N. HECHAM(1) ; K. BEgHDADI(2); S. NOUIOUA(1) ; L. AIT AISSA(1) M. KEDIHA(1) ; S. ASSAMI(1) ; A. BENDIB(2) ; L. ALI PACHA(1); M. TAZIR(1). Service de neurologie, Service d’imagerie médicale, CHU Mustapha Bacha, Alger.
Abstract : Multiple sclerosis (MS) is a chronic progressive neurological disease and the most frequent CNS disabling. The first episode of MS is called clinically isolated syndrome (CIS). The purpose of this study was to describe the clinical and evolutionary characteristics in the short term of the patients seen in the early stage of the disease. Among the 127 recruited patients, 107 (84%) were women. The mean age of onset was 32.5 years (95% CI = [30,7- 34.2]). The onset mode was mono symptomatic in the majority (79,5%) and signs were dominated by motor disorders (44,1% of cases). 67.7% of patients had presented a second clinical event with a majority (72.1%) during the first year. The median time between the first and the second event was 17 months (95% CI = [13.72 to 20.27]). Multiple logistic regression analysis revealed no predictive clinical or demographic factor in the occurrence of the second event. The early brain MRI responded to Barkhof criteria in 94% of patients. In addition, the medullary MRI performed in 49 patients showed abnormalities in 71% of cases A little more than half of our patients (54.45%) were converted to radiological SEP at their second MRI scan performed 3 months after reference MRI. Among these patients 38% had new T2 lesions and 25% had new enhancing lesions after gadolinium injection. 87 patients had received early treatment with immunomodulators (interferon) after being diagnosed with MS within an average of 6.6 months. We found a 41.2% reduction in relapse rate after 1 year of treatment, 61 patients exceeded 2 years of treatment and 57.4% were free of relapses. For the EDSS, there was no significant difference between the EDSS before and after treatment, however, 70.5% of patients had stabilization or reduction of their disability. It is impossible to determine precisely the prognosis for a given patient at the beginning of the disease using clinical markers. Nevertheless, our data confirm the positive effect of early DMT in RR MS in accordance with previous studies.
Key-words : CIS, MS, MRI, immunomodulators, evolution
Résumé : La sclérose en plaques (SEP) est l’affection neurologique chronique évolutive et handicapante la plus fréquente du SNC. La première poussée de la SEP est dénommée syndrome cliniquement isolé (SCI ou CIS). Le but de cette étude était de décrire les caractéristiques cliniques et évolutives à court terme des patients vus au stade précoce de la maladie. Parmi les 127 patients recrutés, 107 étaient des femmes soit 84%. L’âge moyen des patients à la survenue du premier événement clinique était de 32,5 ans (IC 95% = [30,7- 34,2]). Le mode début mono symptomatique a été retrouvé chez 101 patients (79,5%). Les signes cliniques de début sont dominés principalement par les troubles moteurs dans 44,1% des cas, les troubles sensitifs dans 34,6%, les signes cérébelleux dans 28,3%, la névrite optique rétrobulbaire dans 23,6% et les troubles du tronc cérébral dans 19,7% des cas. 67,7% des patients ont présenté un 2ème événement clinique dont la majorité (72,1%) durant la première année. Et le délai médian entre le 1er et le 2ème événement était de 17 mois (95%CI = [13,72 – 20,27]). L’analyse par régression logistique multiple n’a révélé aucun facteur démographique ou clinique prédictifs dans la survenue du 2ème événement. L’IRM cérébrale de début répondait aux critères de Barkhof dans 94% des cas. Et l’IRM médullaire faite chez 49 patients a montré des anomalies dans 71% des cas. L’IRM cérébrale de contrôle réalisée 03 mois après l’IRM de référence chez 101 patients a confirmé la dissémination temporelle dans 54,45% des cas, avec l’apparition de nouvelles lésions T2 dans 38% des cas et de nouvelles lésions actives dans 25% des cas. 87 patients avaient reçu précocement un traitement par immunomodulateurs (interférons) après avoir posé le diagnostic de SEP dans un délai moyen de 6,6 mois (95% CI = 5,33 – 7,87). Les caractéristiques cliniques des patients étaient semblables à celles des cohortes rapportées dans la littérature et aucune association n’a été retrouvée entre l’âge, le sexe, la présence de BOC, les signes cliniques et la survenue d’un 2ème événement clinique. Nous avons constaté une réduction de 41,2 % du taux de poussées (TAP) après 1 an de traitement, 61 patients ont dépassé 2 ans de traitement et 57,4% étaient libres de poussées. Pour l’EDSS, il n’y avait pas de différence significative entre l’EDSS avant et après traitement, néanmoins 70,5 % des patients avaient une stabilisation ou une amélioration de leur handicap.
