M. TERAHI, Service d’Ophtalmologie, CHU Nafissa Hamoud, Hussein Dey, Alger.
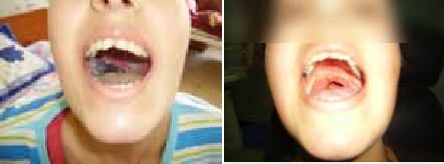
Abstract : Behçet’s disease is a multisystemic vasculitis characterized by intraocular inflammation, oral and genital ulceration, skin lesions as well as many visceral disorders. Corticosteroids and immunosuppressants have long been the basis of treatment. Corticosteroids have an immediate anti-inflammatory effect on acute attacks, but they must be strengthened in the long term by immunosuppressants. Some molecules have more targeted actions such as colchicine that interacts with leukocyte dysfunction or anti-TNFα given the role of this cytokine in the inflammatory process. The management of Behçet’s disease must be multidisciplinary, involving internists and ophthalmologists.
Key-words : Multisystemic vasculitis, blindness, corticosteroids, immunosuppressants, interferon, anti TNFα.
Résumé : La maladie de Behçet est une vascularite multisystémique caractérisée par une inflammation intra-oculaire, des ulcérations orales et génitales, des lésions cutanées ainsi, que de nombreuses atteintes viscérales. Les corticoïdes et les immunosuppresseurs ont longtemps été la base du traitement. Les corticoïdes ont une action anti-inflammatoire immédiate sur les poussées aigues, mais ils doivent être renforcés au long cours par les immunosuppresseurs. Certaines molécules ont des actions plus ciblées comme la colchicine qui interagit avec le dysfonctionnement leucocytaire ou les anti-TNFα compte tenu du rôle de cette cytokine dans le processus inflammatoire. La prise en charge de la maladie de Behçet doit être pluridisciplinaire, associant internistes et ophtalmologistes.
Mots-clés : Vascularite multisystémique, cécité, corticoïdes, immunosuppresseurs, interferon, anti TNFα.
Introduction :
La maladie de Behçet est une maladie chronique évoluant par poussées imprévisibles, spontanément mais partiellement régressives. Son traitement est actuellement purement symptomatique et vise à juguler les poussées et à atténuer les séquelles. Les corticoïdes et les immunosuppresseurs ont longtemps été la base du traitement. Les corticoïdes ont une action anti-inflammatoire immédiate sur les poussées aigues, mais ils doivent être renforcés au long cours par les immunosuppresseurs.
Certaines molécules ont des actions plus ciblées comme la colchicine qui interagit avec le dysfonctionnement leucocytaire ou les anti-TNFα compte tenu du rôle de cette cytokine dans le processus inflammatoire.
L’utilisation plus rationnelle et plus extensive des immunosuppresseurs explique probablement en partie l’amélioration du pronostic visuel.
La prise en charge de la maladie de Behçet doit être pluridisciplinaire, associant internistes et ophtalmologistes.
Un traitement bien adapté permet de réduire le taux de cécité de 75 à 20%, mais pour être efficace ce traitement doit être introduit rapidement.
Clinique :
L’atteinte oculaire de la maladie de Behçet se caractérise par une inflammation des segments antérieurs et postérieurs de l’œil, bilatérale et récurrente (1).
Le patient consulte le plus souvent pour une baisse de l’acuité visuelle ou un simple brouillard visuel ; ces symptômes visuels peuvent être accompagnés de douleurs oculaires et de photophobie (2).
Uvéite Antérieure
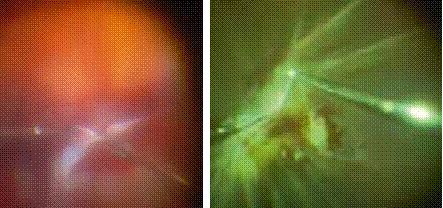
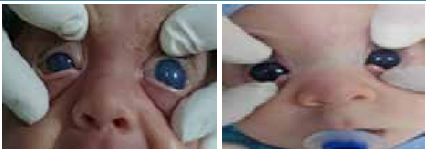
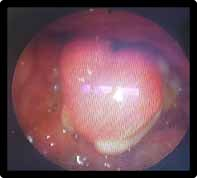
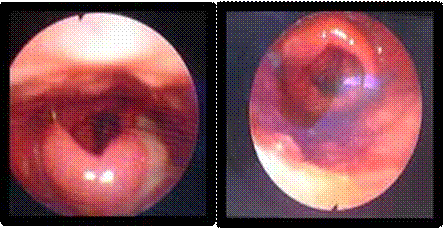
Elle se manifeste par une iridocyclite aiguë non granulomateuse, avec un effet Tyndall important dans la chambre antérieure (figure 1), associé dans un 1/3 des cas à un hy- popion composé de polynucléaires neutrophiles (3).

Uvéite Postérieure
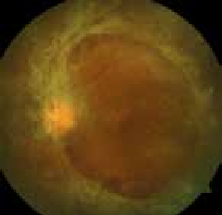
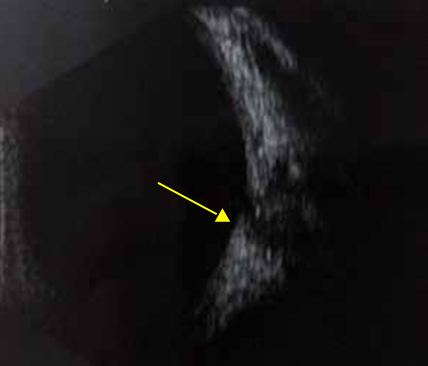
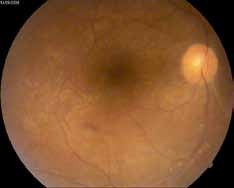
L’uvéite postérieure est la manifestation inflammatoire la plus fréquente et la plus grave. Elle est marquée principalement par une hyalite (figure 2), d’intensité variable, pouvant être responsable d’une baisse importante de l’acuité visuelle avec un fond d’œil complètement masqué (4).
