiatrie, 4-115-A-10, 2001, 10 p.
Gadioux-Madern F, Eschard C. Oeil et soleil. Ann Dermatol Vene- reol 2007 ;134:4S81-4S85
Meynadier J. Photoprotection naturelle. In : Société Française de Photodermatologie. Paris : Arnette ; 2003. p. 125-9.
Césarini J. Génotoxicité UVA-UVB : les systèmes de réparation. In : Société Française de Photodermatologie. Paris : Arnette ; 2003.p. 49-53.
Morison WL. Photoprotection by clothing. Dermatol Ther 2003 ;16:16-22.
Jeanmougin M. Photoprotection vestimentaire. Nouv Dermatol 2005 ; 24:10-3.
Césarini J. Méthodes d’évaluation de la photoprotection externe. Coefficients de protection. In : Société Française de Photodermatolo- gie. Paris : Arnette ; 2003. p. 147-55.
Béani J. Photoprotection et cancers cutanés. In : Société Française de Photodermatologie. Paris : Arnette; 2003. p. 173-80.
Leonard F, Adamski H, Bonnevalle A, Bottlaender A, Bourrain JL, Goujon-Henry C, et al. Étude prospective multicentrique 1991-2001 de la batterie standard des photopatch-tests de la Société Française de Photodermatologie. Ann Dermatol Venereol 2005 ;132:313-20.
Adamski H, Stalder JF. Modalités pratiques de photoprotection de l’enfant. Ann Dermatol Venereol 2007 ;134:4S86-4S87.
Marks R, Foley PA, Jolley D, Knight KR, Harrison J, Thompson SC. The effect of regular sunscreen use on vitamin D levels in an Austra- lian population. Results of a randomized controlled trial. Arch Der- matol 1995;131:415-21.
H. HAMMADI (1), S. ZOBIRI (2),S. DJOUDI (2), Service de Dermatologie, Hôpital Central de l’Armée, Aïn Naâdja, Alger Service de Dermatologie, CHU Mustapha Bacha, Alger.
Abstract : Dermoscopy is a noninvasive, rapid and reproductive technique. It provides a new understanding of clinical morphology of skin lesions. It is based on the use of an optical system which allows an obliquely lighting and eliminates the reflection of the stratum corneum. It also allows, thanks to enlargement, to new semiological criteria. Thus, dermoscopy is an extension of our visual diagnosis, which provides the ability to see through the lesion and not just on the surface. The semiology of dermoscopic images is based on the analysis of the architecture and distribution of melanin in the epidermis and into the dermis. Initially used in melanocytic lesions, it is increasingly recommended in non-melanocytic lesions diagnosis. Dermoscopy is recommended in the most recent guidelines of melanoma management to help the clinician select lesions requiring biopsy or excision to establish a final histopathological diagnosis.
Résumé : La dermoscopie est une technique d’examen non invasive, rapide et reproductive. Elle permet une nouvelle appréhension de la morphologie clinique des lésions cutanées. Elle est basée sur l’utilisation d’un système optique qui permet un éclairage de manière oblique et supprime les reflets de la couche cornée. Elle permet également, grâce à un agrandissement, d’observer de nouveaux critères sémiologiques. Ainsi, la dermoscopie est un prolongement de notre diagnostic visuel, qui offre la possibilité de voir à travers la lésion et pas seulement en surface. La sémiologie des images dermoscopiques repose sur l’analyse de l’architecture et de la distribution de la mélanine dans l’épiderme et dans le derme. Initialement utilisée dans les lésions mélanocytaires, elle est de plus en plus recommandée dans le diagnostic des lésions non mélanocytaires. La dermoscopie est préconisée dans les directives les plus récentes de la prise en charge du mélanome pour aider le clinicien à sélectionner les lésions nécessitant une biopsie ou une excision en vue d’établir un diagnostic histopathologique définitif.
La dermoscopie ou dermatoscopie (connue aussi sous le nom de microscopie de surface ou microscopie par épiluminescence) est une méthode simple, facile et surtout utile dans le diagnostic des lésions pigmentées de la peau, notamment à un stade précoce de mélanome. Le dermoscope permet une visualisation in vivo non invasive de structures cutanées dermo-épidermiques, non accessibles à l’œil nu. Dans les mains d’utilisateurs expérimentés, la dermoscopie peut améliorer la précision du diagnostic du clinicien [1,2], et permet de faire une bonne corrélation anatomo-clinique [3] des lésions cutanées pigmentées et non pigmentées.
Principes de dermoscopie et équipement dermoscopique :
La dermoscopie utilise un microscope portatif appelé dermatoscope (ou dermoscope), qui est équipé d’une lentille grossissante et d’une source de lumière (figure 1).
Son utilisation nécessite un liquide d’immersion qui s’interpose entre la peau et la lame de verre de sa lentille. Ce dispositif permet à l’observateur d’examiner la morphologie de la surface des lésions cutanées.
Quand bien même l’inspection visuelle sans aide (à l’œil nu) de la peau permet au clinicien d’apprécier les caractéristiques morphologiques élémentaires des lésions, telles que leurs tailles, formes, couleurs, contours, la dermoscopie quant à elle, permet de visualiser les structures de la couche cornée et du derme superficiel.
Figure 1 : Quelques modèles de dermoscopes à main (H. P. Soyer, G. Argenziano, R. Hofmann-Wellenhof, R. H. Johr (Eds) Color Atlas of Melanocytic lesions of the Skin). (H. P. Soyer, G. Argenziano, R. Hofmann-Wellenhof, R. H. Johr (Eds) Color Atlas of Melanocytic lesions of the Skin
Aspects physiques/Principes de dermoscopie :
La dermoscopie fournit des informations supplémentaires au-delà de la simple évaluation de la lésion à travers une loupe simple.
Pour comprendre comment la dermoscopie fournit ces informations, il est nécessaire de saisir les principes optiques impliqués, en particulier les interactions de la lumière avec la peau.
La lumière visible est reflétée par la surface de la peau en raison de son indice de réfraction et de sa densité optique plus élevés que ceux de l’air ambiant [4].
La lumière est, soit reflétée à la surface, soit dispersée par le stratum corneum [5]. Par conséquent, les structures cutanées plus profondes ne peuvent pas être visualisées correctement (figure 2).
L’examen des lésions avec dermoscope nécessite un contact direct de la plaque de verre avec la peau, entre lesquelles la présence d’une interface liquide est requise. Cette installation remplace l’interface « peau-air » normale par une interface « peau-liquide ».
Les indices de réfraction dans l’interface « peau-liquide- verre » sont très proches. Cela réduit la réflexion de la lumière et permet de minimiser les reflets ; ce qui rend la couche cornée plus translucide [6] (figure2).
Cette configuration optique permet à l’observateur de voir des structures superficielles de la peau, non accessibles à l’œil nu. Aujourd’hui, La plupart des dermoscopes contiennent des diodes émettrices de lumière pour fournir une illumination, et sont équipés d’une lentille de grossissement.
Figure 2 : La quantité de lumière réfléchie par la surface cutanée est diminuée après utilisation d’un liquide d’immersion, (collection Pr. H. Hammadi)
Structures de base et corrélations histopathologiques en dermoscopie :
La sémiologie des images dermoscopiques est basée surtout sur l’analyse de l’architecture et de la distribution de la mélanine dans l’épiderme et dans le derme, ainsi que sur l’analyse de la disposition et de l’aspect des vaisseaux capillaires cutanés.
Les couleurs : La mélanine est le principal chromophore des lésions pigmentées de la peau. En fonction de sa concentration et de sa profondeur, la couleur perçue par le dermoscope varie du noir au bleu (effet Tyndall) [4].
Elle est noire au niveau des couches superficielles de l’épiderme. La mélanine est perçue comme brun clair à brun foncé dans la jonction dermo-épidermique. Par contre, elle est visualisée en bleu ou en gris dans le derme. L’hémoglobine parait rouge et correspond à des structures vasculaires. La fibrose et la régression sont représentées par la couleur blanche (figure 3)
Figure 3 : La couleur des chromophores de la peau vue en dermoscopie (collection Pr. H. Hammadi)
Réseau pigmenté : Le réseau pigmenté est une structure qui a la forme d’un nid d’abeille, composée de mailles (lignes) pigmentées et d’entre-mailles (trous) hypopigmentées [5,6,7] (figure 4). La réticulation (réseau) représente les crêtes épidermiques qui sont très chargées par le pigment mélanocytaire des kératinocytes ou des mélanocytes. Alors que les entre-mailles hypopigmentées correspondent aux sommets des papilles dermiques pauvres en pigments mélanocytaires. Le réseau pigmenté typique (bénin) est uniforme avec une réticulation régulière, de couleur homogène, qui s’atténue progressivement vers la périphérie (figure 4). Un réseau pigmenté atypique est hétérogène avec des mailles élargies et hétérogènes (couleur et forme), et il s’arrête brusquement en périphérie [8] (figure 5).
Figure 4 : Réseau pigmenté régulier et homogène s’atténuant progressivement en périphérie. (collection Pr. H. Hammadi) Figure 5 : Réseau pigmenté atypique et hétérogène qui s’arrête brusquement en périphérie. (collection Pr. H. Hammadi).
Points : Les points sont des structures de petite taille, inférieure à 0,1 mm de diamètre. Ils peuvent être noirs, brun, gris, ou bleu-gris en fonction de la profondeur du pigment [7,8] (figure 6). Les granules bleu-gris ou bleus (aspect poivré) correspondent à des particules très fines de mélanine, contenues dans les mélanophages, ou libres dans le derme papillaire, ou dans le derme réticulaire [9].
Figure 6 : Image dermoscopique d’un mélanome avec des points en périphérie. (collection Pr. H. Hammadi)
Globules : Les globules sont des structures rondes ou ovales, bien délimitées, qui peuvent être de couleur brune, noire ou rouge. Ils ont un diamètre supérieur à 0,1 mm et correspondent à des thèques mélanocytaires pigmentées ou à un conglomérat de mélanine et/ou à des mélanophages situés à la jonction dermo-épidermique ou dans le derme papillaire [10] (figure 7). Dans les lésions bénignes, les points et les globules ont une forme régulière et sont distribués de façon homogène (souvent au centre de la lésion). Dans le cas des mélanomes, ils sont plutôt de taille et de forme irrégulières, et se trouvent en périphérie de la lésion [10].
Figure 7 : Dermoscopie d’une lésion présentant une architecture globulaire « nævus dermique ». (collection Pr. H. Hammadi)
Stries ramifiées : Les stries ramifiées traduisent un réseau pigmenté altéré et perturbé dans lequel les mailles sont interrompues [7] (figure 8). Elles correspondent histologiquement à des cellules mélanocytaires situées dans des crêtes épidermiques communiquant entre elles.
Figure 8 : Dermoscopie d’une lésion montrant des stries ramifiées (flèche), et des zones sans structures (cercle). (collection Pr. H. Hammadi)
Courants radiaires : Les courants radiaires sont des extensions centrifuges qui partent du centre d’une lésion vers la périphérie [6] (figure 9). Histologiquement, ils représentent des thèques jonctionnelles de mélanocytes pigmentés.
Figure 9 : Dermoscopie d’un nævus de Spitz montrant des pseudopodes (flèche), et des stries (cercle). (collection Pr. H. Hammadi)
Pseudopodes : Les pseudopodes sont des structures formées par une ligne avec une extrémité renflée qui se trouvent en périphérie d’une lésion (figure 9). Ils correspondent à des thèques confluentes ou jonctionnelles de mélanocytes [6,11].
Stries:Les stries correspondent à des courants radiaires ou à des pseudopodes [7,9]. Elles ont la même signification histologique. Les stries peuvent être distribuées de façon symétrique à la périphérie de la lésion évoquant un nævus de Reed (variante pigmentée d’un nævus de Spitz) (figure 9). Au cours d’un mélanome elles sont distribuées de façon segmentaire (une partie de la lésion) [12].
Zones sans structures : Une zone sans structure est une zone qui ne contient pas les structures dermoscopiques de base (globules, réseau pigmenté, etc.) (figure 8). Elle est classiquement hypopigmentée à cause de l’absence ou de la diminution de l’intensité du pigment [7].
Taches d’encre : La tâche d’encre (blotch) est une structure noire foncée qui cache les éléments sous- jacents (figure 10). Elle correspond histologiquement à une concentration très importante de mélanine dans l’épiderme et/ou dans le derme [13].
Figure 10 : Image dermoscopique d’un mélanome avec une zone fortement pigmentée couvrant les structures sous-jacentes. (collection Pr. H. Hammadi)
Zone de régression : La régression est une dépigmentation cicatricielle (plus claire que la peau avoisinante) +/- associée à un peppering (aspect poivré) (figure 11). Elle se traduit histologiquement par de la fibrose et de la mélanose dans le derme (ou dans les mélanophages). Il existe souvent un épaississement de l’épiderme [9,13].
Figure 11 : Dermoscopie d’une régression cicatricielle (flèche) associée à un peppering (cercles). (collection Pr. H. Hammadi)
Voile bleu-blanc : Le voile bleu-blanc est une pigmentation bleuâtre irrégulière et confluente en verre dépoli. Le voile est inhomogène et il occupe une partie de la lésion (figure 12). Il s’agit histologiquement d’une agrégation de cellules fortement pigmentées (mélanocytes atypiques et/ou mélanophages) ou de la mélanine dans le derme, associée à une orthokératose compacte [9,13].
Figure 12 : Dermoscopie d’une lésion montrant un voile bleu-blanc avec des structures de régression. (collection Pr. H. Hammadi)
Pseudo-kystes cornés / Pseudo-comédons : Les pseudo-kystes cornés sont des structures rondes, de couleur blanchâtre ou jaunâtre qui correspondent à des kystes intra-épidermiques remplis de kératine [5,6]. Les pseudo-comédons sont des dépressions de couleur noire ou brune qui correspondent histologiquement à des invaginations de l’épiderme remplies de kératine. Ces deux structures se voient essentiellement au cours des kératoses séborrhéiques [14] (figure 13).