Il est impossible de déterminer précisément le pronostic pour un patient donné au début de la maladie à l’aide de marqueurs cliniques. Néanmoins, nos données confirment l’effet positif des DMT dans les formes rémittentes et sont conformes aux résultats des études précédentes.
Mots-clés : CIS, SEP, IRM, Immunomodulateurs, évolution.
Introduction :
La sclérose en plaques (SEP) est une affection chronique du système nerveux central caractérisée par la survenue de lésions de démyélinisation multifocales, une réaction inflammatoire et une atteinte axonale précoce. C’est une maladie hétérogène dans ses symptômes, dans son évolution générale et dans son pronostic fonctionnel [1,2]. Du fait de la dissémination spatiale des lésions au sein du SNC, la symptomatologie de la SEP est extrêmement variée. Cette variété s’exprime tant dans les manifestations initiales que dans l’évolution de la maladie et ses conséquences [3].
La première poussée de la maladie est dénommée syndrome cliniquement isolé (SCI ou CIS pour Clinically Isolated Syndrome) et elle est monosymptomatique (50 à 70% des cas) ou polysymptomatique. Selon les études et les pays, la présentation clinique varie mais globalement les symptômes sensitifs (45 %), moteurs (20 %) et visuels (17 %) sont les plus fréquents et représentent plus de 70% des tableaux cliniques initiaux [4], par ailleurs, il existe d’autres symptômes ou signes moins perceptibles (dépression, fatigue, etc.), qui peuvent passer inaperçus et qui ne seront diagnostiqués que de manière rétrospective [5,6]. 85% des patients qui ont une SEP ont débuté leur
maladie par un SCI. Par ailleurs, la SEP est variable d’un patient à un autre ; en effet, plus de la moitié d’entre eux évoluent vers un handicap après 15 à 20 ans d’évolution. Dans les études cliniques sur le court terme (deux ans), le risque de nouvelles poussées varie de 17% à 45% suivant les critères d’inclusion des patients. Le risque le plus faible est celui des patients atteints de NORB et le plus élevé est celui des patients atteints de SCI multifocal avec de multiples lésions à l’IRM [7]. Plusieurs études ont été réalisées pour rechercher les facteurs prédictifs cliniques de conversion du SCI en SEP. Cependant, la plupart de ces études ont été faites dans les populations de l’Europe et d’Amérique du nord [8,9].
Enfin, les caractéristiques phénotypiques et évolutives de la SEP différent selon les populations étudiées. Récemment au Maghreb, un profil évolutif particulièrement sévère a été démontré dans plusieurs études au Maroc, en Tunisie et en Algérie [10,11,12,13,14]. Certaines de ces études ont porté sur des cohortes multicentriques, révélant une évolution rapide de la maladie vers le handicap. Concernant l’évolution à court terme des CIS, aucune étude n’a été réalisée en Algérie en dehors de celle de Beghdadi [15].
