F. HADJARAB,K. BOUZID, Oncologie Médicale, Centre Pierre & Marie Curie, Alger.
Abstract : Palliative care is an active, continuous, evolutionary care, coordinated and practiced by a professional pluri-team. They aim, in a comprehensive approach and individualised, to prevent or relieve the physical symptoms, of which pain, but also other symptoms, to anticipate the risks of complications and to take into account the psychological, social and spiritual needs, in the respect of the dignity of then eat person. The palliative care see seeks to ovoid the investigations and the unreasonable treatments and refuse with to cause intentionally death. According to this approach, the patient is regarded as a living being and death like a natural process. The palliative care is addressed to the people reached of evolutionary serious diseases or bringing in to play the vital prognosis or in advanced and final phase, like their family and their close relations. The voluntary ones, formed with the accompaniment and pertaining to associations which select them can supplement, with the agreement of the patient or of his close relations, the action of the medical teams, we quote in this article the various means of palliative care used to improve quality of life of the cancer patients and at the end of the lifetime.
Key-words : Palliative care, disease gravity, pain, pain treatment, quality of life improvement.
Résumé : Les soins palliatifs sont des soins actifs, continus, évolutifs, coordonnés et pratiqués par une équipe pluri professionnelle. Ils ont pour objectif, dans une approche globale et individualisée, de prévenir ou de soulager les symptômes physiques, dont la douleur, mais aussi les autres symptômes, d’anticiper les risques de complications et de prendre en compte les besoins psychologiques, sociaux et spirituels, dans le respect de la dignité de la personne soignée. Les soins palliatifs cherchent à éviter les investigations et les traitements déraisonnables et se refusent à provoquer intentionnellement la mort. Selon cette approche, le patient est considéré comme un être vivant et la mort comme un processus naturel. Les soins palliatifs s’adressent aux personnes atteintes de maladies graves évolutives ou mettant en jeu le pronostic vital ou en phase avancée et terminale, ainsi qu’à leur famille et à leurs proches. Des bénévoles, formés à l’accompagnement et appartenant à des associations qui les sélectionnent peuvent compléter, avec l’accord du malade ou de ses proches, l’action des équipes soignantes, nous citons dans cet article les différents moyens de soins palliatifs utilisés pour améliorer la qualité de vie des patients cancéreux et en fin de vie.
Mots-clés : Soins palliatifs, gravité de la maladie, douleur, traitement de la douleur, amélioration de la qualité de vie.
Définition des soins palliatifs :
Les soins palliatifs sont des soins associés aux traitements de la maladie. Ils ont pour objectif de préserver la qualité de vie, de soulager les douleurs physiques et tous les autres symptômes gênants sources de souffrance ou d’inconfort tels que les vomissements, les essoufflements, la confusion mentale, etc., mais aussi de prendre en compte la souffrance psychologique, sociale ou spirituelle du malade et de ses proches.
L’objectif des soins palliatifs : n’est pas de guérir, mais de préserver la qualité de vie des patients et de leur famille face aux symptômes et aux conséquences d’une maladie grave et potentiellement mortelle. La démarche des soins palliatifs vise ainsi à sauvegarder la dignité de la personne et à éviter les traitements et examens médicaux déraisonnables. Ces soins peuvent se mettre en place à domicile, en institution ou à l’hôpital.
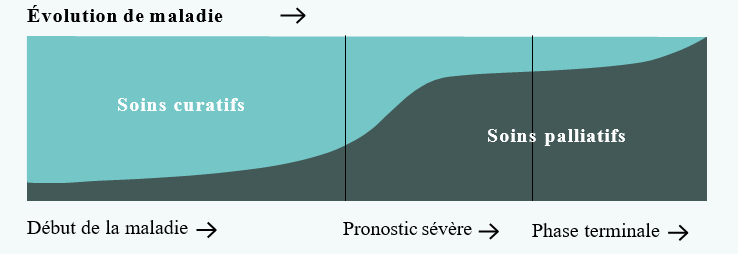
À qui s’adressent les soins palliatifs ? Les soins palliatifs s’adressent soit aux adultes atteints de maladies graves évolutives, mettant en jeu le pronostic vital ou en phase avancée et terminale, soit aux personnes dont la vie prend fin dans le grand âge.