Figure 13 : Dermoscopie d’une kératose séborrhéique, montrant des montrant des pseudo-kystes et des pseudo-comédons. (collection Pr. H. Hammadi)
Fissures / aspect cérébriforme : Des fissures se voient dans les lésions de kératose séborrhéique. Cet aspect est dû à des invaginations linéaires de l’épiderme remplies de kératine. De multiples fissures donnent un aspect cérébriforme [14] (figure 14).
Figure 14 : Des fissures au cours dans une kératose séborrhéique, donnant un aspect cérébriforme. (collection Pr. H. Hammadi)
Structures ressemblant à des empreintes digitales : Les kératoses séborrhéiques débutantes (et les lentigos actiniques) peuvent présenter des crêtes linéaires parallèles donnant un aspect d’empreintes digitales [14] (figure 15).
Bordure mordillée : Les kératoses séborrhéiques planes (surtout du visage) ont une bordure concave qui respecte l’orifice folliculaire. Cela se traduit par un aspect mordillé en périphérie de la lésion [14] (figure16).
Figure 15 : Aspect dermoscopique d’empreintes digitales dans une kératose séborrhéique débutante. (collection Pr. H. Hammadi) Figure 16 : Aspect mordillé en périphérie de la lésion : Image dermoscopique d’une kératose séborrhéique du visage. (collection Pr. H. Hammadi)
Structures en forme de feuilles : Les structures ressemblant à des feuilles sont des lignes radiaires ayant une base commune situées à la périphérie d’une lésion, de couleur brune ou bleu-gris [5,15]. Leur distribution rappelle la forme des doigts de la main (figure 17). Cette structure est assez spécifique d’un carcinome basocellulaire.
Histologiquement, elle correspond aux nodules épithéliaux pigmentés.
Figure 17 : Dermoscopie d’un carcinome basocellulaire avec de multiples zones en forme de feuille en périphérie. (collection Pr. H. Hammadi)
Structures en forme de roues dentées : Les structures en forme de roues dentées sont des structures en lignes radiaires qui convergent vers un point central, de couleur gris-bleu ou brune [15,16]. Cet aspect est très spécifique d’un carcinome basocellulaire (figure 18). Cela correspond à des thèques de cellules tumorales qui s’étendent à partir d’un épithélium folliculaire.
Figure 18 : Structures en forme de roue dentée dans un carcinome baso-cellulaire. (collection Pr. H. Hammadi)
Nids ovoïdes : Les nids ovoïdes sont des structures ovoïdes bien délimitées, confluentes, plus grandes que les globules, distribuées irrégulièrement dans la lésion [15,16] (figure 19). En l’absence d’un réseau pigmenté, les nids ovoïdes sont très suggestifs du diagnostic d’un carcinome basocellulaire.
Figure 19 : Dermoscopie d’un carcinome basocellulaire montrant des nids ovoïdes. (collection Pr. H. Hammadi)
Globules bleu-gris multiples : Les globules multiples bleu-gris sont des structures bien délimitées qui, en l’absence de réseau pigmenté, sont très suggestives d’un carcinome basocellulaire [15,16] (figure 20).
Figure 20 : Image dermoscopique d’une vascularisation en tronc d’arbre, très caractéristique d’un carcinome basocellulaire. Notant la présence de globules multiples bleu-gris. (collection Pr. H. Hammadi)
Patron vasculaire : Certaines structures vasculaires peuvent être visualisées dans les lésions pigmentaires. Elles peuvent se présenter sous formes de virgules, de points d’exclamation et de vaisseaux en forme d’épingle à cheveux [16]. La présence de plusieurs types de vascularisation dans une lésion mélanocytaire, et leur distribution hétérogène, sont en faveur d’une lésion maligne. La vascularisation en tronc d’arbre est très caractéristique d’un carcinome basocellulaire [15,16] (figure 20).
Diagnostics différentiels des lésions pigmentaires de la peau :
La démarche diagnostique des lésions pigmentaires noires en dermoscopie comprend deux niveaux de décision (Stolz) [7] (figure 21) :
Figure 21 : Procédure en deux niveaux pour le diagnostic différentiel des lésions pigmentées [7]. (Argenziano G, Soyer HP, De Giorgi V, et al. Dermoscopy : a Tutorial. 1st edn : EDRA, 2000
Le premier niveau a pour but de différencier les lésions mélanocytaires des lésions non mélanocytaires. Si une des structures évocatrices est présente, la lésion est considérée comme d’origine mélanocytaire.
Le second niveau vise à distinguer, au sein des lésions mélanocytaires, celles qui sont bénignes de celles qui sont malignes (mélanome).
Les structures à rechercher dans le premier niveau de décision (lésion d’origine mélanocytaire) sont : le réseau pigmenté (nævus jonctionnel), les globules agrégés (nævus dermique), une zone bleue diffuse homogène (nævus bleu) (figure 22), des stries (naevus de spitz), pseudo-réseau (visage), ou un patron parallèle (les paumes et les plantes).
Si ce n’est pas le cas, on procède à l’étape suivante qui consiste à répondre successivement aux questions suivantes [7] (figure 23) :
Figure 22 : Lésion bleue homogène diffuse, correspondant à un nævus bleu. (collection Pr. H. Hammadi)Figure 23 : Algorithme pour la différentiation entre une lésion mélanocytaire et non mélanocytaire [7]. (Argenziano G, Soyer HP, De Giorgi V, et al. Dermoscopy : a Tutorial. 1st edn : EDRA, 2000.)
Retrouve-t-on dansla lésion des pseudo- comédons et des pseudo-kystes cornés ? Dans ce cas la lésion doit être considérée comme kératose séborrhéique,
Existe-t-il de télangiectasies en forme de tronc d’arbre, de structures en forme de feuilles d’érable, de nids ovoïdes, de globules bleu-gris multiples, de structures en roue dentée ou d’une ulcération ? Si c’est le cas, en l’absence d’un réseau pigmenté la lésion doit être considérée comme un carcinome basocellulaire. Si on ne retrouve aucune de ces structures, on doit passer à la question suivante.
Retrouve-t-on la présence de globules rouges, rouges-bleu ou noirs ? la lésion doit être considérée comme angiomateuse (hémangiome ou angiokératome) (figure 24).
Figure 24 : Dermoscopie d’un hémangiome : lacunes de couleur rouge (collection Pr. H. Hammadi)
Si les réponses à toutes ces questions sont négatives, la lésion doit quand même être considérée comme lésion mélanocytaire.
Lorsque la lésion est considérée comme lésion mélanocytaire, on doit déterminer si elle est bénigne, suspecte ou maligne (le second niveau de décision). Pour cela de multiples algorithmes ont été élaborés.
Les algorithmes les plus utilisés pour l’analyse des patrons sont : la règle ABCD de dermoscopie [17], la liste italienne en 7 points [18], la méthode de Menzies [6], l’analyse des patrons révisée [8,19], et la liste autrichienne en 3 points.
La règle ABCD publié par Stolz en 1991 attribue un score aux quatre critères : l’asymétrie, les bordures, les couleurs, et les structures dermoscopiques, puis chacun d’entre eux est multiplié par un facteur défini. On additionne le tout pour obtenir un score total, permettant de classer la lésion en bénigne, suspecte ou maligne.
La liste italienne en 07 points (G. Argenziano) est considérée comme ayant le plus de sensibilité entre les mains de non-experts.
Elle est basée sur l’évaluation de 7 critères dont 3 majeurs, et 4 critères mineurs (tableau). Un score final de 3 ou plus est en faveur d’un mélanome.
Cet algorithme ne s’applique pas aux lésions pigmentées des paumes des plantes et au visage où d’autres critères de sémiologie dermoscopique sont utilisés.
La méthode de Menzies évalue la lésion en deux étapes. D’abord la symétrie et la couleur ou on considère toutes les lésions qui sont à la fois symétriques et qui ont une seule couleur, comme bénignes. Dans un deuxième temps on évalue les autres critères dermoscopiques.
La liste autrichienne en 3 points est une approche simple, avec une sensibilité élevée et une spécificité modérée. Elle comporte 3 critères : l’asymétrie, un réseau atypique, et des structures bleu-blanc. Une lésion pigmentée qui comporte deux de ces trois critères doit être excisée.
Un score final de 3 ou plus est en faveur d’un mélanome
Conclusion :
La dermoscopie complète l’examen clinique des lésions pigmentaires. Réalisée par un médecin formé à la sémiologie des images dermoscopiques, elle augmente significativement la performance du diagnostic de mélanome. Elle permet d’éviter les exérèses inutiles pour des lésions bénignes, et elle augmente le nombre d’exérèses de mélanomes à un stade précoce.
Références :
Bafounta, M.L., Beauchet, A., Aegerter, P. & Saiag, P., Is dermoscopy (epiluminescence microscopy) useful for the diagnosis of melanoma? Results of a meta-analysis using techniques adapted to the evaluation of diagnostic tests. Arch Dermatol, 2001, 137, 1343–50.
Kittler, H., Pehamberger, H., Wolff, K. & Binder, M., Diagnostic accuracy of dermoscopy.2002, Lancet Oncol, 3, 159–65.
Benvenuto-Andrade, C., Dusza, S.W., Agero, A.L., Scope, A., Rajad- hyaksha, M., Halpern, A.C. & Marghoob, A.A., Differences between polarized light dermoscopy and immersion contact dermoscopy for the evaluation of skin lesions. 2007, Arch Dermatol, 143, 329–38.
Anderson, R.R. & Parrish, J.A., The optics of human skin. J Invest Dermatol, 1981,77, 13–19.
Stolz W, Braun-Falco O, Bilek P, Landthaler M. Farbatlas der Der- matoskopie.1. ed. Berlin: Blackwell Wissenschaft, 1993.
Menzies SW, Crotty KA, Ingvar C, Mc Carthy W H. An Atlas of surface microscopy of pigmented skin lesions. Lac Graw Hill Austra- lia, 1996.
Argenziano G, Soyer HP, De Giorgi V, et al. Dermoscopy: a Tutorial. 1st Ed. : EDRA, 2000.
Pehamberger H, Steiner A, Wolff K. In vivo epiluminescence mi- croscopy of pigmented skin lesions. I. Pattern analysis of pigmented skin lesions. J Am Acad Dermatol 1987; 17:571-83.
Soyer HP, Smolle J, Hödl S, Pachernegg H, Kerl H. Surface micros- copy: A new approach to the diagnosis of cutaneous pigmented turia of the pseudopod in surface microscopy. Arch Dermatol 1995; 131:436-40.
Argenziano G, Scalvenzi M, Staibano S, et al. Dermatoscopic pit falls in differentiating pigmented Spitz naevus from cutaneous melanomas. Br J Dermatol 1999; 141 : 788-93
Yadav S, Vossaert KA, Kopf AW, Silverman M, Grin-Jorgensen C. Histopathologic correlates of structures seen on dermoscopy (epilu- minescence microscopy). Am J Dermatopathol 1993; 15:297-305.
Braun RP, Robinovitz H, Krischer J, et al. Dermoscopy of pig- mented seborrheic keratosis. Arch Dermatol 1987; 17 : 584-91.
Braun RP, Rabinovitz H, Oliviero M, et al. Dermatoscopie des lésions pigmentées; Ann Dermatol Venereol 2002;129:187–202
Nachbar F, Stolz W, Merkle T, Cognetta AB, Vogt T, Landthaler Met al. The ABCD rule of dermatoscopy. High prospective value in the diagnosis of doubtful melanocytic skin lesions. J Am Acad Der- matol 1994; 30:551-9.
Argenziano G, Fabbrocini G, Carli P, De Giorgi V, Sammarco E, Delfino M. Epiluminescence microscopy for the diagnosis of doubtful melanocytic skin lesions. Comparison of the ABCD rule of derma- toscopy and a new 7- point checklist based on pattern analysis. Arch Dermatol 1998; 134:1563-70.
Stolz W, Landthaler M. Classification, diagnosis and differential diagnosis of malignant melanoma. Chirurg 1994; 65:145-52.
A. KHELIL (1), A. CHIALI (1), H. MESSID (2), ZM. MAKRELOUF (1), NH. MAHMOUDI (1),A. SERRADJ (1), I. BENKAIDALI (3) Service de dermatologie-Vénéréologie, CHU Benaouda Benzerdjeb, Oran Service d’Épidémiologie et de Médecine Préventive, CHU Benaouda Benzerdjeb, Oran Service de dermatologie-Vénéréologie, CHU Mustapha Bacha, Alger.
Abstract : Introduction. Melanoma is a malignant skin tumor, infrequent it is however responsible for a very high mortality rate (75%). Our study aims to describe the epidemiological characteristics of the clinical, anatomo-pathological and evolutionary aspects of MM cases in Oran. Patients and methods. This is a retrospective study carried out on MM patients treated in the Dermatology Department of Benaouda Benzerdjeb CHU and 1st Novembre EHU of Oran, over a period of 15 years. Results. Plantar location of MM was predominant (78%) as well as acrolentiinous (32%) and nodular (30%) forms. Vital status of MM cases described mortality in more than half of the cases (51%). Tables (1, 2) summarize the main patients characteristics with MM and tumor. Conclusion. Our study had shown poor prognosis characteristics of MM in our population by the high frequency of thick MM, the existence of some anatomo-clinical forms with a reserved prognosis and a fairly high mortality rate.