Patients et méthodes :
Le recrutement des patients s’effectuait de Juillet 2012 à juillet 2016 à la consultation hebdomadaire de SEP multidisciplinaire du CHU Mustapha. Les patients étaient ensuite répertoriés au fur et à mesure, sur une base de données comportant les éléments cliniques avec l’évaluation du handicap en utilisant l’EDSS [16], les éléments paracliniques, évolutifs et les thérapeutiques de fond utilisées. Sont inclus dans l’étude tous les patients ayant présenté des symptômes compatibles avec une maladie inflammatoire démyélinisante dont le diagnostic correspondait à celui de SEP, et SEP possible, selon la classification de Mc Donald (2005 et 2010) [18,19]. La catégorie « SEP » possible incluait les patients ayant présenté un épisode clinique isolé (CIS) compatible avec une SEP sans les critères de dissémination temporelle et/ou spatiale. Durant le suivi nous avons colligé 400 patients dont 127 ont été vus au stade précoce de la maladie (CIS correspondant à la première poussée de SEP). Tous les patients ont été suivis durant une année et plus ; ils ont tous eu une IRM cérébrale complétée ou non d’une IRM médullaire. Une IRM cérébrale de contrôle a été réalisée à 3 mois après l’IRM de base chez les patients ayant présenté un seul événement neurologique très suggestif de SEP. Dans notre travail, nous avons décrit les caractéristiques cliniques de ces derniers, soit des 127 patients vus au stade précoce de la maladie. Nous avons essayé de rechercher le délai entre les 2 premières poussées, avec l’identification des facteurs prédictifs cliniques du risque de survenue d’un 2ème événement clinique. Et enfin, nous avons évalué la réponse à court terme des traitements immunomodulateurs en se basant sur la réduction du taux de poussées et sur l’absence de la progression du handicap (EDSS).
Les méthodes statistiques appliquées sont le test de Khi2 et de Fischer pour la comparaison des variables qualitatives. La probabilité de la survenue du 2ème événement a été calculée par la méthode de Kaplan Meier. L’analyse multivariée a été utilisée afin d’étudier l’interrelation entre plusieurs variables et d’estimer le ou les facteurs de risque les plus prédictifs pour l’ensemble des analyses effectuées. Une valeur du seuil de significativité p du test < 0,05 était considérée comme statistiquement significative. Les analyses ont été menées sur les logiciels « SPSS version 19 » et « Med Calc Statistical Software version 14.8.1 ».
Résultats :
1. Caractéristiques démographiques de la cohorte de patients CIS :
Parmi les 127 patients recrutés, 107 étaient des femmes soit 84%. L’âge moyen des patients à la survenue du premier événement clinique était de 32,5 ans (IC 95% = [30,7- 34,2]) avec un minimum de 14 ans, un maximum de 56 ans et un âge médian de 33 ans (tableau 1). La durée de la maladie (depuis l’apparition des premiers symptomes jusqu’au dernier examen) varie entre 1 et 4 ans avec une médiane de 3 ans. Le délai diagnostic varie entre 1 et 9 mois avec une moyenne de 4,09 mois (IC 95%= [3,64-4,55]) et une médiane de 4 mois. Chez 100 patients (79%), le diagnostic a été retenu dans les 3 à 6 mois après le début de la maladie.
| Caractéristiques | No = 127 patients | |
| Sexe n (%) | ||
| Hommes | 20 (15,8) | |
| Femmes | 107 (84,2) | |
| Age de début (ans) | ||
| Moyenne | 32,48 (95%CI= 30,7-34,2) | |
| Extrêmes | 14- 56 | |
| Durée de la maladie (ans) | 2,74 (95%CI= 2,54-2,94) | |
| Délai diagnostic (mois) | 4,09 (95%CI= 3,64-4,55) | |
| Intervalle P1-P2 | ||
| (Kaplan Meier) (mois) | ||
| Moyenne | 26,8 (95%CI = 22,74-30,85) | |
| Médiane | 17 (95%CI = 13,72-20,27) | |
| Interféron No | 87 | |
| Délai du TRT moyenne | 6,6 ±5,94 | |
| (mois) | ||
| Durée du TRT moyenne (ans) (extrême) | 1,99 ± 0,8 (1-4) |
Le mode de début monosymptomatique a été retrouvé chez 101 patients (79,5%). Les signes cliniques de début sont dominés principalement par les troubles moteurs chez 56 patients (44,1%), les troubles sensitifs chez 44 patients (34,6%), les signes cérébelleux chez 36 patients (28,3%), la névrite optique rétrobulbaire chez 30 patients (23,6%) et les troubles du tronc cérébral chez 25 patients (19,7%).