Vascularites rétiniennes
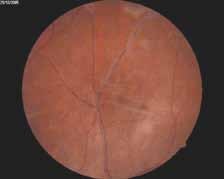
La vascularite rétinienne est fréquente. Elle peut être veineuse le plus souvent, mais aussi artérielle et se traduit par des lésions de périphlébites, de périartérites et de thromboses vasculaires (5).
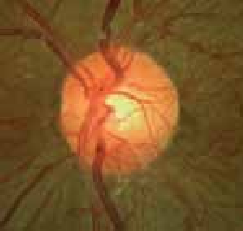
On observe le plus souvent une périphlébite occlusive, caractérisée par un engainement de la paroi veineuse réalisant un aspect de « brume » floconneuse blanchâtre entourant la colonne sanguine. L’atteinte est segmentaire (figure 3).
Examens complémentaires :
Le Laser Cell Flare Meter
Cet appareil permet de réaliser une mesure quantitative du taux de protéines présentes dans l’humeur aqueuse et donc d’assurer une meilleure surveillance des patients en dépistant une augmentation infraclinique du flare pouvant précéder une authentique rechute (2.3).
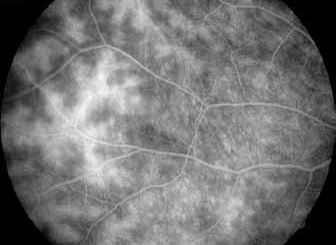
L’angiographie à la Fluorescéine
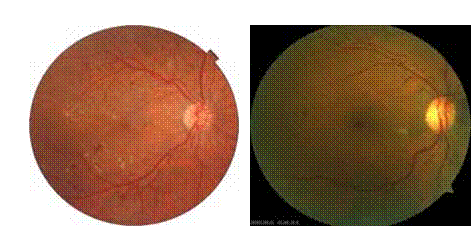
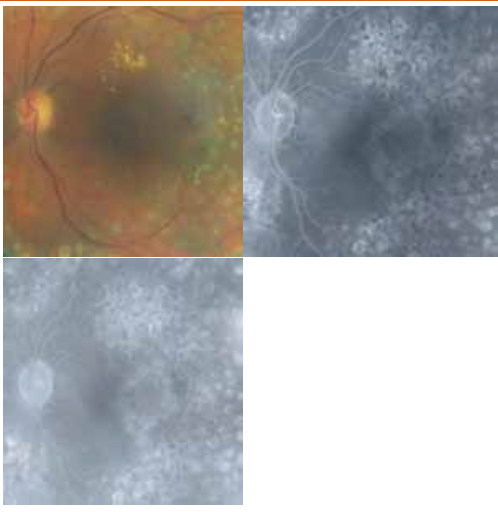
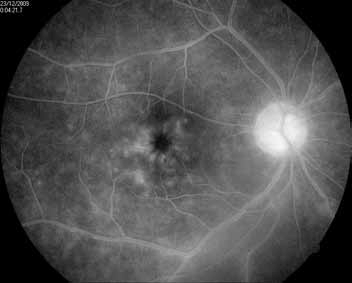
L’angiographie à la fluorescéine est un examen capital car elle permet de faire le bilan de l’atteinte rétinienne, et de retrouver des anomalies infra cliniques dans 6% des cas à type de vascularites rétiniennes (figure 4).
Par ailleurs l’angiographie peut objectiver, un œdème maculaire (figure 5)
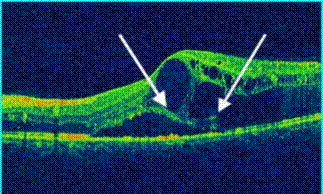
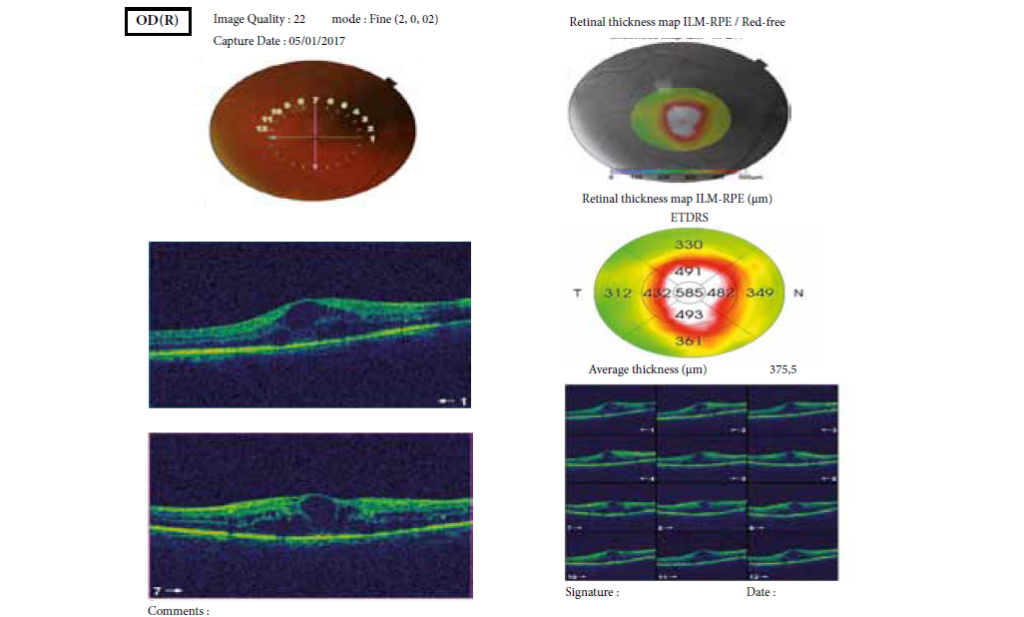
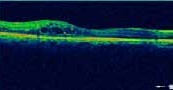
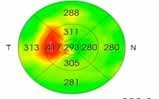
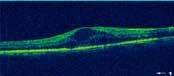
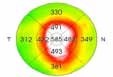
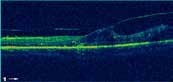
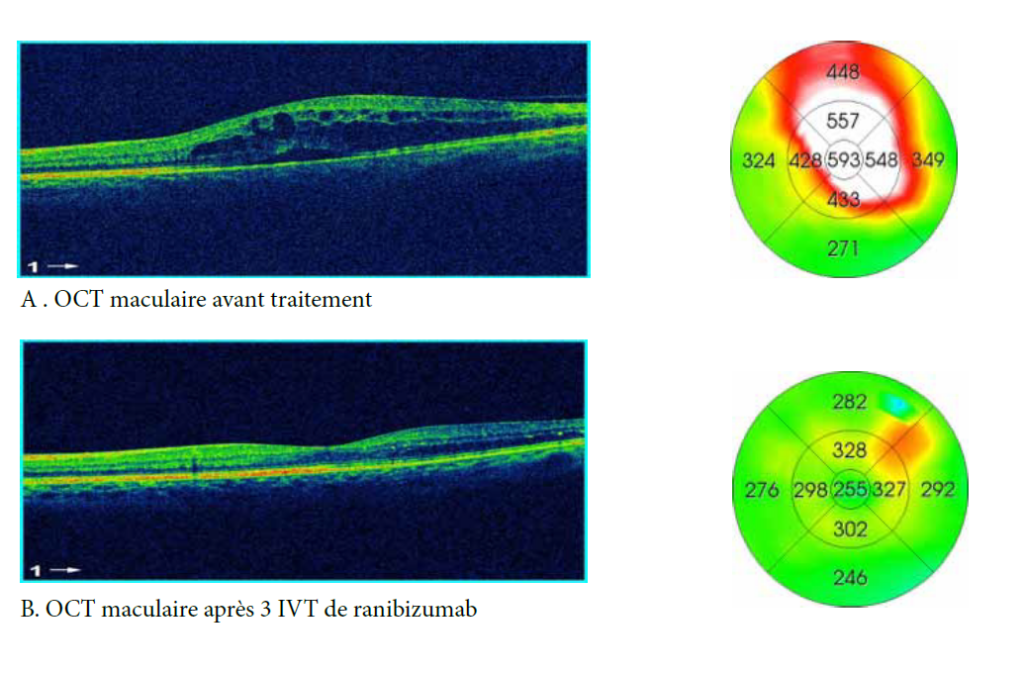
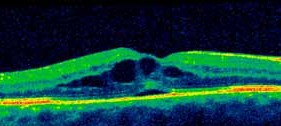