Résumé : Introduction. Le mélanome (MM) est une tumeur cutanée maligne, peu fréquente. Cependant responsable d’un taux de mortalité très élevé. Notre étude a pour objectif de décrire les caractéristiques épidémiologiques des aspects cliniques, anatomo-pathologiques et évolutifs des cas de MM à Oran. Patients et méthodes. Il s’agit d’une étude rétrospective réalisée auprès des cas de MM pris en charge au service de Dermatologie du CHU Benaouda Benzerdjeb d’Oran, et de l’EHU 1er Novembre d’Oran sur une période de 15 ans. Résultats. La localisation plantaire des MM était prédominante (78 %) ainsi que pour ses formes acrolentigineuses (32 %) et nodulaires (30 %). Le statut vital des cas de MM avait décrit une mortalité chez plus de la moitié des cas (51 %). Les tableaux (1, 2) résument les principales caractéristiques des patients atteints de MM et de la tumeur. Conclusion. Notre étude avait mis en évidence des caractéristiques de mauvais pronostic du MM chez notre population par la grande fréquence des MM épais, l’existence de certaines formes anatomo-cliniques à pronostic réservé et un taux de mortalité assez élevé.
Le mélanome (MM) est une prolifération tumorale maligne qui se développe aux dépens des mélanocytes épidermiques. Il ne représente que 5 à 7 % des cancers cutanés. Cependant, il est responsable de 75 % des décès causés par ces cancers. Il est de bon pronostic s’il est détecté tôt alors que l’épaisseur tumorale est encore faible puisqu’on peut guérir un MM enlevé chirurgicalement à un stade précoce [1,2,3,4].
Dans le monde, l’incidence annuelle du MM atteint des sommets chez les blancs en Australie (40/100.000), mais très faible dans les pays où les sujets sont à peau noire ou jaune [5]. Aux États-Unis, l’incidence du MM est très élevée chez la population de race blanche : 21,6/100.000. Entre 1992 et 2005, 70.000 MM invasifs étaient diagnostiqués. On estime qu’un américain sur cinq aura au moins un cancer cutané au cours de sa vie (OMS 2009) et qu’un individu meurt d’un MM toutes les heures. Actuellement, la mortalité tend à se stabiliser en Amérique du Nord [6,7,8,9].
En Europe : Une augmentation importante de l’incidence et de la mortalité est enregistrée dans tous les pays depuis les années 1950 [10]. En Afrique : l’incidence du MM est relativement rare, malgré un ensoleillement intense, et en raison du phototype foncé de la population.
Le MM semble favorisé par l’immunodépression, l’albinisme et l’exposition solaire [11,12,13]. Dans les pays au sud de la méditerranée, l’incidence du MM serait de 0,4/100.000 habitants [14]. Au Maghreb les constats sont : 82 cas recensés sur une période de 19 ans au Maroc ; 131 cas sur une période de 10 ans en Tunisie et 30 cas sur 13 ans à Tunis [15,16,17].
La littérature disponible actuellement concernant l’origine de la maladie est déduite des études réalisées sur les populations des pays au nord de la méditerranée à faible ensoleillement, et à population blanche (blond vénitien), au phénotype et à culture différents des nôtres [5].
L’absence d’études comportant une méthodologie dans notre pays ainsi que dans tous les pays du Maghreb sur le MM, ranime l’interrogation sur la situation du MM en Algérie et précisément au sein de la population d’Oran et nous ont conduits à entreprendre cette étude.
Notre étude a pour objectif de décrire les caractéristiques épidémiologiques des aspects cliniques, anatomo-pathologiques et évolutifs des cas de MM à Oran.
Patients et méthodes
Il s’agit d’une étude d’observation, type descriptive, rétrospective, réalisée auprès de tous les cas de MM diagnostiqués et pris en charge au service de Dermatologie du CHU Benaouda Benzerdjeb d’Oran, et de l’EHU 1er Novembre d’Oran, durant une période de 15 ans (1er janvier 1998 au 31 décembre 2012).
Les critères d’inclusion : ce sont tous les cas de MM confirmés par l’étude anatomo-pathologique dont l’âge ≥ 18 ans, quel que soit le sexe et quel que soit le lieu d’origine et de la résidence.
Les critères d’exclusion : ce sont les dossiers médicaux incomplets et les cas de MM métastatiques sans tumeurs primitives.
Pour l’analyse des variables, nous avons utilisé le logiciel EPI-INFO- version 6.
Résultats
Un total de 100 cas de MM était colligé. Le MM avait touché avec prédilection la femme (54 %) avec un sex- ratio (H/F) = 0,85. L’âge moyen global de survenue au diagnostic était de 59 ± 3,2 ans et la tranche d’âge la plus atteinte était celle de 50 à 79 ans (63 %). Le phototype III était assez fréquent (40 %), et l’exposition solaire chronique était prédominante (78 %). La notion de traumatisme était décrite au siège du MM dans 41 % des cas. La localisation plantaire était majoritaire (78 %) et l’aspect ulcéro-bourgeonnant était rencontré chez la moitié des cas (50 %) (figure 1). Une nette prédominance des MM épais (Breslow > 1 mm : 95,7 %) était notée. Plus de 73% des cas avaient présenté une ulcération. Les formes acrolentigineuses (32 %) (figure 2) et nodulaires (30 %) étaient prédominantes. Plus de 40 % des cas s’étaient présentés avec des localisations secondaires au moment du diagnostic. Le statut vital des cas de MM avait décrit un taux de mortalité assez élevé (51 %).
Figure 1 : Mélanome d’aspect ulcéro-bourgeonnant et pigmenté de siège plantaire chez une femme de 79 ans Iconographie du service de Dermatologie-Vénéréologie CHU OranFigure 2 : Mélanome type acrolentigineux du talon chez un homme de 44 ans Iconographie du service de Dermatologie-Vénéréologie CHU Oran
Discussion
Le MM correspond à une prolifération maligne de mélanocytes normalement présents le long de la jonction
dermo-épidermique. Il peut soit apparaître de novo (70-80 %), soit résulter de la transformation maligne d’un nævus [18,19].
Dans les pays occidentaux, l’atteinte féminine du MM est prédominante. Elle est estimée entre 52,8 % à 55 % et un sex-ratio entre 0,8 et 0,98 [20,21,22,23].
De même que pour la Tunisie et le Maroc (60,6 % et 65 % respectivement) avec un sex-ratio similaire au Maroc (0,8) [24,25].
Rejoignant ainsi les résultats de notre cohorte. Par contre, une particularité était notée au Mali ; marquée par une prédominance masculine (56 %) [14].
La tranche d’âge la plus représentée dans notre cohorte était celle de 50 à 79 ans. Des résultats similaires sont décrits par d’autres auteurs dans des études internationales (50 à 74 ans) [14,22]. Pour l’âge moyen au moment du diagnostic, les résultats de notre cohorte ainsi que ceux des pays voisins, Maroc et Tunisie, concluent à une atteinte fréquente de l’adulte d’âge moyen (5ème décennie de la vie) [25,26].
Alors que cette moyenne d’âge est un peu plus élevée en France et au Mali (60,6 ± 15,9 et 60,37 ± 11,02 respectivement) [14,21].
La couleur de la peau joue un rôle important dans la survenue du MM. Les données d’actualisation de la littérature confirment que le risque de MM cutané est augmenté chez les sujets ayant un phototype cutané de type I ou II. Par contre, il est plus de dix fois moins fréquent chez les phénotypes foncés [27,28,29].
Le phototype III qui est naturellement protégé par la présence de la mélanine au niveau de la peau était prédominant dans notre cohorte. Confortant ainsi les résultats Maghrébins, où les phototypes foncés étaient fréquents : en Algérie, le phototype III et IV ; en Tunisie le phototype III (52,8 %) et au Maroc le phototype IV (73,9 %) [24,25,30].
L’action du soleil sur la peau est cumulative. L’apparition des effets chroniques dépend de deux facteurs : la dose totale de photons reçue et la qualité de photo-protection naturelle de l’individu [31]. L’exposition solaire chronique qui parait être protectrice d’après les données de la littérature, était retrouvée chez 78 % de notre population de par l’habitat ensoleillé, les habitudes de vie (tâches ménagères en plein soleil) et la nécessité professionnelle (cultivateurs).
La notion de traumatisme était fréquemment rapportée dans notre cohorte. L’hypothèse des traumatismes bien établie dans la survenue des tumeurs cutanées, reste très discutée. Pour la majorité des auteurs, le traumatisme ne serait en aucun cas à l’origine de la genèse du MM mais il attirerait surtout l’attention des malades sur un MM déjà constitué [32,33,34].
Comme il est décrit dans notre cohorte, la localisation plantaire du MM est fréquemment décrite voire prédominante dans les études Africaines et Maghrébines : 100 % au Mali, 70 % en Tunisie, 62 % en côte d’ivoire et 50 % en Algérie [35,25]. La grande majorité de ces pays partagent certaines habitudes comportementales telles que la marche à pieds nus qui expose aux microtraumatismes répétés au siège de la tumeur.
Au Maroc, Chraïbi décrit un aspect tumoral similaire à celui retrouvé dans notre cohorte où la tumeur est habituellement ulcéro-bourgeonnante [36].
L’épaisseur tumorale selon Breslow est un facteur indépendant, prédictif du risque de métastases ganglionnaires régionales, de récidives précoces et de décès [37,38].
Parmi les facteurs histo-pronostiques du MM présents dans notre cohorte sont : l’épaisseur tumorale selon Breslow et l’ulcération. Nous avons noté une fréquence très élevée des MM épais (> 1 mm). Du fait d’un retard diagnostique en rapport avec une consultation tardive dépassant pour la majorité des cas les 18 mois, comme il est noté dans toutes les séries maghrébines [25,26,30].
Probablement en rapport avec certaines croyances trbales, un manque d’information de la population sur les dangers du MM, ou il pourrait s’agir tout simplement d’un problème de proximité des centres de soins pour certains pays.
Nos résultats n’avaient pas rejoint les données de la littérature, qui confirment que les MM < 1 mm sont en hausse, que leur incidence (1988-1999) a doublé, et que la proportion des MM identifiés à un stade avancé avec des métastases a diminué [28,39,40].
L’ulcération selon Breslow peut fausser la mesure de l’épaisseur tumorale des MM qu’ils fins soient ou épais. Elle était présente chez la majorité des cas de notre cohorte, ce qui rejoint les résultats d’une étude Marocaine où elle est décrite par El Haouari (60 % des cas) [25]. Par contre, les chiffres français sont meilleurs et trouvent que la majorité des MM ne présentent pas d’ulcération (77,7 %), comme il est décrit dans le travail de MAS [21].
La classification anatomo-clinique des MM tente de résumer leurs profils évolutifs à cinq grandes variétés : le MM superficiel extensif (SSM), le MM nodulaire (NM), le lentigo malin de Dubreuilh (LM); le MM acral lentigineux (ALM), le MM des muqueuses (MLM) [41]. Dans la littérature française, nous retrouvons une fréquence assez élevée des formes anatomo-cliniques superficielles
du MM (SSM : 60 à 70 % des cas), contre une faible fréquence des MM épais (MM nodulaire : 10 à 20 % des cas) [42].
Témoins du diagnostic précoce et des résultats des campagnes d’information et de dépistage sur le MM.
En Afrique et au Maghreb, Swan et Samaila rapportent que les MM sont peu fréquents, de diagnostic tardif mais souvent épais de type nodulaire ou acrolentigineux [43,44].
Ce qui rejoint les résultats de notre cohorte, où une fréquence assez élevée des formes épaisses, à mauvais pronostic (acrolentigineuse et nodulaire) était retrouvée. Le MM prend naissance à la jonction dermo-épidermique et suit une progression biphasique. Il évolue dans une
première phase « horizontalement » au-dessus de la membrane basale sans risque métastatique et, dans une deuxième phase « verticalement » pénétrant profondément le derme, à haut risque métastatique [41].
Compte tenu des données de la littérature, Rivers et Gandini confirment que la proportion des MM métastasés d’emblée a diminué : 11 % en 1988-1990 versus 5 % en 1997-1999 [45,46].
Plus de 40 % des cas de notre cohorte avaient présenté des localisations secondaires au moment du diagnostic. Probablement favorisées, par la localisation relativement cachée de certains MM (plantaires, dorsales et muqueuses), ainsi que par la fréquence élevée des formes anatomo-cliniques épaisses. Ceci pourrait témoigner de la méconnaissance ou de la négligence des patients devant une lésion asymptomatique conduisant manifestement à un retard au diagnostic ; et pourrait également expliquer le taux élevé des décès dépassant la moitié des cas de notre cohorte. En sachant que la majorité des cas avaient consulté à un stade avancé de la maladie.
Limites de l’étude
Notre étude était menée dans une structure hospitalière et était donc sujette à des biais de sélection. Elle était rétrospective et en conséquence exposée à des biais de collecte des données (informations mentionnées sur les dossiers médicaux) ainsi que des biais de mémorisation quant à l’exactitude des données de l’interrogatoire.
Conclusion
Les MM épais, les formes anatomo-cliniques à mauvais pronostic et les taux de mortalité assez élevés étaient des facteurs de risque qui avaient caractérisé les MM chez notre population. L’ensemble de nos résultats se rapprochaient de ceux retrouvés dans les séries maghrébines et africaines et mériteraient la mise en œuvre d’une stratégie de prévention. Puisque la détermination des facteurs de risque propres à chaque population peut aider à lutter contre le MM et à améliorer la prise en charge des patients.
Tableau 1 : Répartition des principales caractéristiques des cas de mélanomesTableau 2 : Répartition des caractéristiques cliniques et anatomiques de mélanome
Références :
Cummins DL, Cummins JM, Pantle H and al. Cutaneous malignant melanoma. Mayo Clin Proc 2006; 81(4): 500-507.