À quels moments de la maladie ? Les soins palliatifs ne sont pas réservés aux malades en fin de vie. Ils sont mis en place au cours de différentes périodes de la maladie. Peuvent être pratiqués tôt dans la maladie, pour aider à mieux la vivre et pour anticiper les difficultés qui pourraient survenir. Ils sont alors associés à des traitements dont l’objectif est de guérir ou de ralentir l’évolution de la maladie (chimiothérapie, radiothérapie, etc.). Enfin, lorsque l’évolution de la maladie est inéluctable et que le décès se rapproche, les soins palliatifs deviennent une priorité.
« Aidées par les équipes de soins palliatifs ».
Quels professionnels concernés par les recommandations ?
Les professionnels concernés sont les médecins généralistes et spécialistes, les infirmier(e)s, les aides soignant(e) s, les kinésithérapeutes, les psychologues, les assistantes sociales, les ergothérapeutes. Des bénévoles formés à l’accompagnement et appartenant à des associations. Où mettre en œuvre les soins palliatifs ? : Ils peuvent être mis en œuvre à domicile, dans les unités de soins palliatifs, dans les établissements de santé en cours, moyen et long séjour, dans les structures destinées aux personnes âgées avec le soutien des unités mobiles de soins palliatifs.
3.Les unités de soins palliatifs (USP) :
sont des services spécialisés en soins palliatifs situés à l’hôpital. Elles prennent en charge les situations les plus compliquées et sont également des lieux de recherche et de formation des soignants.
2. Les équipes mobiles de soins palliatifs
(EMSP) : sont des équipes pluridisciplinaires (médecins généralistes formés pour les soins palliatifs, infirmières, psychologues) qui se déplacent au lit du malade et/ou auprès des soignants.
Les lits identifiés en soins palliatifs (LISP) : sont installés dans des services hospitaliers qui sont confrontés à des fins de vie fréquentes …
4. Les réseaux de soins palliatifs :
Ils apportent conseil, soutien, appui et formation aux équipes soignantes du domicile (infirmières, médecin traitant, services de soins infirmiers à domicile, etc.),
Ils travaillent avec les structures médico-sociales (maison de retraite, etc.),
Ils organisent le retour à domicile dans de bonnes conditions (mise en place des ressources humaines et financières nécessaires, etc.),
Ils prévoient un lit de repli vers un lieu d’hospitalisation si jamais la situation à domicile devient trop difficile.
5. Les services d’hospitalisation à domicile
(HAD) : L’hospitalisation à domicile est mise en place sur prescription médicale lorsque la personne souhaite rester à son domicile et qu’elle nécessite beaucoup de soins et beaucoup de passages de soignants. L’équipe du service d’hospitalisation à domicile travaille en lien avec le médecin traitant et les autres soignants.
Là aussi le rôle du généraliste est capital, car il prend en charge ce genre de patients surtout en fin de vie à leur domicile, en collaboration étroite avec le médecin spécialiste, l’oncologue exerçant à l’hôpital (traitement de la douleur, infections pulmonaires, urinaires, infections cutanées, escarres, dénutrition, perfusions, injection d’antibiotiques …).
À Alger, il existe une structure de HAD à el Biar (Dar El Hakim), il faut savoir que depuis sa création, un grand bénéfice et aide a été apporté pour nos malades en phase palliative du traitement et en fin de vie.
Quels sont les principes de prise en charge des patients en soins palliatifs ?
Le respect du confort, du libre arbitre et de la dignité.La prise en compte de la souffrance globale du patient : (écoute, communication, réconfort, respect de l’autre).
L’évaluation et le suivi de l’état psychique du patient : en soins palliatifs, le patient vit une phase d’angoisse intense quand il se voit confronté à l’approche de sa mort. La perte de sa confiance dans l’avenir, de ses repères, angoissé, révolté, plus vulnérable et plus influençable. Le rôle de l’équipe soignante est de reconnaître cette crise existentielle à partir des symptômes exprimés par le patient (anxiété, insomnies, douleur, etc.).
La qualité de l’accompagnement et de l’abord relationnel du patient et de ses proches nécessitent une disponibilité particulière de l’équipe soignante. Ils visent à signifier d’emblée au patient que sa dignité et son confort seront respectés par les différents professionnels qui interviennent dans la prise en charge.L’information et la communication avec le patient et ses proches.La prise en charge de la phase terminale et de l’agonie : la phase terminale est à anticiper, par le choix du lieu de soins le plus adapté au patient, par la mise à la disposition de l’équipe soignante ou éventuellement des proches, des médicaments nécessaires pour le soulager.