La Tomographie en Cohérence Optique (OCT) L’OCT est indiquée essentiellement dans l’œdème maculaire compliquant les atteintes du segment postérieur (figure 6). Elle permet de mesurer de manière reproductible l’épaisseur maculaire, afin de suivre l’évolution de cet œdème et donc d’adapter les différents traitements (4.5).
Traitement :
Les corticoïdes
La corticothérapie systémique est indiquée en cas de poussée inflammatoire. Elle fait appel à la méthylprednisolone à la dose 10mg/kg/j sans dépasser 1g/j, en perfusion 3 heures pendant 3 jours, relayée par la prednisone (6).
On utilisera pour le relai, la prednisone (Cortancyl®) plutôt que la prednisolone (Solupred®) en raison d’une meilleure biodisponibilité. La prednisone a de plus l’intérêt d’être un corticoïde de demi-vie biologique courte ayant une faible action inhibitrice sur l’axe hypothalamo-hypophyso-surrénalien.
La dose utilisée est de 0,5 à 1,5mg/kg/jour en une seule prise le matin et ce, pendant 1 mois voire 2 mois, cette dose est diminuée progressivement par paliers de 10 % toutes les 1 à 2 semaines avec un palier de 1 mois dans la décroissance lorsqu’on arrive à mi-dose de la prescription initiale jusqu’à obtenir une dose seuil de 5 à 10 mg/j. Une dégression trop rapide prédispose aux rechutes. La prise un jour sur deux permet de réduire les effets secondaires.
Les japonais n’utilisent pas de corticoïdes par voie générale au long cours dans le Behçet oculaire, car certaines études rétrospectives auraient montré un pronostic fonctionnel à long terme meilleur chez les patients n’ayant pas eu de corticoïdes.
La corticothérapie à fortes doses permet de juguler la réaction inflammatoire en 1 à 2 semaines et donc d’améliorer l’acuité visuelle, mais est insuffisante pour espacer les rechutes : un traitement de fond par les immunosup-presseurs est nécessaire.