Burgeiro A, Mollinedo F, Oliveira PJ. Ipilimumab and vemurafenib: two different routes for targeting melanoma. Curr Cancer Drug Targets. 2013;13(8):879-894]
Survie des patients atteints de cancers en France : étude des registres du réseau Francim, éditions Springer 2007.
Sober AJ, Fitzpatrick TB, Mihm MC et al. Early recognition of cutaneous melanoma. JAMA 1979; 242: 2795-9.
Bonnet-Blanc JM. Tumeurs cutanées épithéliales et mélaniques : Mélanomes. Ann Dermatol-Vénéréol 2008 ; 11s ; Vol135, n°11s ; pp.F147- F135.
Rouhani P, Hu S, Kirsner RS. Melanoma in Hispanic and black Americans. Cancer Control. 2008;15:248-253.
Pollack LA, Li J, Berkowitz Z et al. Melanoma survival in the United States, 1992 to 2005. J Am Acad Dermatol 2011; 65 (5 Suppl 1):S78-86.
Cabos-Siguier B. Étude de deux facteurs de transcription impliqués dans le développement des mélanomes et la formation de métastases : N Oct-3 et HIF-2a. Caractérisation structurale, recherche de partenaires et étude de leur interaction avec l’ADN. Doctorat de l’Université de Toulouse. Soutenue le 22 juillet 2009
Bataille V. Génétique et épidémiologie du mélanome. Ann Dermato Venereol. 2006 ; 133: 56-62.
De Vries E, Coebergh JW. Cutaneous malignant melanoma in Europe. Eur J Cancer 2004;40:2355-66.
Adegbidi H, Yedomon H, Atadokpede F and al. Skin cancers at the National University Hospital of Cotonou from 1985 to 2004. Int J Dermatol. 2007; 46(Suppl 1):26-29.
ME, Ebughe G. Cutaneous cancers in Calabar, Southern Nigeria. Dermatol Online J. 2009;15(4):11
Seleye-Fubara D, Etebu EN. Histological review of melano-carcinoma in Port Harcourt. Niger J Clin Pract. 2005; 8(2):110-113.
Coulibaly DK. Aspects épidémio-cliniques et histo-pathologiques du mélanome malin dans le service de dermatologie du CNAM, Bamako (Mali).Thèse soutenue 2008.
Ammar-Khodja A, Benkaidali I, Zouhair K, Lazrak S, EL Ouazani T, Lakhdar H, Fazaa B, Bouassida S, Kamoun MR, Zahaf A, Benosman A, Nouira R, Zili J, Doss N. Mélanome malin (MM) au Maghreb. Ann Dermatol Vénéréol 2003 ; 130(4) : 29-31.
El-Ouazzani T, Lakhdar H. le mélanome malin : expérience ma- rocaine. 20è Congrès de l’Association des Dermatologues et Syphili- graphes de langue française, Montréal : 8-11 juin 1992. Poster 1.
Gharbi R. Malignant in Tunisia. 17th wold Congress of Dermato- logy Berlin 24-29 Mai 1987. Volume of Abrastracts, part II; 213.
Lin j, Goto Y, Takata M. Polyclonality of BRAF mutations in pri- mary melanoma and the selection of mutant alleles during progression J Natl Cancer Inst., 2009 ; 101 :1423-7.
DI Cesare MP, Antunes A, Truchelet F. Melanome. Encycl Méd Chir, Dermatologie ; 98-595-A-10,2000:15p.
MAS L. Rôle du médecin généraliste dans le diagnostic du méla- nome : étude sur une base de population du nord-est de la France en 2008. Thèse présentée et soutenue publiquement le 22 octobre 2010.
Comte J, Debarre J, Defossez G et coll. Le mélanome de la peau. In- cidence et mortalité des cancers en Poitou-Charentes. Octobre 2012.
Les cancers en France en 2013. Epidémiologie des cancers ; Epi- démiologie du mélanome cutané. INCa 2013. Les données du cancer
; p53-57.
Mseddi M, Marrekchi S, Abdelmaksoud W and al. Epidemio-cli- nical profil of skin cancer in southern Tunisia. Tunisie médicale. ISSN 0041-4131. 2007; 85: 505-508.
El Haouari. A. les mélanomes cutanés et muqueux : A propos de 23 cas. Fès (Maroc). Thèse 2012.
Ammar Khodja A ;Benkaidali I(Service de dermatologie, CHU Mustapha.Alger.) Zouhair.K, Lazrak.S, (service de dermatologie.CHU Ibn Rochd Casablanca). Kammoun MR, Zahaf A, (service de der- matologie Tunisiens). Mélanome malin (MM) au Maghreb. La revue Médico Pharmaceutique no 32, 3éme Trim. 2004.
Bliss JM, Ford D, Swerdlow AJ et al. Risk of cutaneous melanoma associated with pigmentation characteristics and freckling: systematic overview of 10 case-control studies. International Melanoma Analysis Group (IMAGE) [archive], Int J Cancer, 1995; 62:367-76.
HAS. Stratégie de Diagnostic Précoce du Mélanome; Recomman- dation en Santé Publique Rapport D’évaluation ; Service évaluation médico-économique et santé publique Octobre 2006.
Bataille V, De Vries E. Melanoma-Part 1: epidemiology, risk fac- tors, and prevention [archive], BMJ, 2008;337:a2249.
Hammadi H, Serradj A, Bouadjar B, Ammar-Khodja A, Benkai- dali I et al. Profil du mélanome en Algérie: Etude multicentrique. La 9ème journée nationale de la Société Algérienne de Dermatologie Esthétique et Cosmétique. Alger avril 2013.
Zerguine R. Skin and the sun. Batna J Med Sci 2015; 2: x-x.
Kaskel P, Kind P, Sander S and al. Trauma and melanoma formation
: a true association? British Journal of Dermatology 2000; 143: 749-53.
Phan A, Touzet S, Dalle S and al. Acral lentiginous melanoma :
histopathological prognostic features of 121 cases. British Journal of Dermatology 2007; 157, pp311-318.
Phan A. Le mélanome acro-lentigineux: Etude rétrospective épi- démiologique, clinique et anatomo-pathologique, et recherche de fac- teurs pronostiques d’un sous-type rare de mélanome. Thèse soutenue publiquement le 04 mai 2011.
Boudghène-Stambouli O, Belbachir A. Le mélanome au Maghreb. Ann Dermatol vénéréol ; 1997, 124: 559.
Chraibi.R. Mélanomes au Maroc : Aspects épidemio-cliniques des cancers cutanés des extrémités. Med Maghreb 2008; 23 (160) 43-45.
Schmidt CR, Panageas KS, Coit DG and al. An increased number of sentinel lymph nodes is associated with advanced Breslow depth and lymphovascular invasion inpatients with primary melanoma. Ann Surg Oncol. 2009;16(4):948-52.
Willi JP, Matter M, Buchegger F et al. Sentinel lymph node invol- vement and a high Breslow index are independent factors of risk for early relapse of melanoma. 2007; 46(6):244-51.
Benoit-Covern. C. Evolution de l’incidence du mélanome en Seine-Maritime sur une période de 10 ans [thèse de médecine]. Rouen
: faculté mixte de médecine et de pharmacie de Rouen; 2003.
Halna JM, Grandadam M, Buemi A. Étude épidémiologique des cancers cutanés basée sur la population d’un département français de 1988 à 1996. Résultats du registre des cancers du Haut Rhin, 2000. Nouv Dermatol 2000;19(1):48-45.
Saurat J-H, Lipsker D, Thomas L et al. Dermatologie et infections sexuellement transmissibles. Elsevier Masson SAS; 2017 (pages 687- 688), ISBN : 978-2-294-74649-9. e-ISBN : 978-2-294-74882-0.
Item149-Tumeurs cutanées épithéliales et mélaniques : méla- nomes. Critères cliniques et histopathologiques du pronostic. Ann Dermatol vénéréol; 2012:139, A150-A157.
Swan MC, Hudson DA. Malignant melanoma in South Africans of mixed ancestry: a retrospective analysis. Melanoma Research 2003;13(4):415-419.
Samaila MOA, Rafindadi A H. Pattern of Cutaneous Malignant Melanoma in Zaria, Nigeria. Ann Afr Med 2006; 5(1):16-19.
Rivers JK. Is there more than one road to melanoma? Lancet 2004; 363: 728-30.
Gandini S, Sera F, Cattaruzza MS and al. Meta-analysis of risk factors for cutaneous melanoma: I. Common and atypical naevi. Eur J Cancer 2005;41:28-44.
H. HAMMADI, Service de Dermatologie, Hôpital Central de l’Armée, Aïn Naâdja, Alger
Abstract : Cutaneous carcinomas are the most common cancers of adults [1], they are mainly divided into basal cell carcinomas and squamous cell carcinoma (cutaneous). Cutaneous squamous cell carcinoma affects the skin and mucous membranes where their aggressive evolution is often more marked and may be life-threatening because of their metastatic capacity. Cutaneous squamous cell carcinoma affects the elderly and is the most common skin cancer after basal cell carcinoma, outpacing melanoma in men [2]. During the course of the CBC the evolution is essentially local with a good prognosis in the majority of the cases. The treatment of cutaneous carcinomas is primarily surgical, other alternatives can be considered depending on the depth, the size of the tumor. Dermoscopy is a new tool that provides immediate results very beneficial for the medical decision in dermatological practice. The prevention of cutaneous carcinomas is based on photoprotection and regular monitoring of patients at risk.
Résumé : Les carcinomes cutanés sont les cancers les plus fréquents de l’adulte [1], ils se divisent principalement en carcinomes basocellulaires (CBC) et carcinomes épidermoïdes cutanés (CEC). Les CEC touchent la peau et les muqueuses, où leur évolution agressive est souvent plus marquée, pouvant menacer le pronostic vital par leur capacité métastatique. Les carcinomes épidermoïdes cutanés (CEC) touchent les sujets âgés et sont les plus fréquents des cancers cutanés, après les carcinomes basocellulaires, devançant le mélanome chez l’homme [2]. Au cours du CBC, l’évolution est essentiellement locale avec un bon pronostic dans la majorité des cas. Le traitement des carcinomes cutanés est avant tout chirurgical, d’autres alternatives peuvent être envisageables en fonction de la profondeur, et de la taille de la tumeur. La dermoscopie est un nouvel outil qui fournit des résultats immédiats très bénéfiques pour la décision médicale en pratique dermatologique. La prévention des carcinomes cutanés repose sur la photoprotection et sur le suivi régulier des patients à risque.
Introduction/ Épidémiologie : À la différence du CBC, le CEC se développe le plus souvent sur des kératoses actiniques (KA) précancéreuses, d’où l’importance d’un diagnostic précoce à ce stade. Les données publiées en Europe, montrent une incidence standardisée des carcinomes épidermoïdes qui varie entre 10 à 20/100.000 chez l’homme, et 5 à 10/100.000 chez la femme. Une étude prospective en Champagne-Ardenne (France) [3] estime l’incidence annuelle brute du CEC à 30/100.000 dans la population générale. Elle est 4 fois inférieure à celle du carcinome basocellulaire.
Facteurs de risque de survenue des CEC et de leurs précurseurs :
Le CEC se développe dans la majorité dans cas en zones cutanées photo-exposées, à partir d’une kératoses actiniques dont il partage les facteurs de risque. Le risque de développer un CEC est manifestement plus important chez les phototypes clairs. Certains co-carcinogènes sont corrélés à des localisations particulières : tabac et CEC de la lèvre inférieure, HPV et CEC génitaux ou anaux.
Les CEC surviennent également avec une fréquence accrue chez les immunodéprimés : patients transplantés, sidéens, etc., ainsi qu’au cours de certaines génodermatoses.
Plus rarement, le CEC survient sur une ancienne cicatrice, une ulcération chronique, une inflammation cutanée chronique (ex. : maladie de Verneuil, radiodermite), une maladie de Bowen, ou de novo.
Formes anatomocliniques des carcinomes épidermoïdes et de leurs précurseurs :
Kératoses actiniques :
Elles réalisent de petites plaques érythémateuses ou pigmentées, rugueuses au toucher (figure 1). Le diagnostic histologique d’une kératose actinique repose sur la présence d’anomalies kératinocytaires variables : perte de la polarité avec modification de l’architecture épidermique, atypies cytonucléaires, cellules dyskératosiques, acantholyse [4].
L’épiderme peut être aminci ou hyperplasique. La lésion de KA est presque toujours associée à une élastose solaire dans le derme. Sur le plan cytologique, les anomalies sont discrètes et n’intéressent pas la totalité de l’épaisseur épidermique, ni les annexes. On ne parle de carcinome in situ que lorsque ces anomalies sont associées, marquées et intéressent la totalité de l’épaisseur épidermique [4].
Figure 1 : Kératose actinique non pigmentée : lésions érythémateuses rugueuses. Collection Pr. H. Hammadi.
Carcinomes épidermoïdes non invasifs (maladie de Bowen, kératoacanthome) :
Les carcinomes épidermoïdes, regroupent les tumeurs malignes cutanées primitives exprimant une différenciation malpighienne et distinctes des autres tumeurs épithéliales cutanées primitives. Selon la présence ou non d’un franchissement de la membrane basale épidermique et le niveau d’infiltration du derme, on distingue : le carcinome micro-invasif limité au derme superficiel, et le carcinome invasif infiltrant le derme moyen et/ou profond, voire l’hypoderme et les tissus sous-cutanés.
Maladie de Bowen : Elle siège sur la peau exposée ou non au soleil incluant la face, les membres inférieurs et les zones périunguéales ou sur les muqueuses génitales. Sur le plan anatomopathologique, [5]. L’épiderme est hyperplasique, désorganisé dans son architecture, constitué sur toute son épaisseur de kératinocytes atypiques. Elle évolue, après plusieurs années, en carcinome épidermoïde dans 3 à 28 % des cas selon les études.