• Le délai de survenue du 2ème événement :
Parmi les 127 patients, 86 (67,7%) ont présenté un 2ème événement clinique dont la majorité (72,1%) durant la première année. 21 patients (24,4%) durant la 2ème année et 3 patients seulement (3,5%) ont eu un 2ème événement après la troisième année. La probabilité de la survenue du 2èmeévénement a été calculée par la méthode de Kaplan Meier. Le délai médian entre P1-P2 était de 17 mois (95% CI = [13,72 – 20,27]) (figure 1). L’analyse multivariée a été utilisée afin d’étudier l’interrelation entre plusieurs variables et d’estimer le ou les facteurs de risque les plus prédictifs. Les variables incluses ont été : l’âge de début, le sexe, les signes cliniques de début (troubles moteurs, cérébelleux, l’atteinte du TC et l’atteinte polysymptomatique) et la présence de bandes oligoclonales dans le LCR. L’analyse par régression logistique multiple n’a révélé aucun facteur démographique ou clinique prédictif dans la survenue du 2ème événement.
• IRM cérébrale et médullaire :
L’IRM cérébrale de début répondait aux critères de Barkhof chez 119 patients soit 94%. L’IRM médullaire faite chez 49 patients a montré des anomalies chez 35 patients (71%), et était normale chez 14 patients (29%). L’IRM cérébrale de contrôle réalisée 03 mois après l’IRM de référence faite chez 101 patients a
confirmé la dissémination temporelle chez 55 patients (54,45%), avec l’apparition de nouvelles lésions T2 chez 39 patients (38%) avec de nouvelles lésions actives chez 26 patients (25%).
• Étude du LCR :
Les bandes oligoclonales BOC étaient présentes chez 98 patients (78,4%).
1. Évaluation du traitement par interféron :
87 patients avaient reçu un traitement par immunomodulateurs (interférons) après avoir posé le diagnostic de SEP dans un délai moyen de 6,6 mois (95% CI = 5,33 – 7,87) avec un minimum de 1 mois et un maximum de 24 mois. La durée moyenne de traitement était de 1,99 ans (95% CI = 1,82 – 2,16) avec des extrêmes allant de 1 à 4 ans. 61 patients. (70%) avaient une durée de traitement supérieure ou égale à 2 ans.
2.1. Évaluation du nombre de poussées :
Nous avons constaté une réduction de 41,2 % du taux de poussées (TAP) après 1 an de traitement.
Concernant l’EDSS, il n’y avait pas de différence significative entre l’EDSS avant et après traitement (Tableau 2).
| Avant traitement | Après traitement | P valeur | |
| Taux annuel de poussées | 1,31 ± 0,05 | 0,54 ± 0,08 | 0,001 |
| EDSS | 1,56 ± 0,90 | 1,55 ± 0,12 | 0,915 |
Sur les 87 patients, 61 ont dépassé 2 ans de traitement : 35 (57,4%) étaient libres de poussées, 23 (37,7%) avaient fait entre 1 et 2 poussées et 3 patients seulement (4,9%) avaient fait plus de 2 poussées (Tableau3).
| Nombre de poussées | Avant traite- ment | 2 ans après traitement |
| 0 | 35 (57,4%) | |
| 1 | 41 (67,2%) | 18 (29,5%) |
| 2 | 20 (32,8%) | 5 (8,2%) |
| >2 | 3 (4,9%) |
2.2. Évaluation du score EDSS :
Le tableau 4 résume l’évolution des scores EDSS à moyen terme (à 1 an puis à 2 ans). Une stabilité correspondait à une fluctuation du score de plus ou moins 0,5 point au maximum. L’aggravation du score EDSS correspondait à une augmentation persistante d’au moins 1. Une amélioration correspondait à une diminution d’au moins 1 point du score EDSS.