Une corticothérapie locale est également nécessaire lors des poussées et repose sur la dexaméthasone en collyre, avec des instillations fréquentes quand le Tyndall de l’humeur aqueuse est dense, de manière à éviter les synéchies irido-cristalliniennes.
Dans les formes à participation antérieure prédominante, une corticothérapie locale sous forme d’injections latérobulbaires ou sous conjonctivales peut être adjointe. Cette corticothérapie locale sera associée à un cycloplégique.
La surveillance de la corticothérapie par voie générale doit être rigoureuse, cette surveillance est liée aux complications bien connues de ce traitement.
Au plan clinique, il est indispensable de surveiller le poids, la tension artérielle, et la survenue d’effets secondaires osseux (ostéoporose cortisonique) métaboliques (diabète induit, prise de poids), cutanés (faciès cushingoïde, vergetures irréversibles), vasculaires (rétention hydrosodée avec risque d’hypertension artérielle et d’œdème), et infectieux (infections pyogènes, parasitoses, mycoses, viroses).
Au plan biologique, une détermination mensuelle de la kaliémie, de la glycémie, et de la calcémie sont nécessaires (11).
De plus, un régime pauvre en glucides d’absorption rapide est nécessaire pour éviter la prise de poids trop importante. Un régime désodé est également indispensable jusqu’à la prescription d’une dose inférieure ou égale à 20 mg.
Il est habituel de prescrire systématiquement une supplémentation en calcium (1g/jour pour compenser la diminution de l’absorption intestinale), associée à une supplémentation en vitamine D (400 à 800 unités par jour). Ces mesures associées au maintien d’un exercice physique régulier visent à prévenir l’ostéoporose cortisonique.
Par ailleurs, il n’est pas nécessaire de prescrire systématiquement un traitement antiulcéreux type pansement gastrique, antiacide, ou inhibiteur de la pompe à protons), car les corticoïdes ne provoquent pas d’ulcère gastroduodénal mais sont susceptibles de retarder la cicatrisation d’un ulcère en évolution.
Les immunosuppresseurs :
• Le Chlorambucil (Chloraminophene®)
Le chlorambucil est un agent alkylant qui agit en inhibant l’ADN et la prolifération cellulaire.
C’est le premier immunosuppresseur à avoir été utilisé dans la maladie de Behçet, il est prescrit à la dose de 0,1 à 0,2 mg/kg/jour.
Une dose d’entretien peut être nécessaire pendant en moyenne 18 mois avec des fenêtres thérapeutiques de 2 à 6 mois, selon la tolérance hématologique.
Les effets secondaires du chlorambucil sont essentiellement hématologiques, en effet il existe un risque de leucémie secondaire chez les patients ayant une dose cumulative de 1.300 mg. Les autres effets secondaires décrits sont des cancers gastriques et cutanés, la stérilité et des infections sévères.
Sa toxicité hématologique limite son utilisation (14).
• Le Cyclophosphamide (Endoxan®)
Le cyclophosphamide est un agent alkylant de la famille des moutardes azotées, c’est une des substances immunosuppressives les plus puissantes, il agit en inhibant la prolifération des cellules T et B par inhibition de l’ADN en faisant des liaisons covalentes entre les molécules.
Il est prescrit à la dose de 8 à 12 mg/kg/j ou de 750 à 1000 mg/m2/j en perfusion intraveineuse, en cures mensuelles pendant les 9 premiers mois. Ces cures doivent être espacées s’il apparaît une intolérance hématologique (pancytopénie) ou d’autres complications, qu’elles soient rénales ou infectieuses.
Par la suite, un traitement d’entretien de trois à cinq cures par an est utile.
Certains auteurs préfèrent la forme orale, à raison de 1 à 2 mg/kg/j (18).
Les résultats à long terme obtenus avec le cyclophosphamide seraient meilleurs par rapport à ceux du chlorambucil (6).
Les effets secondaires du traitement sont une alopécie toujours transitoire mais fréquente. Une aménorrhée définitive peut survenir ce qui est un frein à l’utilisation de ce produit chez des femmes jeunes qui doivent être informées de cette complication éventuelle. Chez l’homme, une azoospermie définitive est fréquente.
Une autre complication possible est la cystite hématurique liée à l’élimination du produit sous forme d’un catabolite toxique pour la muqueuse vésicale ; elle peut être évitée par une réhydratation.
De plus, les patients ayant reçu un traitement prolongé par le cyclophosphamide doivent être surveillés pour dépister un cancer de la vessie, des analyses d’urine pour dépister une hématurie devraient être pratiquées tous les 3 ou 6 mois même après arrêt du cyclophosphamide. Des pneumopathies interstitielles, des nécroses myocardiques et des atteintes hépatiques ainsi qu’un syndrome de sécrétion inappropriée d’hormone antidiurétique ont été décrites, mais c’est surtout la toxicité hématologique et les risques infectieux liés à la puissante immunosuppression induite par ce traitement qui dominent les effets secondaires. Ceci justifie une surveillance de l’hémogramme (13).
Les complications tumorales sont communes à tous les immunosuppresseurs à type de cancers cutanés, maladie de Kaposi, lymphomes non Hodgkiniens de même que des leucémies aigues.
Enfin le cyclophosphamide est tératogène et l’allaitement est contre indiqué.
• L’Azathioprine (Imurel®)
L’azathioprine est un antimétabolite qui se transforme après ingestion en 6-mercaptopurine, un puissant inhibiteur de la purine synthétase, il agit en inhibant la réplication de l’ADN et la prolifération clonale des lymphocytes au moment de l’induction de la réponse im- munitaire et affecte toutes les cellules immunitaires et la réponse humorale (7).