Kératoacanthome : Le diagnostic de kératoacanthome repose classiquement sur l’association de critères cliniques et histopathologiques : il s’agit d’une tumeur d’apparition rapide, centrée par un cratère kératosique caractérisée par une organisation générale symétrique de la lésion autour du cratère central occupé par de la kératine. L’index mitotique est faible et la régression spontanée se fait en 2 à 4 mois.
Carcinomes épidermoïdes cutanés invasifs :
Les aspects anatomocliniques du CEC invasif sont polymorphes. Ils diffèrent par leurs morphologies cliniques, leurs aspects histologiques et par leurs comportements évolutifs.
Dans sa forme classique le CEC se présente sous forme d’une lésion ulcéro-végétante. Il s’agit d’une tumeur à la fois saillante et infiltrante, à surface irrégulière, siège d’une ulcération à fond bourgeonnant et saignotant (figure 2). Cassarino et coll. estiment le risque métastatique des carcinomes invasifs qui se développent sur une kératose actinique à 0,5 %. Histologiquement, la lésion montre des kératinocytes atypiques qui forment des bourgeons irréguliers, infiltrant le derme, et volontiers en connexion avec l’épiderme.
Les classifications disponibles dans la littérature correspondent à des descriptions d’auteurs, nous proposons de classer les CEC en deux groupes en fonction de leur pouvoir métastatique [6] ;
Le premier groupe est celui des CEC à faible pouvoir métastatique. Il comporte : le CEC ulcéro-végétant (la forme commune), le CEC verruqueux, le CEC à cellules fusiformes, le CEC métatypique, et le CEC mixte.
Le second groupe dont le pouvoir métastatique est plus important est représenté par : le CEC acantholytique, le CEC muco-épidermoïde et le CEC desmoplastique.
Figure 2 : Carcinome épidermoïde cutané : lésion ulcéreuse du scalpe. Collection Pr. H. Hammadi.
L’apport de la dermoscopie dans le diagnostic du CEC :
La dermatoscopie ou dermoscopie est une méthode simple, facile et surtout utile dans le diagnostic des lésions tumorales de la peau. Initialement utilisée dans les lésions mélanocytaires, elle est de plus en plus recommandée dans le diagnostic des lésions non mélanocytaires. Le dermatoscope permet une visualisation in vivo, non invasive de structures cutanées dermo-épidermiques, non accessibles à l’œil nu. Dans les mains d’utilisateurs expérimentés, la dermoscopie peut améliorer la précision du diagnostic du clinicien et permet de faire une bonne corrélation anatomo-clinique des lésions cutanées pigmentées et non pigmentées.
Les kératoses actiniques non pigmentées sur le visage montrent quatre caractéristiques dermoscopiques (patron en fraise) [7] (figure 3) :
Un fond d’érythème épargnant les follicules pileux, formant un «pseudo-réseau» rose à rouge (cercles blancs sur un fond rouge),
Squames blanc-jaunâtres,
Vascularisation fine (droite ou glomérulaire) entre les orifices pilaires,
Les ouvertures folliculaires entourées d’un halo blanc et rempli d’un bouchon kératosique jaunâtre (cercle blanc entourant une motte jaune)
Le pseudo-réseau est une structure retrouvée essentiellement au niveau du visage à cause de l’anatomie très différente de cette partie du corps. La peau du visage présente en effet des crêtes épidermiques aplaties avec une grande concentration d’ostiums folliculaires, des glandes sébacées et de structures annexielles.
Les entre-mailles hypopigmentées du pseudo-réseau correspondent à la présence des ostiums folliculaires et des structures annexielles. Dermoscopiquement, le diagnostic de la kératose actinique pigmentée est basé sur la présence d’un pseudo-réseau brun, noir ou gris situé entre les ouvertures ostiales, rempli de kératine, et la limite nette de la lésion.
Le CEC révèle généralement une masse centrale de kératine et une ulcération entourée par des vaisseaux [7]. Il présente fréquemment des patrons vasculaires similaires au kératoacanthome (en boucle ou en lignes sinueuses, pointillé, enroulé) [7]. L’ulcération et les croûtes de sang sont distribuées irrégulièrement sur la surface de la tumeur (figure 4). Il est possible de trouver le patron en fraise en contiguïté avec la lésion du CEC.
Figure 3 : Image dermoscopique d’une kératose actinique. Pseudo réseau rouge : Réseau rouge mêlé à de petites zones blanches arrondies correspondant aux ouvertures folliculaires. Collection Pr. H. Hammadi.Figure 4 : Image dermoscopique d’un carcinome épidermoide. Vaisseaux en épingle à cheveux ( vaisseaux en boucles tordues) + Ulcération. Collection Pr. H. Hammadi.
Pronostic des carcinomes épidermoïdes
La survenue d’une rechute ou de métastases au cours des CEC est le plus souvent liée soit à une prise en charge initiale inadaptée ou tardive (délai de consultation très long), soit en rapport avec des formes anatomo- cliniques agressives. Le risque métastatique des CEC en peau photoexposée est estimé à 2,3 % à 5 ans et à 5,2 % après 5 ans d’évolution.
Il n’existe pas de classification pronostique satisfaisante des CEC primitifs, car les études rétrospectives ne permettent pas de savoir si les variantes morphologiques sont un paramètre pronostique indépendant, notamment de l’épaisseur tumorale et de la profondeur d’invasion. Le groupe de travail sur le carcinome épidermoïde cutané de la Société Française de Dermatologie [6] propose la classification pronostique suivante (tableau 1).
Critères
Groupe 1 : à faible risque
Groupe 2 : à risque significatif
Cliniques
Primitif vs récidive Degré d’infiltration clinique Symptômes neurologiques d’envahissement
Statut immunitaire
Taille (diamètre) en fonction de la localisation
Primitif Absence Non
Immunocompétent < 10 mm en zone R+ < 20 mm en zone R-
Récidive Adhérence au plan profond Oui
Immunodéprimé 10 mm en zone R+ 20 mm en zone R-
Anatomopathologiques
Envahissement périnerveux Degré de différenciation cellulaire
Formes histologiques
Profondeur (niveau de Clark) Épaisseur
Non Bon
CEC commun, verruqueux, fusiforme (hors zone irradiée), mixte ou métatypique
Tableau 1 : Classification pronostique des carcinomes épidermoïdes cutanés.
CEC : carcinome épidermoïde cutané. Zone R+ : Zone à risque significatif (R+) : les zones péri-orificielles (nez, lèvres, oreille externe, paupières), les zones non insolées (périnée, sacrum, plantes des pieds, ongles), ou sur ra- diodermite, cicatrice de brûlure, inflammation chronique, ulcères chroniques. Zone R- : Zone à risque bas : autres localisations de l’extrémité céphalique, du tronc et des membres (zones photo-exposées).
Les carcinomes épidermoïdes peuvent être responsables de métastases locales, régionales ou à distance entraînant le décès. La dissémination par voie lymphatique est responsable de 80% des localisations métastatiques.
Traitement des carcinomes cutanés et des lésions précancéreuses [6] :
Il existe plusieurs alternatives thérapeutiques pour le traitement des carcinomes cutanés et des lésions pré-cancéreuses : la chirurgie, la radiothérapie, la cryochirurgie, le curetage électrocoagulation, la photothérapie dynamique, l’imiquimod topique, et le 5-fluoro- uraciletopique.
Le type de la tumeur, sa taille, son caractère récidivant et sa localisation sont des éléments déterminants dans le choix de la thérapeutique. La prise en charge des patients
doit tenir compte, en plus de l’objectif curatif, d’un confort nécessaire et d’un résultat esthétique acceptable [6].
Cryothérapie et cryochirurgie :
Il s’agit de méthodes de destruction tissulaire par le froid par l’azote liquide. Elle consiste à vaporiser de l’azote liquide sur une lésion dans le but d’obtenir une congélation. La cryothérapie est une méthode simple, rapide et peu coûteuse.
Curetage–électrocoagulation :
Le curetage–électrocoagulation est une technique de destruction rapide, qui comporte, une détersion de la lésion à la curette sous anesthésie locale, puis une coagulation à l’aide d’un bistouri électrique.
Lasers :
Le laser CO2, provoque par effet thermique, une vaporisation non sélective des tissus, avec perte de substance et nécrose de coagulation. C’est une méthode plus coûteuse, et moins accessible que le curetage- électrocoagulation.
Fluoro-uracile :
Le 5-FUcrème 5 % dispose d’une AMM pour le traitement des kératoses actiniques et des carcinomes in situ (CIS). Les effets indésirables les plus fréquemment observés sont des réactions locales (douleurs, prurit, brûlure au point d’application).
Imiquimod :
L’imiquimod crème à 5 % est commercialisé en sachets, permettant de couvrir une zone cutanée de 10-20 cm2 (un sachet/jour maximum). Les réactions locales sont fréquentes et peuvent conduire à espacer les applications.
Photothérapie dynamique (PDT) :
La PDT est un traitement non invasif destiné à détruire sélectivement par la lumière des cellules pathologiques ayant accumulé une substance photosensibilisante. Les molécules actuellement les plus utilisées par voie topique sont l’acide 5-aminolévulinique (5- ALA) et le méthyle aminolévulinate. Récemment, une variante de la PDT a été proposée (Daylight PDT), dans laquelle la crème méthyle aminolévulinate (MAL) est appliquée sur la peau et les patients s’exposent par la suite au soleil pendant 2 heures (avec un écran solaire).
Radiothérapie :
L’indication de la radiothérapie doit être posée avec précaution et adaptée selon : l’âge, l’autonomie, l’éloignement géographique du patient, la localisation et la taille du carcinome, la disponibilité et la spécificité du plateau technique du service de radiothérapie.
La difficulté de l’indication de la radiothérapie réside dans les séquelles radiques secondaires qui rendent difficile la prise en charge chirurgicale ultérieure.
Traitement chirurgical des carcinomes cutanés :
La chirurgie est le traitement de choix de ces tumeurs. Elle présente l’avantage de fournir une pièce d’exérèse permettant la confirmation histologique du diagnostic et la vérification de la qualité de l’exérèse, et d’obtenir un taux très élevé de contrôle local et la guérison d’une grande majorité de patients.
Elle reste le traitement de référence, auquel tous les autres traitements non chirurgicaux du carcinome épidermoïde cutané (CEC) doivent être comparés.
Chimiothérapie par voie générale des carcinomes cutanés :
La chimiothérapie a une place limitée, réduite aux échecs de la chirurgie et de la radiothérapie, liée à la mise en œuvre inadéquate ou trop tardive de ces traitements. Dans le guide du NHRMC [6], le cisplatine est la chimiothérapie de référence. La littérature sur ce sujet est très pauvre, réduite à quelques séries de petite taille et à des cas isolés.
Prévention des CEC et de leurs pré-curseurs :
La survenue d’un CEC et des kératoses actiniques est directement liée à l’exposition solaire et donc fonction de la latitude et du mode de vie. Donc, la réduction de la prévalence des KA et des CEC nécessite une meilleure prise de conscience pour améliorer les comportements de protection solaire.
Les patients qui ont eu un carcinome épidermoïde forment un groupe à haut risque d’avoir un autre carcinome. Ainsi, 52 % d’entre eux auront un autre cancer cutané dans les 5 ans.
Trakatelli [8] recommande la surveillance pendant 5 ans de ces sujets à risque, mais sans précision sur le rythme de la surveillance.
Le carcinome basocellulaire CBC
Introduction/ Épidémiologie
Les CBC sont aussi les cancers cutanés les plus fréquents, en particulier chez les sujets de phototype clair. Ces tumeurs sont souvent localisées sur les zones photoexposées et peuvent entraîner un lourd préjudice fonctionnel et esthétique. En France Le taux d’incidence dans le Haut-Rhin était de 100/ 100.000 habitants par an [2]. En Australie, l’incidence est augmentée à 400 cas pour 100.000 habitants par an. La plupart des CBC surviennent après 50 ans (âge moyen au tour de 65 ans) ; sans prédominance de sexe. Le CBC ne survient pas sur une lésion précancéreuse.
Facteurs étiologiques [10] :
Le soleil : 80 % d’entre eux surviennent sur des zones photoexposées. Deux types d’expositions solaires peuvent être néfastes : les expositions intermittentes aiguës sur une peau non préparée (coups de soleil sur une courte période de vacances), ou l’exposition chronique (expositions répétées sur de longues années au rayonnement solaire).
Prédisposition génétique : le CBC est plus fréquent chez les sujets à peau claire (phototype I et II)
Les autres agents carcinogènes : Ce sont essentiellement les rayons ultra-violets (cabines UV, photothérapie).
Les greffés d’organes font plus de CBC que la population générale du fait de l’immunosuppression.
Il existe des maladies congénitales rares prédisposant à leur développement comme le xeroderma pigmentosum, et le syndrome des hamartomes basocellulaires.
Aspects clinques des carcinomes basocellulaires [10] :
Cliniquement le CBC survient dans les zones photoexposées dans plus de 85% des cas. Le CBC donne rarement des métastases, mais il possède un potentiel invasif localement, pouvant entraîner une destruction tissulaire importante. Il n’est jamais localisé sur les muqueuses.
Il existe plusieurs variétés anatomo-cliniques de gravité variable.
Le CBC nodulaire (60 % des cas). Papule arrondie translucide et télangiectasique qui va s’étaler progressivement (figure 5). Il siège n’importe où, mais il est plus fréquent dans les zones photoexposées (90 %).
Le CBC superficiel ou pagétoïde. Plaque érythémateuse plane, bien limitée à extension très lente.