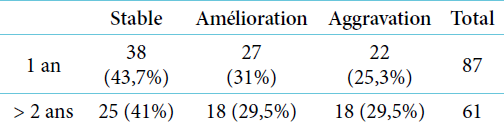
De façon globale, à 2 ans, 70,5 % des patients avaient une stabilisation ou une amélioration de leur handicap. (Tableau 4)
Discussion :
Les caractéristiques cliniques des patients étaient semblables à celles des cohortes rapportées dans la littérature. L’âge moyen de notre série était de 32,5 ans, comparé à 32,6 ans dans l’étude menée par Liu [20], 29 ans dans l’étude de Wing [21] et 25,5 ans dans l’étude réalisée par Alroughani et al. en 2012 [22]. Dans la présente étude, 67,7% des patients ont présenté un 2ème événement clinique pendant la période de suivi. D’Alessandro et al [9], Alroughani et al [23] et Ruet et al [24], ont rapporté la probabilité de survenue à 2 ans, d’un 2ème événement clinique à 36%, 60,8% et 53,5% respectivement. Tous étaient au-dessous du taux de 77% observé dans le groupe traité par placebo dans l’étude de l’essai CHAMPS en 2002 [25]. La majorité de nos patients étaient des femmes, mais concernant le sexe il n’y avait pas de différence dans le taux de ceux qui ont fait un 2ème événement clinique et ceux qui ne l’ont pas fait, confirmant les données déjà publiées par Alroughani en 2012 [23] et Mowry et al en 2009 [26]. L’identification des facteurs prédictifs du risque de survenue d’un 2ème événement clinique peut être bénéfique, surtout sur le plan thérapeutique, car il a été démontré qu’un traitement précoce pourrait retarder dans le temps la survenue d’un 2ème événement clinique et réduire ainsi la fréquence de conversion des CIS en SEP cliniquement définie [27,28,29].
Diverses études ont été menées pour identifier ces facteurs de risque. Cependant, la plupart de ces études ont été faites dans les populations de l’Europe et d’Amérique présentés un 2ème événement entre 3 et 19 mois [23]. En France, Confavreux et al. en 2003 [30] qui ont étudié l’histoire naturelle de 1.215 patients, ont constaté que le délai médian d’un 2ème événement clinique était de 21mois. En 2011, Ruet a retrouvé un délai moyen de 15 à 17 mois [24]. Debouverie et al. ont rapporté un délai entre les 2 premières poussées court chez les patients originaires du Maghreb par rapport aux Européens [31]. Les signes cliniques de début ne semblent pas affecter le délai de survenue d’un 2ème événement clinique. Les essais cliniques effectués sur les patients CIS n’ont pas montré de différences dans les signes cliniques de début et l’apparition d’un 2ème événement clinique [32] [27] [28] [33].
Tintoré et coll. ont constaté que les facteurs démographiques et cliniques sont des facteurs à impact faible pour développer un 2ème événement [34]. Dans certaines études, quelques signes cliniques de début ont été associés à la survenue précoce d’un 2ème événement. Ainsi, dans l’étude de Ruet et al. [24], l’atteinte sphinctérienne était le seul signe clinique de début constituant un facteur de risque d’un 2ème événement. Dans l’étude de Wing et al. [21], les patients ayant présenté au début une atteinte cérébelleuse, avaient une tendance à avoir un 2ème événement dans un intervalle de temps plus court.
Dans notre étude, aucune association n’a été retrouvée entre l’âge, le sexe, la présence de BOC et la survenue d’un 2ème événement clinique. Alroughani et al. [23] ont rapporté que l’âge de début jeune était un facteur prédictif de la survenue d’un 2ème événement dans les 2 ans. Concernant la présence de BOC, aucune différence significative n’a été retrouvée entre la présence ou l’absence de BOC et le délai de survenue d’un 2ème événement dans 2 études récentes [20,21]. Beghdadi [15] a retrouvé dans son étude que tous les paramètres IRM (le nombre de lésions T2, la topographie et l’activité lésionnelle), avaient une valeur prédictive positive dans la conversion du SCI en SEP selon les critères de Mc Donald, augmentant significativement le risque d’une deuxième poussée. De ce fait, l’IRM est actuellement l’outil le plus puissant pour prédire la conversion en SEP cliniquement définie [34].
Dans notre travail comme dans l’étude de Beghdadi [15], l’IRM cérébrale de début était pathologique dans tous les cas et répondait aux critères de Barkhof chez 94% des patients. Un peu plus de la moitié de nos patients (54,45%) se sont convertis en SEP radiologique à leur 2ème IRM de contrôle réalisée 3 mois après l’IRM de référence, dont 38% avaient de nouvelles lésions T2 et 25% avaient de nouvelles lésions rehaussées après injection de gadolinium.