Après administration par voie orale, l’azathioprine est rapidement et presque complètement absorbée avec un taux plasmatique maximal atteint en 2 heures.
Elle est rapidement métabolisée par trois voies enzymatiques compétitives. Son métabolite actif est le dérivé nitro-imidazole de la 6-mercapyopurine. La thiopurine méthyltransférase et la xanthine oxydase conduisent à des métabolites inactifs.
L’hypoxanthine guanine phosphoribosyltransférase catabolise la 6-mercaptoprine en métabolites nucléotiques de la 6-thioguanine qui sont les principaux responsables de la toxicité de l’azathioprine.
Les déficits en xanthine oxydase et en hypoxanthine guanine phosphoribosyltransférase sont rares au contraire du déficit en thiopurine méthyltransférase qui est présent à l’état homozygote chez 0,3% de la population et présent à l’état hétérozygote chez 11,1%.
L’activité érythrocytaire de la thiopurine méthyltransférase reflète le génotype. Son déficit entraine un détournement du catabolisme de l’azathioprine vers la voie de la xanthine guanine phosphoryltransférase, d’où l’accumulation de 6-thioguanine nucléotide et le risque de myélotoxicité. Plusieurs études ont montré une corrélation entre l’activité de la thiopurine méthyltransférase érythrocytaire et les effets secondaires de l’azathioprine, ce qui conduit des équipes à la contrôler systématiquement avant la mise en route d’un traitement par azathioprine.
Utilisée à la dose de 1 à 2,5 mg/kg/j ; l’azathioprine diminue les rechutes oculaires.
Elle est généralement associée à de faibles doses de prednisone et/ou à la ciclosporine.
Lors d’une étude prospective, randomisée, l’efficacité de l’azathioprine (2,5mg/kg/j) comparée au placebo, a été évaluée chez 73 patients avec un suivi sur 2 ans.
Au terme de cette étude les 25 patients qui n’avaient pas d’atteinte oculaire initiale, ont présenté moins de manifestations oculaires sous azathioprine que sous placebo. Les 48 patients qui avaient une atteinte oculaire initiale ont présenté moins d’uvéites et d’hypopion avec le traitement immunosuppresseur.
On a également observé une diminution des récidives des manifestations buccales, génitales et articulaires lors de l’utilisation de l’azathioprine.
L’azathioprine est relativement bien tolérée, les effets secondaires les plus fréquents sont l’anorexie et les nausées. L’hépatotoxicité, la leucopénie et la thrombocytopénie sont relativement rares (14).
Bien que tératogène chez l’animal, dans l’espèce humaine, des centaines d’observations de grossesses exposées permettent d’écarter à ce jour, l’hypothèse d’une augmentation importante du risque malformatif de l’azathioprine sur le fœtus.
Les immunomodulateurs :
• La Colchicine
La colchicine est un alcaloïde tricyclique qui agit en
inhibant la migration et la division des polynucléaires neutrophiles, en diminuant plus particulièrement leur chimiotactisme, modifiant leur adhésivité, et en interférant avec la libération de leucokines.
Elle peut être utilisée en association avec les autres traitements dans toutes les formes oculaires de la maladie de Behçet et permettrait de réduire les doses des immunosuppresseurs.
Elle prévient les récurrences de la maladie, mais n’est pas suffisamment efficace pour traiter seule une poussée inflammatoire aiguë.
Prescrite à la dose de 1 à 2mg/j, elle est systématiquement utilisée dans le traitement des manifestations cutanéomuqueuses et permet de juguler l’aphtose buccogénitale.
Elle est également active sur les lésions articulaires de faible gravité.
Sa bonne tolérance et l’absence de surveillance biologique font de la colchicine l’un des traitements de base les plus couramment utilisés.
• La Ciclosporine A
Isolée d’un métabolite fungique, la ciclosporine se lie dans l’organisme à des protéines intracellulaires, les immunophilines. Il en résulte une inhibition de la transcription des gènes de nombreuses cytokines et notamment de l’IL-2. Elle n’a pas d’action myélotoxique et un effet immunosuppresseur purement suspensif.
Elle est utilisée à la dose de 5 à 10 mg/kg/j dans la prévention des rechutes oculaires qu’elle réduirait de près de 70%. La dose de 10 mg/kg/j est généralement mal tolérée. Sa toxicité rénale peut nécessiter une diminution des doses qui n’est pas sans effet sur la survenue des rechutes. Cette néphrotoxicité limite son utilisation en monothérapie. Par conséquent, il peut être judicieux de commencer par des doses de 5 mg/kg/j et, si l’inflammation oculaire ne régresse que partiellement, on peut y adjoindre une faible dose de prednisone ou l’associer à l’azathioprine dans les uvéites sévères.
Utilisée en association aux corticoïdes, elle permet l’amélioration ou la stabilisation de l’acuité visuelle dans 75% des cas.
Plusieurs études ont évalué l’efficacité de la ciclosporine dans la maladie de Behçet en particulier dans le traitement des lésions oculaires avec des résultats satisfaisants.
L’efficacité de la ciclosporine à la dose de 5mg/kg/j comparée au cyclophosposphamide à la dose de 1.000 mg en injection intraveineuse une fois par mois, a été évaluée
chez 23 patients présentant une uvéite évolutive. Une amélioration de l’acuité visuelle a été obtenue au bout de 6 mois chez les patients prenant de la ciclosporine. Au bout de deux ans de traitement, aucune différence significative n’a été observée entre les deux groupes.