Le CBC sclérodermiforme. Une plaque blanchâtre dure, aux limites imprécises qui ressemble à une cicatrice
Les formes pigmentées ou tatouées : Comme le nom l’indique, ces lésions contiennent un fort pigment brun ou noir et sont plus fréquentes chez les personnes à la peau foncée
3. Les formes ulcéreuses
Lesformestérébrantes.
Figure 5 : Aspect clinique d’un carcinome basocellulaire nodulaire. Collection Pr. H. Hammadi.
Anatomie-pathologique : Typiquement, le CBC est formé d’amas cellulaires compacts de petites cellules basophiles à limites nettes, à disposition périphérique palissadique. Ils peuvent s’associer à une certaine fibrose du derme. Les formes infiltrantes ou sclérodermiformes sont associées à un stroma dense et fibreux.
Dermoscopie du carcinome basocellulaire :
L’algorithme dermoscopique du CBC repose sur l’absence de réseau pigmenté associé à au moins l’un des six critères suivants : Les nids ovoïdes, l’aspect en roue dentée, l’aspect en feuille d’érable, la vascularisation en tronc d’arbre, les globules gris-bleu et la présence d’une ulcération [11].
Le CBC tatoué peut poser un problème de diagnostic différentiel avec le mélanome : en effet les zones rouge-blanchâtre avec des vaisseaux irréguliers, les lignes blanches, et les points brun-gris sont rencontrés dans les mélanomes régressifs [12]. La maladie de Bowen pigmentée est un autre diagnostic différentiel possible avec le CBC pigmenté. La présence de vaisseaux glomérulaires et des points brun-gris alignés oriente vers le diagnostic de maladie de Bowen pigmentée [12].
Cette méthode diagnostique permet entre autre de reconnaître les sous types de CBC (les CBC superficiels des non superficiels), lors de la décision thérapeutique qui dépend de manière significative du type histologique (traitement chirurgical ou non).
Plusieurs critères dermoscopiques ont été signalés comme étant plus fréquent dans les CBC superficiels que dans les CBC nodulaires, alors que d’autres se voient le plus souvent dans les tumeurs non superficiels [13].
Lallas et al. proposent les éléments dermoscopiques suivants pour établir le diagnostic de CBC superficiel [14] : présence de feuilles d’érable, de fines et courtes télangiectasies et l’absence de vaisseaux en tronc d’arbre, de nids ovoïdes gris-bleu et d’ulcération (sensibilité de 81,9 % et spécificité de 81,8 %) (Figure 6). Cet algorithme dermoscopique permet d’identifier de façon plus aisée les CBC superficiels.
Figure 6 : Dermoscopie d’un carcinome basocellulaire montrant des nids ovoïdes et d’une vascularisation en tronc d’arbre très caractéristique. Collection Pr. H. Hammadi.
Évolution / Pronostic de carcinome basocellulaire [15] :
Le CBC n’entraîne que rarement des métastases, mais il a un potentiel invasif local pouvant entraîner des destructions tissulaires voisines osseuses ou viscérales. L’évolution locale des CBC est caractérisée par l’extension progressive dans les tissus adjacents (derme, fascia, périoste, périchondre).
Le pronostic du CBC dépend du risque d’extension locale et de la fréquence des récidives. Ce risque est lié également aux difficultés thérapeutiques. Les facteurs pronostiques sont cliniques (la localisation, la taille, les limites ) et histologiques (les formes sclérodermiformes et infiltrant sont de mauvais pronostic).
En pratique, trois groupes pronostiques sont individualisés :
• Le groupe de bon pronostic :
CBC superficiels primaires et tumeur de Pinkus,
CBC nodulaires primaires de moins de 1 cm sur la zone à risque intermédiaire de récidive et de moins de 2 cm sur la zone à bas risque de récidive ;
• Le groupe de pronostic intermédiaire :
Les CBC superficiels récidivés,
Les CBC nodulaires de moins de 1 cm sur zone à haut risque de récidive, de plus de 1 cm sur une zone à risque, intermédiaire de récidive et de plus de 2 cm sur zone à bas risque de récidive ;
• Le groupe de mauvais pronostic :
Les CBC sclérodermiformes ou mal limités et les formes histologiquement agressives,
Les CBC récidivants (sauf les formes superficielles),
Les CBC nodulaires sur zone à haut risque de récidive et de taille supérieure à 1 cm.
Traitement du carcinome basocellulaire [15] :
Le traitement de choix est la chirurgie car elle permet un contrôle histologique de la pièce d’exérèse. Les marges d’exérèse varieront de quelques millimètres à un centimètre en fonction des paramètres pronostiques.
D’autres options thérapeutiques sont possibles comme :
La cryochirurgie : Cette méthode consiste à détruire la tumeur par une congélation forte à l’azote liquide ou au protoxyde d’azote.
La radiothérapie : en cas de contre-indication à la chirurgie
La photothérapie dynamique topique : elle est intéressante dans les CBC superficiels multiples ou étendus. Les tumeurs superficielles multiples peuvent être traitées par une chimiothérapie locale (5-fluorouracile) ou par immunothérapie locale (Imiquimod). Une chimiothérapie est rarement nécessaire.
Traitement préventif du carcinome basocellulaire:
Il repose sur la photoprotection et le suivi régulier des malades à risques. Un suivi semestriel la première année, puis annuel durant toute la vie est recommandé.
Références :
Depinho RA. The age of cancer. Nature 2000 ; 408 :248-254.
Halna JM, Grandadam M, Buerni A. Epidemiologic study of skin cancers from french population (1988-1996). Report of cancer Regis- tration from Haut Rhin. Nouv Dermatol 2000 ;19:48-55.
Bernard P, Derancourt C, Arnoult-Coudoux E, Picot R, Delvincourt C. [Skin cancer diagnosis by dermatologists in the region of Champagne- Ardenne : a prospective study]. Ann Dermatol Venereol 2001 ;128:883-7.
Cockerell CJ. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”). J Am Acad Dermatol 2000 ;42:11-7.
Rinker MH, Fenske NA, Scalf LA, Glass LF. Histologic variants of squa- mous cell carcinoma of the skin. Cancer Control 2001 ; 8 :354-563.
Carcinome épidermoïde cutané (carcinome spinocellulaire) : Recom- mandations de pratique clinique pour la prise en charge diagnostique et thérapeutique, Ann Dermatol Venereol (2009), 136, S189-S242.
Zalaudek I, Cameron. A, Rosendahl C., Actinic keratosis, Bowen’s disease, keratoacanthoma, and squamous cell carcinoma, in Ashfaq A Marghoob, Josep Malvehy, Ralph P Braun, Atlas of Dermoscopy, Second Edition, imforma healthcare (2012) ; p48-57.
Trakatelli M, Ulrich C, del Marmol V, Euvrard S, Stockfleth E, Abeni D. Epidemiology of non melanoma skin cancer (NMSC) in Europe : accurate and comparable data are needed for effective public health monitoring and interventions. Br J Dermatol 2007 ;156 (Suppl 3):1-7.
StaplesM, Marks R, Giles F. Trends in the incidence of non melano- cytic skin cancer (NMSC) treated in Australia 1985-1995 : are primary prevention programs starting to have an effect? Int J Cancer 1998 ; 78:144-8.
J.-C. Guillaume, B. Guillot, Carcinomes basocellulaires EMC (El- sevier Masson SAS, Paris) ; 6e édition ; 12-5, 654-660
Altamura et al, Dermatoscopy of basal cell carcinoma : Morpholo- gic variability of global and local features and accuracy of diagnosis J Am Acad Dermatol, 2010 ; 62, 1, p 67-75E. Conde Montero, L. Thomas, S. Dalle, Carcinome basocellulaire tatoué, Ann Dermatol Venereol (2013) 140, 485—486Delaunay J, et al. Carcinome basocellulaire superficiel versus carci- nome basocellulaire nodulaire. Ann Dermatol Venereol (2014)
Lallas A, Tzellos T, Kyrgidis A, Apalla Z, Zalaudek I, Karatolias A, et al. Accuracy of dermoscopic criteria for discriminating super-fi- cial from other subtypes of basal cell carcinoma. J Am Acad Dermatol 2014 ;70:303—11
L. TAIBI-BERRAH, Service de Dermatologie, CHU Mustapha Bacha, Alger.
Abstract : Primary cutaneous lymphomas PCL are specifically cutaneous B-cell and T-cell lymphomas. They represent the most common extranodal localization after digestive lymphomas. The incidence of these pathologies considered to be rare appears to be increasing [1]. The last two decades have been marked by the recognition of the specificity of the PCL in relation to their systemic equivalents and the origin of more and more detailed classifications, albeit complex but much more precise. Fortunately, a constructive dialogue between clinical specialists, skin pathologists and hematology has led to the adoption, under the aegis of the EORTC (European organization of Research and treatment of Cancer) and the WHO (World Health Organization), of a common classification WHO 2008 updated in 2016 [2,3], of all hematopoietic tumors, recognizing cutaneous lymphomas as specific entities. The broad field of cutaneous lymphomas [table I] does not allow to approach all of them or detailed all their characteristics through in this review. This article, though not exhaustive, is a didactic and up-to-date review of the main anatomo-clinical entities of LCP encountered in dermatological practice.
Key-words : Mycosis fungoides, Sésary syndrome, CD30+ cutaneous lymphomas, follicular center-cell cutaneous B-cell lymphoma, marginal zone cutaneous B-cell lymphoma, pri- mary cutaneous diffuse large B-cell lymphoma Leg type.
Résumé : Les lymphomes cutanés primitifs « LCP » regroupent des lymphomes B et T spécifiquement cutanés. Ils représentent la localisation extra-ganglionnaire la plus fréquente après les lymphomes digestifs. L’incidence de ces pathologies considérées comme rares semble en augmentation [1]. Les deux dernières décennies ont été marquées par la reconnaissance de la spécificité des LCP par rapport à leurs équivalents systémiques et à l’origine de classifications de plus en plus détaillées, certes complexes mais bien plus précises. Fort heureusement, un dialogue constructif entre les spécialistes cliniciens et pathologistes de la peau, et de l’hématologie a conduit à adopter, sous l’égide de l’EORTC (European Organisation of Research and Traitement of Cancer) et de l’OMS, une classification commune OMS 2008 actualisée en 2016 [2,3], de l’ensemble des tumeurs hématopoïétiques, reconnaissant les lymphomes cutanés comme entités spécifiques. Le large champ des lymphomes cutanés [Tableau I], ne permet pas de tous les aborder ou de détailler l’ensemble de leurs caractéristiques à travers la revue ci-après. Cet article cependant, bien que non exhaustif, constitue une revue didactique et actualisée des principales entités anatomo-cliniques de LCP rencontrées en pratique dermatologique.
Mots-clés : Mycosis fongoïde, Syndrome de Sésary, Lymphomes cutanés CD30+, Lymphome B centrofolliculaire, Lymphome cutané de la zone marginale, Lymphome à grandes cellules de type membre inférieur.
Définition des lymphomes cutanés primitifs :
Les LCP sont des proliférations clonales de lymphocytes matures, localisées et limitées à la peau avec absence de localisation extra-cutanée au moment du diagnostic. Ils répondent ainsi nécessairement aux stades N0 et M0 de la classification TNM des lymphomes cutanés [4,5].
Ils constituent un groupe hétérogène d’entités aux tableaux cliniques, histologiques et évolutifs très variés.
Leur diagnostic repose sur une confrontation anatomo-clinique, complétée par des études immunophénotypiques, voire moléculaires et génotypiques.
Contrairement aux lymphomes systémiques où prédominent les proliférations de phénotype B, ce sont les lymphomes T qui sont les plus fréquents dans la peau.
Lymphomes à cellules T et NK matures
Lymphomes de bas grade : – Mycosis fongoïde ( MF ) et variantes : • MF pilotrope • MF pagétoïde • MF chalazodermique – Lymphoproliférations T cutanées CD30+ : • Papulose lymphomatoïde • Lymphome T anaplasique cutané primitif : – Lymphome T sous-cutané à type de panniculite ( TRC alpha-bêta ) – Lymphome T de type hydroavacciniforme
Lymphomes de haut grade : – Syndrome de Sézary – Leucémie/lymphome T de l’adulte ( HTLV-1 ) – Lymphome T gamma-delta cutané primitif ( cutané-sous-cutané ) – Lymphome NK/T extraganglionnaire de type nasal Entités provisoires : – Lymphome T cutané primitif pléomorphe à petites/moyennes cellules – Lymphome T cutané primitif CD8+ Cytotoxique épidermotrope agressif
Lymphomes à cellules B matures
Lymphomes de bas grade : – Lymphome centrofolliculaire cutané primitif – Lymphome B de la zone marginale de type MALT extraganglion- naire cutané
Lymphomes de haut grade : – Lymphome B diffus à grandes cellules, NOS : • Lymphome B diffus à grandes cellules cutané primitif de type jambes • granulomatose lymphomatoÏde – Lymphome B intravasculaire
Syndromes lymphoprolifératifs associés à l’immunodépression
– Syndrome lymphoprolifératif associé à une maladie dysimmunitaire — Syndrome lymphoprolifératif post-transplantation
Leucémie aiguё myéloÏde et hémopathies à précurseurs myéloÏdes
– Tumeur à cellules plasmacytoÏdes dendritiques blastiques
MALT : mucosa-associated lymphoid tissue ; NOS : not otherwise specified ; NK ; natural killer.
Tableau 1 : Principaux lymphomes et hémopathies de présentation habituellement ou fréquemment cutanée primitive, extraits de la classification OMS 2008. (Les entités jugées provisoires en italique.)
Le mycosis fongoïde (MF) :
Le mycosis fongoïde est un lymphome T cutané épidermotrope (LTCE). Il est le plus fréquent de tous les LCP représentant jusqu’à 40 % des cas [6].