L’application des critères de Mc Donald de dissémination dans le temps et dans l’espace a certainement influencé la forte proportion de SEP radiologiques. Gómez-Moreno et al. ont appliqué la version révisée de 2010 des critères de Mc Donald sur 67 CIS ; après moins de 2 ans de suivi, 74% ont développé une SEP radiologique. Ils ont conclu que le critère DIT à l’aide d’une IRM unique pourrait améliorer la précision du diagnostic précoce de la SEP dans ce groupe de patients présentant un seul événement clinique [35]. Dans l’étude d’Alroughani et al. [23], 37 patients sur 59 ont développé une SEP radiologique sur une IRM de contrôle réalisée entre 3 et 6 mois après l’IRM de référence.
Il convient de noter que la plupart des études sont en accord avec la notion que l’impact des immunomodulateurs actuels appelés par les anglophones « Disease Modifying Treatment (DMT) » est probablement le résultat de la réduction de l’effet anti-inflammatoire au début de la maladie plutôt d’un effet sur la progression [36,37]. Les caractéristiques des patients étaient semblables à celles des cohortes rapportées dans la littérature en ce qui concerne le taux annuel de poussées (TAP), et la proportion des répondeurs (environ 60%) [38,39]. En basant la réponse du traitement sur la réduction du taux de poussées et sur l’absence de la progression du handicap (EDSS), environ 70,5% de nos patients ont eu une stabilisation ou une amélioration de leur handicap et 57,4% étaient libres de poussées après 2 ans de traitement. Nos données confirment l’effet positif des DMT dans les formes rémittentes documentées par les essais cliniques et sont conformes aux résultats des études précédentes. En fait, le pourcentage des répondeurs était proche de celui rapporté par Waubant et al. [38] (68,3%), Portaccio et al [40] (72%) et un peu plus faible que celui de Fromont et al [41] qui était de 87%. En pratique, les interférons ne réduisent pas le taux de poussées à 100%, et les critères de réponse au traitement basé sur les poussées ont des limites. Dans la SEP, il y a une réduction spontanée du nombre de poussées dans le temps [30]. Cependant dans notre étude, les patients sont nouvellement diagnostiqués avec une courte durée de la maladie, ce qui pourrait limiter l’impact de ce biais sur nos résultats. Ainsi, l’évaluation de ces traitements devrait se faire aussi sur les données d’imagerie. Ces données nous manquaient et devraient être prises en compte dans de futurs travaux.
Conclusion :
Il est impossible de déterminer précisément le pronostic pour un patient donné au début de la maladie à l’aide de marqueurs cliniques [42,43]. La même chose s’applique probablement à la réponse aux traitements [44]. Néanmoins, nos données confirment l’effet positif des DMT dans les formes rémittentes et sont conformes aux résultats des études précédentes. D’autres marqueurs non cliniques, tels que ceux donnés par l’IRM et surtout les marqueurs pharmacogénétiques pourraient aider à répondre à la question. Des efforts supplémentaires sont nécessaires pour mieux identifier les patients qui répondent au traitement, en intégrant la clinique, l’IRM et les marqueurs pharmacogénétiques.
Références
- Confavreux C, Compston A. The naturel history of multiple sclerosis. In: McAlpine’s multiple sclerosis – 4thedition.Compston A ed. London: Churchil Livingstone Elsevier. 2006; p. 138-276.
- Confavreux C, Compston, A. The natural history of multiple sclerosis. In: Compston A, McAlpine’s Multiple Sclerosis. 4th ed. London: Churchill Livingstone Elsevier; 2006: 183-272.
- Ouallet JC; Brochet B. Aspects cliniques, physiopathologiques et thérapeutiques de la sclérose en plaques. EMC-Neurologie 1. 2004; 415–457.
- Weinshenker BG. The natural history of multiple sclerosis: update 1998. Semin Neurol. 1998; 18(3):301-7. Review.
- Lerdal A, Celius EG, Krupp L, Dahl AA. A prospective study of patterns of fatigue in multiple sclerosis. Eur J Neurol. 2007 Dec;14(12):1338-43.
- Siegert RJ, Abernethy DA. Depression in multiple sclerosis: a re- view. J Neurol Neurosurg Psychiatry. 2005 Apr; 76(4):469-75.