Une autre étude rétrospective a montré que l’association de la ciclosporine avec une corticothérapie était plus efficace que lorsqu’elle était utilisée seule.
Dans une autre série, l’association de la ciclosporine à la prednisolone chez 22 patients a montré une amélioration ou une stabilisation de l’acuité visuelle chez 95% des malades. Chez tous les patients, ce traitement a permis une diminution des réactions inflammatoires au niveau des lésions oculaires et une amélioration des autres manifestations.
Les effets secondaires de la ciclosporine sont nombreux et notamment rénaux, à type de tubulopathies proximales habituellement réversibles. Plus rarement, il peut y avoir une fibrose interstitielle irréversible.
Une hypertension artérielle peut être observée pouvant justifier la prescription d’antihypertenseurs en s’abstenant de prescrire des diurétiques.
De plus la ciclosporine a été accusée de favoriser la survenue de complications neurologiques au cours de la maladie de Behçet, notamment chez les patients ayant une localisation neurologique préexistante.
D’autres effets secondaires sont possibles tels qu’une hypertrichose, un tremblement, une microangiopathie thrombotique, une hypertrophie gingivale.
Enfin, de nombreuses interactions médicamenteuses en limitent l’utilisation et justifient la surveillance des taux sériques de ciclosporine.
Les agents biologiques :
- Les Anticorps monoclonaux anti-Tnfα L’anticorps le plus utilisé dans ce contexte est l’infliximab (Remicade®), c’est un anticorps partiellement humanisé ou la partie variable de l’anticorps vient de la souris alors que le reste de la molécule est d’origine humaine.
Cet anticorps est un puissant immunomodulateur, il se fixe avec une forte affinité au TNFα libre ou lié à un récepteur. Il possède également une activité cytolytique. Sa demi-vie est de 14 jours (8).
Il est depuis quelques années utilisé avec succès dans le traitement des phénomènes inflammatoires sévères comme la maladie de Crohn et l’arthrite rhumatoïde, c’est d’ailleurs dans ce contexte que l’on avait observé un effet positif sur l’uvéite antérieure HLA-B27 qui a mené à son usage dans l’inflammation oculaire.
Il est utilisé à la dose de 5mg/kg en perfusion lente intraveineuse toutes les 8 semaines (9).
Dans le traitement de l’atteinte oculaire sévère de la maladie de Behçet, les résultats sont très satisfaisants avec amélioration spectaculaire de l’inflammation oculaire aux premières heures, contrôle de la vascularite et de la nécrose rétinienne au cours des 2 premières semaines ainsi qu’une réduction des rechutes oculaires par un facteur 5.
Ces effets positifs dans le Behçet oculaire ont été rapportés par plusieurs auteurs, d’autres études ont rapportés l’efficacité de l’infliximab non seulement dans les uvéites réfractaires de la maladie de Behçet mais également dans d’autres uvéites réfractaires, d’autre origine. L’infliximab est indiqué au cours des manifestations oculaires initiales devant une inflammation du segment postérieur en cas d’atteinte unilatérale avec une acuité visuelle inférieure à 2/10, ou en cas d’atteinte bilatérale avec menace visuelle.
Il est également indiqué lorsque surviennent 2 ou plusieurs rechutes par an, ou en cas d’intolérance à des doses appropriées d’azathioprine et/ou d’interféron.
Ce traitement doit se faire en collaboration étroite avec le service de médecine interne. En effet plusieurs cas de tuberculose disséminée ont été induits par l’infliximab, le dépistage de la tuberculose est de ce fait systématique. D’autres risques sont moins importants tels que la sclérose en plaques, l’insuffisance cardiaque congestive, ainsi que la production d’auto-anticorps car l’infliximab n’est pas un anticorps totalement humanisé (15).
• L’interféron α2A
Les interférons sont de petits peptides d’environ 160 acides aminés qui peuvent être synthétisés par la plupart des cellules et qui ont des propriétés antivirales antipro-lifératives et immunomodulatrices.
On distingue les interférons α : d’origine macrophagique, β : d’origine fibroblastique et γ : d’origine lymphocytaire T (10).
L’INF α recombinant issu du génie génétique est largement utilisé pour ses propriétés antivirales au cours de l’hépatite B active, pour ses propriétés antitumorales au cours des syndromes myéloprolifératifs de certaines néoplasies, et est largement utilisé pour ses propriétés immunomodulatrices au cours de la maladie de Behçet et au cours d’uvéites réfractaires incluant les uvéites dues à la maladie de Behçet.
Une méta-analyse a repris 36 études incluant 338 patients atteints de la maladie de Behçet dont 182 présentaient une uvéite récurrente, chaque étude relatait de façon rétrospective un petit nombre de cas.
Bien que cette méta-analyse montre l’intérêt indiscutable de l’INF α au cours des différentes atteintes de la maladie de Behçet, et en particulier son action bénéfique sur la diminution de la fréquence et de la gravite des uvéites, seuls de tous petits groupes de patients ont été évalués de façon rigoureuse.
Le seuil de corticodépendance est significativement abaissé lorsque l’INF α est utilisé en association (12).
L’INF α utilisé est l’INF α2a (Roferon®). Les doses prescrites varient entre 3 et 9 millions d’unités par jour, 3 fois par semaine, la plupart des auteurs s’accordent pour prescrire les doses utilisées au cours du traitement des hépatites soit 3 millions d’unités 3 fois par semaine (23.24). L’effet secondaire quasiconstant est un syndrome grippal qui nécessite la prescription associée de paracétamol. Un effet secondaire redoutable est neuropsychiatrique avec un syndrome dépressif accompagné d’idées suicidaires.