Sa pathogénie, mal connue, en fait essentiellement une maladie par accumulation clonale de lymphocytes dans la peau, puis une affection tumorale aux stades avancés.
A. Présentation clinique du mycosis fongoïde classique :
Le MF affecte généralement les sujets adultes d’âge moyen, avec une légère prédominance masculine.
Ce lymphome de bas grade de malignité évolue classiquement en trois phases successives identifiables à l’examen dermatologique : plaques non infiltrées, plaques infiltrées, tumeurs.
Les plaques de début sont érythémateuses et finement squameuses, rouge bistre, souples, à surface fripée, à disposition arciforme, bien limitées, parfois encochées (Photo 1) de dimensions variables de un à une dizaine de centimètres
Photo 1 : Mycosis Fongoïde Débutant (Dr TAIBI, service de dermatologue du CHU Mustapha Bacha, Alger)
Les plaques infiltrées représentent l’évolution des précédentes. L’épaississement progressif en profondeur est bien perçu à la palpation des lésions. Elles peuvent s’éroder ou s’ulcérer. (Photo 2).
Photo 2 : Mycosis Fongoïde en plaques infiltrées (Dr TAIBI, service de dermatologue du CHU Mustapha Bacha, Alger)
Ces éléments sont initialement localisés en priorité sur les zones non photo-exposées : fesses, flancs, haut des cuisses, creux axillaires.
Les signes fonctionnels sont dominés par un prurit d’intensité variable ou par des sensations douloureuses, notamment aux stades d’érosions [7].
L’état général est conservé. Les aires ganglionnaires sont habituellement libres.
Le mycosis fongoïde suit en principe une progression très lente, s’étendant sur des années voire des décennies.
Les lésions nodulaires ou tumorales, souvent ulcérées, apparaissent dans de rares cas sur les plaques infiltrées. Le pronostic s’altère progressivement à la fois en qualité et en espérance de vie. Cette aggravation clinique est souvent contemporaine d’une transformation histologique. Les muqueuses peuvent être touchées à ce stade [8].
L’érythrodermie (érythème atteignant 90 % ou plus de la surface corporelle) chez un nombre très restreint de patients, ou encore une atteinte ganglionnaire voire viscérale spécifique peuvent apparaître.
B. Diagnostic histopathologique et immunopathologique du mycosis fongoïde :
Les biopsies cutanées doivent être multiples et sur des sites différents. Réalisées au punch de 4 mm et facilement répétitives, ou à la lame de bistouri classique.
Le fragment est à fixer dans le formol. Il est recommandé de faire une biopsie à l’état frais ou rapidement congelée pour l’étude de clonalité (PCR ou FISH).
Le diagnostic histopathologique de mycosis fongoïde repose sur la présence d’un infiltrat lymphocytaire du derme superficiel caractérisé par des lymphocytes atypiques et un épidermotropisme [9].
Les atypies des lymphocytes sont définies par la taille du noyau, un peu plus grande que celle d’un noyau de lymphocyte normal, et par les irrégularités du contour nucléaire cérébriforme. À noter que les cellules sont de taille petite à moyenne.
L’épidermotropisme est défini par la migration épidermique de lymphocytes atypiques, disposés en « file indienne » dans la couche basale épidermique ou regroupés en amas appelés « abcès de Pautrier ».
L’immunohistochimie du mycosis fongoïde souligne la nature T auxiliaire CD3+, CD4+ des lymphocytes atypiques dans 90% des cas. Cependant, d’authentiques mycosis fongoïde CD8+ sont décrits.
Sur le plan moléculaire, l’interprétation d’un clone cutané lésionnel dominant doit être intégrée au contexte clinique, histologique et immunologique, clonalité n’étant pas synonyme de malignité.
Enfin, des signes dits de ‘’transformation’’ signant l’aggravation du pronostic, peuvent être retrouvés aux stades avancés, avec apparition d’un pourcentage significatif supérieur à 25 % de cellules de grande taille, exprimant souvent l’antigène CD30 [8].
Formes anatomo-cliniques du mycosis fongoïde :La variante pilotrope est la plus fréquente. Le pilotropisme lymphocytaire autour des follicules pileux est à l’origine de lésions acnéiformes, comédonniennes ou alopéciantes, particulièrement au niveau des sourcils [10].
La variante pagétoïde, rare et réputée très indolente, prend l’aspect de plaques érythémato-squameuses. Les cellules néoplasiques sont toutes localisées dans l’épiderme, rappelant l’image histologique de la maladie de Paget mammaire [11].
La variante granulomateuse chalazodermique, encore plus rare, et d’évolution très chronique, se développe dans les grands plis sous forme de plaques évoluant vers une hyperlaxité cutanée. L’infiltrat lymphocytaire T est associé à des cellules macrophagiques multinucléées et une destruction du réseau élastinique [12].
Diagnostics différentiels du mycosis fongoïde :
Aux stades initiaux, il est souvent délicat d’éliminer des affections cutanées inflammatoires chroniques lymphocytaires bénignes, fréquentes en pratique dermatologique courante et peu spécifiques : eczématiformes ou psoriasiformes (contacts, médicaments).
Le diagnostic reste souvent hésitant pendant longtemps. C’est dire l’importance de revoir régulièrement les patients atteints d’infiltrats cliniquement et histologiquement mal définis et douteux pour réévaluer clinique, histologie et immunohistochimie [13].
Aux stades plus avancés, notamment tumoraux ou érythrodermiques, le diagnostic différentiel doit essentiellement écarter les autres lymphomes cutanés primitifs.
C. Évolution du mycosis fongoïde :
Au stade débutant, la maladie est considérée indolente, car le traitement permet souvent d’obtenir une rémission clinique. Cependant, le taux de rechute est important et la maladie a une évolution chronique, plus ou moins contrôlée par le traitement.
La maladie progresse dans environ 20 % des cas, avec une extension qui fixe le pronostic et doit être évaluée selon le système de « staging » en vigueur [14]. Au stade de transformation en lymphome T à grandes cellules, la survie spécifique à 5 ans estimée à 20 % [14]. Le traitement devient souvent plus difficile et de moins en moins efficace.
Le syndrome de SEZARY :
Le syndrome de Sézary (SS) est aussi un LTCE. Il doit être systématiquement évoqué en cas d’érythrodermie. Il a pendant longtemps été considéré comme la forme leucémique du mycosis fongoïde avec lequel il partage un aspect cytologique commun : la « cellule de Sézary ».
Présentation clinique du syndrome de Sézary :Albert Sézary décrivait en 1938 un syndrome associant une érythrodermie, des adénopathies périphériques et des cellules atypiques dans le sang.
Le syndrome de Sézary est assez rare, représentant environ 6 % des lymphomes T cutanés selon les données européennes [15]. Il atteint les sujets adultes et plutôt âgés, au-delà de 50-60 ans.
Il se caractérise cliniquement par des signes témoignant de l’infiltration lymphomateuse diffuse de la peau (Photo 3) :
Une érythrodermie sèche infiltrée très prurigineuse, avec desquamation diffuse et un aspect en « drapé » du tronc.
Une kératodermie palmo-plantaire avec épaississement et irrégularité des ongles : onychodystrophie.
Un ectropion et un aspect léonin traduisant la forte infiltration du visage.
Une alopécie squameuse.
Des adénopathies superficielles.
Photo 3 : Syndrome de Sézary : Erythrodermie infiltrée et pigmentée (Dr TAIBI, service de dermatologue du CHU Mustapha Bacha, Alger)
B. Histologie et immunophénotypage cutanés du syndrome de Sézary [16]
L’infiltrat lymphocytaire dermique périvasculaire est fréquemment associé à un épidermotropisme et composé de cellules atypiques de phénotype T CD4+, avec parfois des trous phénotypiques sous forme d’une perte de CD7, plus rarement de CD2 ou CD5. Les cellules expriment fortement PD1.
A. Cytologie et immunophénotypage sanguin :
Le frottis sanguin identifie les « cellules de Sézary » de morphologie particulière : de taille petite à moyenne, avec typiquement des noyaux incisurés à contours irréguliers, « cérébriformes ».
La cytométrie de flux permet le phénotypage des lymphocytes sanguins néoplasiques. Le phénotype est T, CD4+ avec parfois des pertes d’antigènes T, CD7 plus souvent que CD2. La perte de CD26 et l’expression aberrante de CD158k/KIR3DL2 sont très utiles pour le diagnostic positif.
B. Biologie moléculaire du syndrome de Sézary :
Présence fréquente d’un clone T cutané et sanguin identique.
C. Synthèses des critères diagnostiques du syndrome de Sézary [17].
Le diagnostic d’un syndrome de Sézary peut être retenu, selon les critères WHO 2008-2016 en vigueur sur:
La présence dans le sang d’un nombre de cellules de Sézary > 1.000/mm3,
Ou d’un clone sanguin cytogénétiquement anormal,
Ou d’une hyperlymphocytose clonale,Ou d’un ratio CD4/CD8 >10,
Ou de cellules circulantes avec un phénotype aberrant, associées à la présence d’un clone T cutané identique dans le sang et la peau.
D. Diagnostic différentiel du syndrome de Sézary :
Il se pose principalement au début avec une dermatose inflammatoire érythrodermique : psoriasis érythrodermique, dermatite atopique ou certaines toxidermies, en particulier le syndrome DRESS (drug rash with eosino- philia and systemic symptoms).
E. Évolution et traitement :
Le syndrome de Sézary est un lymphome agressif avec une médiane de survie à 5 ans de l’ordre de 20%.
Lymphomes T cutanés primitifs CD30+ :
papulose lymphomatoïde et lymphome T cutané primitif à grandes cellules anaplasiques Ce sont les plus fréquents des lymphomes T cutanés non épidermotropes. Leur pronostic est en général excellent, très différent de celui des lymphomes ganglionnaires de même histologie. Un bilan d’extension est donc indispensable [18].
Ce groupe représente un spectre de maladies, avec à une extrémité la papulose lymphomatoïde (PL) et à l’autre le lymphome anaplasique à grandes cellules cutané primitif LCP-GCA.
Photo 4 : Lymphome T cutané primitif anaplasique (Dr TAIBI, service de dermatologue du CHU Mustapha Bacha, Alger).
A. Présentation clinique et associations pathologiques :
La papulose lymphomatoïde se présente sous la forme de papules ou de petites tumeurs brunes violacées, d’apparition rapide, évoluant vers l’ulcération et la nécrose, solitaires ou disséminées. L’évolution se fait typiquement en poussées entrecoupées de rémissions. Une régression spontanée, partielle ou complète survient dans 30 % des cas, dans les mois qui suivent.
Le LCP-GCA réalise une tumeur généralement solitaire (Photo 4), parfois ulcérée, lentement progressive atteignant des dimensions importantes supérieures à 10 cm de diamètre. Une tendance à la régression peut être observée mais elle reste volontiers partielle pour les tumeurs volumineuses. Un envahissement ganglionnaire locorégional peut survenir, sans pour autant modifier le pronostic de la maladie mais imposant la recherche systématique d’un lymphome T anaplasique systémique. Comme pour la papulose lymphomatoïde, l’évolution est indolente.
A. Histopathologie et immunomarquages :
Un infiltrat dense dermique voire hypodermique fait de grands lymphocytes.
Grandes cellules de morphologie variable majoritairement anaplasiques.
Ces cellules expriment majoritairement l’antigène CD30 mais aussi CD3+, et en général CD4+. ALK n’est en principe pas exprimé.
Différentes variantes histopathologiques de papulose lymphomatoïde sont décrites et classées du Type A au Type E.
A. Diagnostic différentiel :
Dans un contexte de mycosis fongoïde (MF), celui du diagnostic différentiel entre une papulose lymphomatoïde CD30+ associée au MF et un MF transformé exprimant CD30.
Une localisation secondaire de lymphome T anaplasique systémique. L’expression d’ALK et le bilan d’extension pourront faire la part entre les deux entités cutanées primitive et systémique de pronostics totalement opposés.
B. Évolution et pronostic :
Le pronostic global est très favorable en termes de survie (90 % à 10 ans). Il est surtout lié à la possible association à un autre lymphome qu’il faut toujours rechercher, notamment un mycosis fongoïde, un lymphome T anaplasique ganglionnaire, mais également un lymphome B, plus souvent alors un lymphome de Hodgkin qu’une LLC [1].
Lymphomes B cutanés primitifs :
A. Introduction et définitions :
Le paysage des lymphomes B cutanés primitifs est moins complexe que celui des lymphomes T, puisqu’il existe classiquement 3 principaux types, dont deux sont de bas grade, et un plus agressif. La reconnaissance d’une quatrième entité a fait porter le nombre de lymphomes B cutanés primitifs à quatre selon la classification WHO 2016 [3].
Il est important de préciser qu’il n’existe pas de continuum entre les lymphomes B cutanés de bas grade et les formes agressives à grandes cellules. Le processus de transformation de formes de bas grade en lymphome agressif semble effectivement un évènement exceptionnel dans les lymphomes B cutanés primitifs, contrairement à leurs homologues ganglionnaires.
Lymphome B cutané primitif centrofolliculaire :Le lymphome B cutané centrofolliculaire (LBCP-CF) représente environ 40 % des lymphomes B cutanés primitifs. Il atteint les adultes autour de la cinquantaine [20].
C. Présentation clinique [22]
Les lésions sont des papules ou des nodules érythémateux fixes, parfois multiples mais alors le plus souvent regroupés dans une même région cutanée. Les localisations préférentielles sont l’extrémité céphalique et le tronc (Photos 5 et 6).
Il n’y a le plus souvent aucun signe fonctionnel. Les aires ganglionnaires sont libres. Le bilan biologique est en général normal en particulier la NFS et les LDH.