- Miller D, Barkhof F, Montalban X, Thompson A, Filippi M. Clini- cally isolated syndromes suggestive of multiple sclerosis, part I: natu- ral history, pathogenesis, diagnosis, and prognosis. Lancet Neurol. 2005; 4:281-8.
- Ruet A, Deloire MS, Ouallet JC, Molinier S, Brochet B. Predictive factors for multiple sclerosis in patients with clinically isolated spinal cord syndrome. Mult Scler. 2011 Mar; 17(3):312-8
- D’Alessandro R, Vignatelli L, Lugaresi A, Baldin E, Granella F, et al. Risk of multiple sclerosis following clinically isolated syndrome: a 4-year prospective study. J Neurol. 2013 Jun; 260(6):1583-93.
- Barka Z. Prévalence, formes cliniques, évolution, et traitement de la sclérose en plaques dans la région de Tlemcen. Thèse de doctorat en sciences médicales (2013). Faculté de médecine de Tlemcen.
- Araqi-Houssaini A, Lahlou I, Benkadmir Y, Elotmani H, Hajjaj I, et al. Multiple sclerosis severity score in a cohort of Moroccan pa- tients. Mult Scler 2014 May ; 20(6) :764-5.
- Hecham N, Nouioua S, Sifi Y, Toubal N, Aissa LA et al. Multiple sclerosis: progression rate and severity in a multicenter cohort from Algeria. Mult Scler. 2014 Dec; 20(14):1923-4.
- Sidhom Y, Damak M, Riahi A, Hizem Y, Mrissa R, et al. Clinical features and disability progression in multiple sclerosis in Tunisia: do we really have a more aggressive disease course? J NeurolSci. 2014 Aug 15 ; 343(1-2) :110-4.
- Sidhom Y, Maillart E, Tezenas du Montcel S, Kacem I, Lubetzki C, et al. Fast multiple sclerosis progression in North Africans: Both genetics and environment matter. Neurology. 2017 Mar 28; 88(13):1218-1225.
- Baghdadi K. Apport de l’imagerie par résonnance magnétique conventionnelle dans le syndrome clinique isolé suggestif de la pre- mière poussée de la sclérose en plaques. Thèse de doctorat en sciences médicales faculté de médecine d’Alger (2016).
- Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983 Nov; 33(11):1444-52.
- Roxburgh RH, Seaman SR, Masterman T, Hensiek AE, Sawcer SJ, et al. Multiple Sclerosis Severity Score: using disability and disease du- ration to rate disease severity. Neurology. 2005 Apr 12; 64(7):1144-51.
- Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the «Mc- Donald Criteria». Ann Neurol. 2005 Dec; 58(6):840-6. Review
- Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDo- nald criteria. Ann Neurol. 2011 Feb; 69(2):292-302
- Liu Y, Duan Y, Yu C, Qin W, Chen H, Dong H, Ye J, Butzkueven H, Li K. Clinical isolated syndrome: a 3-year follow-up study in China. Clin Neurol Neurosurg. 2011 Oct; 113(8):658-60.
- Wing AC, Vasconcelos CC, Calvet J, Papais-Alvarenga RM, Thuler LC. Risk factors for convertion to clinically defined multiple sclerosis after clinically isolated syndrome in a racially mixed Brazilian cohort. Clin Neurol Neurosurg. 2016 Jul; 146:40-4.
- Alroughani R, Ashkanani A, Lamdhade S: Clinical characteris- tics of multiple sclerosis in Kuwait: data from the new MS registry of Amiri Hospital. Int J Neurosci 2012; 122: 82.
- Alroughani R, Al Hashel J, Lamdhade S, Ahmed SF. Predictors of Conversion to Multiple Sclerosis in Patients with Clinical Isolated Syndrome Using the 2010 Revised McDonald Criteria. ISRN Neurol. 2012; 2012:792192.
- Ruet A, Deloire MS, Ouallet JC, Molinier S, Brochet B. Predictive factors for multiple sclerosis in patients with clinically isolated spinal cord syndrome. Mult Scler. 2011 Mar; 17(3):312-8.
- Predictors of short-term disease activity following a first clinical demyelinating event: analysis of the CHAMPS placebo group. Mult Scler. 2002 Oct; 8(5):405-9.