Une leucopénie, une thrombopénie et des manifestations cutanées à type de prurit, d’alopécie peuvent survenir.
Biologiquement des anticorps sériques divers peuvent apparaitre, en général sans émergence de manifestations systémiques, cependant des thyroïdites, des lupus induits ont été décrits. Par ailleurs la survenue de sarcoïdose est possible.
Les effets secondaires ophtalmologiques sont une rétinopathie avec nodules cotonneux, hémorragies rétiniennes en flammèches, œdème rétinien, oblitérations artériolaires ou veineuses. Des neuropathies optiques ischémiques ont été décrites.
Les traitements intraoculaires
• Triamcinolone en injections intravitréennes
Il existe plusieurs études pilotes mais aucune étude randomisée portant sur l’utilisation intravitréenne de la triamcinolone acétonide dans le traitement de l’œdème maculaire compliquant la maladie de Behçet.
La triamcinolone permet d’améliorer rapidement la fonction visuelle et de diminuer l’œdème maculaire, mais cet effet est temporaire, et une réinjection devient nécessaire 3 à 6 mois plus tard.
L’injection s’effectue par l’administration de 0,1 ml de triamcinolone à 40 mg/ml à 4 mm du limbe. Elle est faite de préférence dans la partie inférieure de l’œil (entre 4 et 8 heures) pendant que le patient regarde vers le haut. Ces injections peuvent également se faire en sous ténonien.
Trois complications ont été décrites : une augmentation transitoire de la pression intraoculaire, l’évolution d’une cataracte et la survenue d’une pseudo-endophtalmie 24 à 72 heures après injection (21.22).
• Implant de Fluocinolone Acetonide
Cet implant est commercialisé aux états unis, il doit être inséré par voie chirurgicale au niveau de la pars plana, et placé de préférence au niveau de la partie inférieure de l’œil.
Il contient de l’acétonide de fluocinolone qui diffuse de façon constante dans l’œil pendant une période de 72 mois (17).
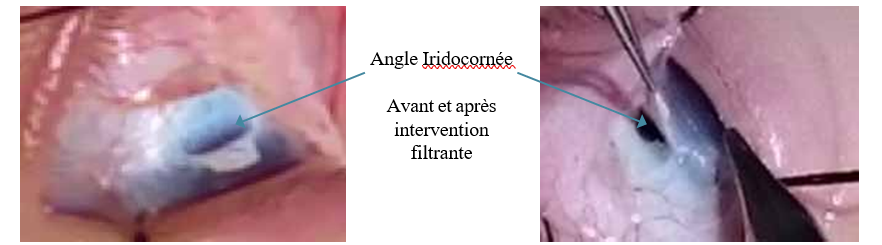
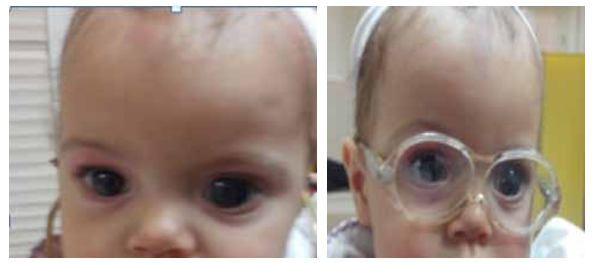
Les résultats rapportés à 34 semaines d’une étude prospective, randomisée, multicentrique et contrôlée portant sur 278 patients avec panuvéite récidivante non infectieuses ont démontré une diminution du taux de rechutes de 51,4% à 6,1% (p<0,001) mais avec une augmentation du taux de rechutes de l’œil controlatéral de 20,3% à 42% , et survenue d’un glaucome secondaire dans 51,1% des cas nécessitant une chirurgie filtrante dans 5,8% des cas et une cataracte secondaire dans 19,8% des cas (19).
• Les Anti-VEGF
De petites études pilotes ont été publiées sur l’utilisation des anti-VEGF dans le traitement des œdèmes maculaires compliquant les uvéites et dans le traitement des néovaisseaux inflammatoires montrant une diminution de l’épaisseur maculaire et une amélioration de l’acuité visuelle, mais l’effet est transitoire et ne dure que 2 à 3 semaines. De plus ces études concernent un petit nombre de patients et le suivi est relativement court (16).
L’anti-VEGF qui a été utilisé est le Bevacizumab (Avas- tin®) qui est commercialisé pour l’usage intraveineux sous forme de flacons de 4 ml à la concentration de 25 mg /ml pour le traitement des cancers colorectaux, il est utilisé dans ce contexte hors AMM. La dose injectée en intravitréen est de 1,25 mg, elle se fait à la pars plana dans le quadrant inféro-temporal à 4mm du limbe.
Traitement des complications oculaires
• Chirurgie de la cataracte
La chirurgie de la cataracte compliquée ne peut être proposée qu’après une rémission d’au moins 3 mois.
L’existence de rechutes pendant l’année qui précède la chirurgie augmente le risque de rechute postopératoire.
Cette chirurgie doit être minutieuse, avec nettoyage du sac capsulaire et mise en place d’un implant cristallinien tout polyméthyl méthacrylate ou à surface héparinée dans le sac capsulaire. Les implants cristalliniens en acrylique sont mal tolérés, responsables d’une exacerbation des phénomènes inflammatoires uvéaux et vitréens. Une corticothérapie systématique périopératoire à la dose de 0,5 mg/kg/j, démarrée une semaine avant la chirurgie permet de réduire le risque de rechute post- opératoire.