Photo 5 : Lymphome B centro-folliculaire nodulaire (Dr TAIBI, service de dermatologue du CHU Mustapha Bacha, Alger)Photo 6 : Lymphome B centro-folliculaire en plaque (Dr TAIBI, service de dermatologue du CHU Mustapha Bacha, Alger)
D. Histopathologie [21]
L’infiltrat néoplasique est dermique ou dermo-hypodermique, épargnant l’épiderme. Sous forme de nappes diffuses ou de nodules, rappelant l’aspect observé dans les lymphomes folliculaires ganglionnaires.
Les cellules néoplasiques des LBCP-CF rappellent les éléments constituant le centre germinatif des follicules lymphoïdes activés. Il s’agit d’un mélange de centrocytes (petits ou grands lymphocytes à noyaux clivés), mêlés à des centroblastes et des immunoblastes.
Contrairement à leurs équivalents ganglionnaires, les LBCP-CF ne sont pas classés en grades.
E. Immunohistochimie :
Les cellules tumorales ont un phénotype centro-germinatif CD20+ et BCL6+ et dans la majorité des cas n’expriment pas BCL2 contrairement à l’équivalent folliculaire ganglionnaire [23].
F.Diagnostic différentiel :
L’un des principaux problèmes qui se posent, est de faire le diagnostic différentiel entre un lymphome cutané primitif et un envahissement cutané secondaire par un lymphome B systémique [25]. Il faudra donc éliminer :
Une leucémie lymphoïde chronique avec infiltrat cutané.
Un lymphome folliculaire ganglionnaire : surtout en cas de positivité du BCL2. La recherche d’une translocation T(14 ; 18) par FISH peut être utile [25].
Un lymphome B à grandes cellules cutané primitif ou secondaire plus péjoratif : certains LBCP-CF sont constitués de grands centrocytes et/ou centroblastes et immunoblastes. La recherche de la mutation de MYD88 peut être préconisée, puisqu’elle est assez fréquente dans les lymphomes de type jambe et exceptionnelle dans les autres entités.
Un bilan d’extension par tomodensitométrie thoraco- abdomino-pelvienne est obligatoire dans tous les cas pour répondre à la définition du lymphome cutané primitif. L’étude de la moelle osseuse est quant à elle optionnelle [24].
G. Pronostic et traitement :
Le PC-FCL a une évolution indolente avec une survie spécifique à 5 ans de 100 %, mais l’évolution progressive des lésions peut former des tumeurs de grande taille invalidantes et affichantes. En cas de lésion unique, le traitement repose sur une radiothérapie ou une exérèse chirurgicale. Si les lésions sont multiples, un traitement par anticorps anti-CD20 ou radiothérapie multi-champs seront à discuter au cas par cas. La récidive est fréquente au cours du suivi, qui doit donc être prolongé et repose principalement sur l’examen clinique [25,26].
Lymphome B cutané de la zone marginale de type MALT (LBCP-ZM) :
Le LBCP-ZM représente environ 40 % des lymphomes B cutanés primitifs. Il atteint les sujets adultes.
A. Présentation clinique :
Les lésions cutanées sont généralement de multiples papules et/ou nodules de quelques millimètres à quelques centimètres de diamètre (photo 7), groupées ou éparses sur les membres ou le tronc. Elles ne sont ni douloureuses ni prurigineuses. Il n’y a pas d’adénopathie. En général, la NFS est normale, et il n’y a pas d’élévation des LDH [26].
Photo 7 : Lymphome B cutané de la Zone marginale type MALT (Dr TAIBI, service de dermatologue du CHU Mustapha Bacha, Alger)
B. Histopathologie :
L’infiltrat dermique contient des follicules lymphoïdes parfois anormaux.
Les lymphocytes B néoplasiques sont petits et ont un phénotype post-germinatif mature CD20+, BCL6-, CD10-, BCL2+, MUM1+.
Il peut exister en périphérie de l’infiltrat des nappes de plasmocytes avec une expression monotypique d’une chaîne légère (plus souvent kappa que lambda).
Les translocations caractérisant les lymphomes B de type MALT systémiques ne sont qu’exceptionnellement retrouvées dans LBCP-ZM [30].
C. Diagnostic différentiel :
Infiltrat lymphocytaire réactionnel pseudo-lymphomateux : les critères de distinction les plus utiles sont la démonstration du caractère clonal de l’infiltrat.
Pseudo-lymphome borrélien : envisagé devant un infiltrat lymphocytaire B à petites cellules d’évolution chronique. Cela justifie d’effectuer une sérologie voire une PCR spécifique de Borrelia burgdoferi, et la mise sous traitement antibiotique d’épreuve au moindre doute en zones d’endémie [26,27].
Les localisations cutanées secondaires de lymphomes ganglionnaires de la zone marginale ou du manteau sont exceptionnelles. Le contexte clinique et les marqueurs utilisés pour le diagnostic dans le ganglion aident beaucoup.
D. Pronostic et traitement :
Le pronostic de ce lymphome est excellent, avec une survie à 5 ans qui peut être estimée à 100 % comme pour le LBCP-CF.
Les modalités du traitement sont très proches de celles du LBCP-CF, reposant sur la radiothérapie ou l’exérèse pour des lésions isolées et la radiothérapie multi-champ ou le rituximab pour des lésions multiples. Les rechutes sont fréquentes.
Lymphome B diffus à grandes cellules cutané primitif, « de type jambe » (LBCP-GC-TJ) :
Il s’agit d’une forme primitivement cutanée de lymphome B diffus à grandes cellules, représentant environ 10% des lymphomes B cutanés primitifs, et 5 % des lymphomes cutanés (GFELC 2013-2016).
Ce lymphome apparaît maintenant comme une entité à part entière dans l’actualisation OMS 2016 [2,3].
Il survient chez des sujets âgés de plus de 70 ans, avec une nette prédominance féminine (sex-ratio de 1:4 environ) [1,28].
A. Présentation clinique :
Les lésions sont des tumeurs rouges violacées, d’évolution rapide vers des dimensions parfois supérieures à 10 cm et souvent ulcérées. Uniques ou multiples. La localisation typique, justifiant l’appellation actuellement admise, est le membre inférieur, mais d’autres régions cutanées peuvent être concernées (Figures).
L’examen clinique doit rechercher une atteinte ganglionnaire qui peut déjà être présente au moment du diagnostic. Le bilan biologique est normal, les LDH peuvent être élevées en cas de forte masse tumorale [27,28].
Photo 8 : Lymphome B cutané à grandes cellules de type jambe (Dr TAIBI, service de dermatologue du CHU Mustapha Bacha, Alger)Photo 9 : Lymphome B cutané à grandes cellules de type jambe siégeant sur l’épaule (Dr TAIBI, service de dermatologue du CHU Mustapha Bacha, Alger);
B. Histopathologie et biologie moléculaire; mutations de MYD88 :
Une prolifération dermique ou dermo-hypodermique dense d’architecture diffuse de grands lymphocytes atypiques à noyaux arrondis : les immunoblastes [29].
Sur le plan phénotypique, il s’agit de cellules B, avec un profil plutôt de type « non centro-germinatif » : BCL2+, MUM1+, BCL6 hétérogène variable.
Il est actuellement admis que l’anomalie moléculaire récurrente ayant une signification diagnostique et pronostique est la mutation de MYD88 [31]. Fréquemment présente dans les LBCP-GC-TJ, elle permet d’éliminer un LBCP-CF et semble associée à un pronostic plus péjoratif.
B. Pronostic du LBCP-GC-TJ :
Ce lymphome a un comportement agressif avec possibles atteintes viscérales, incluant notamment une atteinte cérébrale grevant le pronostic.
Comme pour les autres lymphomes B agressifs, le pronostic a été révolutionné par la thérapie ciblée anti- CD20 (Rituximab) associée à la polychimiothérapie : R-CHOP. Ce traitement a fait basculer la survie spécifique à 3 ans de 45% à environ 85%. Néanmoins, une surveillance clinique prolongée est nécessaire.
Conclusion :
Les lymphomes cutanés offrent un univers multiple et complexe. Leur grande hétérogénéité clinique, immunohistologique et évolutive nécessite une prise en charge très spécialisée, alliant dermatologues et anatomopathologistes mais aussi hématologistes, et reposant sur des réunions de concertations pluridisciplinaires RCP où la confrontation anatomoclinique est obligatoire.
Références
Markova A, Martin A. Weinstock. (2010) Trends in Cutaneous Lymphoma Epidemiology. Clinical Lymphoma, Myeloma & Leuke- mia, Vol. 10, Suppl. 2, S63-S66, 2010
Swerdlow SH, Campo E, Harris NL et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press, Lyon 2008.
Swerdlow SH et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms Blood 2016. 127(20):2375-90.
Kim YH, Willemze R, Pimpinelli N et al. (2007) TNM classification system for primary cutaneous lymphomas other than mycosis fun- goides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 110: 479-84
Olsen, E., et al. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: a proposal of the International So- ciety for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood, 2007. 110(6): p. 1713-22.
Grob JJ (2005) Epidémiologie du Mycosis Fongoïde. Ann Dermatol Vénéréol 132: 5S11-2
Kehn CA, Belongie IP, Shistik G et al. (2007) The diagnosis, sta- ging, and treatment options for mycosis fungoides. Cancer Control 14: 102-11
Vergier B et al (2000). Transformation of Mycosis fungoides: cli- nico-pathological and prognostic features of 45 cases. Blood 2000; 95 :2212-8.
Leshman JS et al, (2010). Folliculotropic mycosis fungoides: single- center study and systematic review. Arch Dermatol 2010; 146 : 607-13.
Steffen C, (2005). Ketro-Goodman disease, Woringer-Koloppdi- sease, and pagetoide reticulosis. Am J Dermatopathol 2005; 27 : 68-85.
Kempf W et al, (2008). Granulomatous mycosis fungoides. A mul- ticenter study of the cutaneous lymphoma task force of the EORTC. Arch Dermatol 2008; 144 : 1609-17.
Beylot-Barry M et al, 2010. Prise en Charge Des Lymphomes T cutanés : Recommandations du groupe français des lymphomes cuta- nés. Ann Dermatol Venerol 2010; 137 : 611-21.
Olsen E et al, (2007). Revision to the staging and classification of Mycosis fungoides and Sezary syndrome: a proposal of the ISCL and the cutaneous lymphoma task force of the EORTC. Blood 2007; 110 : 1713-22.
D’Incan M, Souteyrand P (2009) Lymphome de Sézary et autres lymphomes érythrodermiques. In: Saurat JH. Dermatologie et mala- dies sexuellement transmissibles. Paris: Masson. p. 519-21
Nicolas Ortonne, et al. KIR3DL2 : une nouvelle étape dans l’his- toire du syndrome de Sézary. M/S n° 8-9, vol. 22, août-septembre 2006
Bagot M, Ortonne N, (2012) Lymphomes cutanés: Classification. EMC Dermatologie, 98-680-A-15.
Beylot-BarryM, Vergier B, (2013). Lympho-proliférations cuta- nées CD30+ et Papilose lymphomatoide. Les Lymphomes cutanés sous l’égide du GFELC. Paris: Springer-Verlag; 2013 : 95-110
Kempf W et al, (2011). Consensus recommendations for the treat- ment of primary cutaneous CD30+ lympho-proliferative disorders. Blood 2011; 118 :4024-35
Suarez et al(2013) Primary cutaneous B-cell lymphomas. Part I. Clinical features, diagnosis, and classification. J Am Acad Dermatol Sep2013, 329.e2.
Kempf W, Denisjuk N, Kerl K, Cozzio A, Sander C (2012) Primary cutaneous B-cell lymphomas. Journal of the German Society of Der- matology. JDDG; 2012, 10:12–23
Grange F et al, (2013). Lymphomes B cutanés centrofolliculaires. Les Lymphomes cutanés sous l’égide du GFELC. Paris: Springer-Ver- lag; 2013 : 169-175.
Pham-Ledard A, Cowppli-Bony A et al (2015) Diagnostic and Prognostic Value of BCL2 Rearrangement in 53 Patients With Follicu- lar Lymphoma Presenting as Primary Skin Lesions. Am J Clin Pathol 2015; 143:362-373.
Senff NJ, Kluin-Nelemans HC, Willemze R (2008) Results of bone marrow examination in 275 patients with histological features that suggest an indolent type of cutaneous B-cell lymphoma. Br J Haema- tol 142: 52-6
Kim BK, et al. Clinico-pathologic, immuno-phenotypic, and molecular cytogenetic fluorescence in situ hybridization analysis of primary and secondary cutaneous follicular lymphomas. Am J Surg Pathol. 2005.
Grange F et al, (2013), Lymphomes B cutanés de la zone marginale. Les lymphomes cutanés – sous l’égide du Groupe français d’étude des lymphomes cutanés
Ram-Wolff C, (2016), Lymphomes B cutanés. EMC- Dermatolo- gie. Vol 11; N°4 ; Nov 2016
Grange F, et al. Primary cutaneous diffuse large B-cell lymphoma, leg type: clinico-pathologic features and prognostic analysis in 60 cases. Arch Dermatol. 2007; 143:1144-1150.
Takino H, Li C, Hu S, et al. Primary cutaneous marginal zone B-cell lymphoma: a molecular and clinico-pathological study of cases from Asia, Germany, and the United States. Mod Pathol. 2008;21.
Pham-Ledard A, Beylot-Barry M, Barbe C, Leduc M, Petrella T, Vergier B, Martinez F, Cappellen D, Merlio JP, Grange F. High fre- quency and clinical prognostic value of MYD88 L265P mutation in primary cutaneous diffuse large B-cell lymphoma, leg-type. JAMA Dermatol. 2014;150(11):1173-9.