- Mowry EM, Pesic M, Grimes B, Deen SR, Bacchetti P, Waubant E. Clinical predictors of early second event in patients with clinically isolated syndrome. J Neurol. 2009 Jul; 256(7):1061-6.
- Comi G, Filippi M, Barkhof F, Durelli L, Edan G, Fernández O, et al; Early Treatment of Multiple Sclerosis Study Group. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet. 2001 May 19; 357(9268):1576-82.
- Comi G, Martinelli V, Rodegher M, Moiola L, Bajenaru O, et al. Effect of glatiramer acetate on conversion to clinically definite mul- tiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009 Oct 31; 374(9700):1503-11.
- Freedman MS. Evidence for the efficacy of interferon beta-1b in delaying the onset of clinically definite multiple sclerosis in indivi- duals with clinically isolated syndrome. Ther Adv Neurol Disord. 2014 Nov; 7(6):279-88.
- Confavreux C, Vukusic S, Adeleine P. Early clinical predictors and progression of irreversible disability in multiple sclerosis: an amnesic process. Brain 2003; 126(Pt 4):770-82.
- Debouverie M, Lebrun C, Jeannin S, et al. More severe disability of North Africans vs Europeans with MS in France. Neurology 2007; 68(1): 29-32.
- Jacobs LD, Beck RW, Simon JH, Kinkel RP, Brownscheidle CM,et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N Engl J Med. 2000 Sep 28; 343(13):898-904.
- Nielsen JM, Pohl C, Polman CH, Barkhof F, Freedman MS, et al. MRI characteristics are predictive for CDMS in monofocal, but not in multifocal patients with a clinically isolated syndrome. BMC Neurol. 2009 May 20; 9:19.
- Tintoré M, Rovira À, Río J, Otero-Romero S, Arrambide G, et al. Defining high, medium and low impact prognostic factors for deve- loping multiple sclerosis. Brain. 2015 Jul; 138(Pt 7):1863-74.
- Gómez-Moreno M, Díaz-Sánchez M, Ramos-González A et al. Application of the 2010 McDonald criteria for the diagnosis of mul- tiple sclerosis in a Spanish cohort of patients with clinically isolated syndromes. MultScler. 2012 Jan; 18(1):39-44.
- Shirani A, Zhao Y, Karim ME, Evans C, Kingwell E, et al. Asso- ciation between use of interferon beta and progression of disability in patients with relapsing-remitting multiple sclerosis. JAMA. 2012 Jul18; 308 (3):247-56.
- Healy BC, Engler D, Gholipour T, Weiner H, Bakshi R, Chitnis T. Ac- counting for disease modifying therapy in models of clinical progression in multiple sclerosis. J Neurol Sci. 2011 Apr15; 303 (1-2):109-13.
- Waubant E, Vukusic S, Gignoux L, Dubief FD, Achiti I, et al. Cli- nical characteristics of responders to interferon therapy for relapsing MS. Neurology. 2003 Jul 22; 61(2):184-9.
- Villoslada P, Oksenberg JR, Rio J, Montalban X. Clinical charac- teristics of responders to interferon therapy for relapsing MS. Neuro- logy. 2004 May 11; 62(9):1653.
- Portaccio E, Zipoli V, Siracusa G, Sorbi S, Amato MP. Response to interferon-beta therapy in relapsing-remitting multiple sclero- sis: a comparison of different clinical criteria. Mult Scler. 2006 Jun; 12(3):281-6.
- Fromont A, Debouverie M, Le Teuff G, Quantin C, Binquet C, Moreau T. Clinical parameters to predict response to interferon in relapsing multiple sclerosis. Neuroepidemiology. 2008; 31(3):150-6.
- Moreau T, Confavreux C. [Can the prognosis of multiple scle- rosis be predicted?]. Pathol Biol (Paris). 2000 Mar; 48(2):132-8. Review.
- Sartori A, Abdoli M, Freedman MS. Can we predict benign multiple sclerosis? Results of a 20-year long-term follow-up study. J Neurol. 2017 Jun; 264(6):1068-1075.
- Río J, Auger C, Rovira À. MR Imaging in Monitoring and Predic- ting Treatment Response in Multiple Sclerosis. Neuroimaging Clin N Am. 2017 May; 27(2):277-287