Les immunosuppresseurs sont maintenus. L’association d’une corticothérapie topique en post-opératoire est indispensable.
Les résultats fonctionnels de la chirurgie de la cataracte compliquée sont conditionnés par la sévérité de l’atteinte du pôle postérieur, et notamment par l’atrophie optique et la dégénérescence maculaire post-inflammatoire.
Ainsi l’acuité visuelle post-opératoire serait significativement plus basse par rapport à celle des yeux opérés de cataracte dans les suites d’uvéites idiopathiques.
• Chirurgie vitréorétinienne
Elle est indiquée dans les formes compliquées d’organisation vitréenne. Elle permet d’améliorer la fonction visuelle, et de diminuer de façon significative le nombre et la durée des rechutes en post opératoire (20).
• Traitement de l’ischémie rétinienne
Les territoires d’ischémie rétinienne secondaires aux occlusions veineuses doivent être photocoagulés au laser Argon ou Krypton, en période de rémission.
Ce traitement est généralement bien toléré, parfois une corticothérapie prophylactique peut être nécessaire pour éviter une exacerbation des phénomènes inflammatoires.
Un œdème maculaire peut survenir ou s’aggraver dans les suites d’une photocoagulation rétinienne.
Références :
- BHISITKUL RB, FOSTER CS Diagnosis and ophthalmological features of Behçet’s disease. International ophthalmology clinics 1996; 36 (1): 127 -134.
- BINISTI P. Œil et maladie de Behçet. Rev du Prat 1999; 49: 1999- 2003.
- YATES PA, MICHELSON JB. Behçet disease. International ophthal- mology clinics 2006; 46 (2): 209-233.
- MICHELSON JB. Behçet’s disease. International Ophthalmology clinicis 1990; vol 30 N°4.
- BENTAARIT C et al. Les manifestations articulaires de la maladie de Behçet à propos de 309 cas. Rev médecine interne 2001; 49-55.
- YAZICI H et al. Pratical treatment recommendations for pharma- cotherapy of Behçet’s syndrome. Drugs 1991; 42:796-804.
- YAZICI H et al. A controlled trial of azathioprine in Behçet’s syn- drome. N Engl J Med 1990; 322:281.
- SUHLER EB, SMITH JR, WERTHEIN MS et al. A prospective trial of infliximab therapy for refractory uveitis: preliminary safety and effi- cacy out comes. Arch Ophthalmol 2005; 123 (7): 903-912.
- TOHME A et al. La maladie de Behçet. Press Med 1999 ; 28 :1080- 1083.
- SOBRIN T, KIM EC, CHRISTEN W et al. Infliximab therapy for the treatment of refractory ocular inflammatory disease. Arch ophthalmol 2007; vol 125, N°7.
- REED JB et al. High dose intavenous pulse methylprednisolone hemisuccinate in acute Behçet retinitis. Am J Ophthalmol 1998; 125:410-411.
- PLSKOVA J, GREINER K, FORRESTER JV Interferon-α as an ef- fective treatment for non-infections posterior uveitis and panuveitis. Am J Ophthalmol 2007; 144: 55-61
- OZYAZGAN Y et al. Low dose cyclosporine A versus pulsed cyclophosphamide in Behçet’s syndrome: a single masked trial. Br J Ophthalmol 1992 ; 76:241-243.
- LUSTING MJ, CUNNINGHAM ET. Use of immunosuppressive agents in uveitis. Curr opin ophthalmol 2003; 14: 399-412.
- LINDSTEDT EW, BAARSMA GS, KUIJPERS R VANHGEN PM. Anti-TNF-α therapy for sight threatening uveitis. Br Journal of ophthalmology 2005; 89: 533-536.
- MASKENSEN C, HEINZ C, MATHIAS D. Intravitreal Bevacizu- mab (Avastin) as a treatment for refractory macular edema in patients with uveitis: a pilot study. Retina 2008; volume 28, issue 1.
- KARCORLU M, MUDUN B, OZDEMIR H, BURUMCEK E. In- travitreal triamcinolone for the treatment of cystoid macular edema. Am J Ophthalmol 2004; 138:289-291.
- HIJIKATA K et al. Visual prognosis in Behçet’s disease: effects of cyclophosphamide and colchicine Jpn J Ophthalmol 1978; 16:284-329.
- GOLDSTEIN DA, GODFREY DG, HALL A et al. Intrapressure in patients with uveitis treated with fluocinolone implants. Arch. Ophthalmol 2007; 125:1478-1485.
- FOSTER CS, FARIS BM. Adamandiades Behçet’s disease. Retina and Vitreous: p 2212-2221.
- DEGENRING RF, JONAS JB. Intravitréal injection of triamcino- lone acetonide as treatment for chronic uveitis. Br. J Ophthalmol 2003; 87:361-364.
- DEUTER CME, KÖTTER I, GUERAUDYN I, STUEBIGER N. Interferon alfa-2a: a new treatment option for long lasting refractory cytoide macular edma in uveitis A pilot study. Retina 2006; 26: 78-791.
- DEMIROGLU H. Response to allegations and some consideration on interferon treatment in Behçet disease. British Journal of ophthal- mology 2004; 88: 312.
- BODAGHI B, GENDRON G, WECHSLER B ET AL. Efficacy of interferon alpha in the treatment of refractory and sight threatening uveitis : a retrospective monocentrique study of 45 patients.British Journal of ophthalmology 2007; 91:335-339.