La préparation au deuil : L’instauration d’une bonne communication avec les proches du patient ainsi qu’une information précoce et régulière sur l’évolution de sa maladie.
En pratique, quels sont les soins palliatifs ?
Les soins palliatifs sont des soins actifs qui prennent en compte l’ensemble des besoins de la personne, le soulagement de la douleur et des symptômes gênants.
« Ainsi on qualifie le contrôle de la douleur et des symptômes + soutien psychologique » = soins palliatifs.
Modalités de prise en charge des principaux symptômes : Les symptômes présentés par un patient en soins palliatifs ont comme caractéristiques d’être associés, voire intriqués, surtout quand on approche de la phase terminale…
1. Douleur : Plus de 70 % des personnes à un stade avancé du cancer souffrent de douleurs.
– Évaluation de la douleur : Il est important de demander à chaque patient s’il a mal. (expression du visage, transpiration, pâleur et pouls rapide). Une évaluation précise de la douleur est essentielle pour en identifier les causes. Les questions à poser doivent inclure les suivantes :
Combien de douleurs différentes y a-t-il ? Il est utile de les noter sur une carte du corps. Interrogez le patient sur chacune des douleurs.
Où se trouve la douleur et quelle est la sensation res- sentie ?
Depuis combien de temps souffrez-vous ?
Qu’est-ce qui l’aggrave ou la soulage ?
Des médicaments l’ont-ils jamais soulagé ?
La douleur est-elle pire lors de mouvements ?
Les os ou les articulations sont-ils sensibles ?
Y a-t-il des changements de sensation au niveau de la peau à l’endroit de la douleur ?
Les muscles sont-ils sensibles ou tendus ?
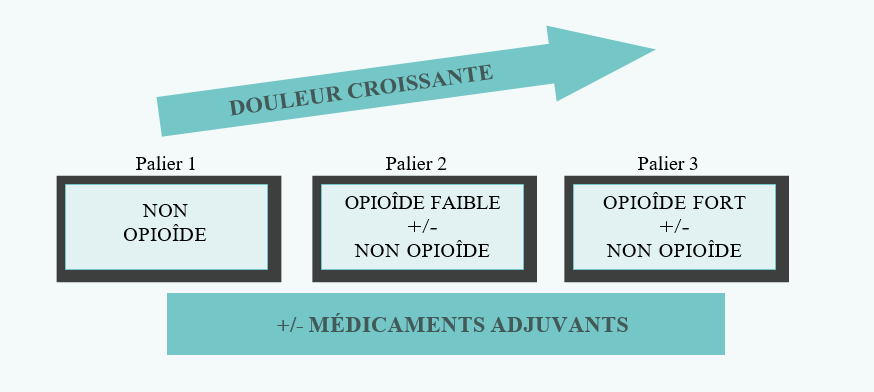
Prescrire : Les analgésiques (médicaments anti-douleur) qui se répartissent en deux groupes :
Non-opioïdes : Ils comprennent le paracétamol (acétaminophène) et les anti-inflammatoires non stéroïdiens (AINS) comme l’aspirine, l’ibuprofène et le diclofénac. Le principal effet secondaire de l’aspirine et des autres AINS est une irritation de l’estomac. Il faut donc qu’ils soient pris, si possible, avec de la nourriture.
Opioïdes : ils comprennent les médicaments similaires à la morphine comme la codéine, le tramadol et la morphine. Les analgésiques doivent être donnés :
Par voie orale : C’est la méthode la plus simple, si le patient ne peut pas avaler de cachets, alors les voies sous-cutanée, rectale ou buccale sont des alternatives.
À heures fixes : Donnez-en à intervalles réguliers selon leur durée d’action, par exemple la codéine 30mg toutes les 4 heures.
Par paliers : Par étape, en fonction de l’accroissement de l’intensité de la douleur (voir ci-après).
Analgésiques non-opioïdes
Dose
Durée de l’effet
Paracétamol
500 mg à 1g 4x/j
4 heures
Aspirine
300 à 600 mgg4x/j
6 heures
Ibuprofen
200 à 400 mg 4x/j
6 à 8 heures
Diclofénac
50 mg 3/j
8 heures
Analgésiques opioïdes
Dose
Durée de l’effet
Codéine (palier 2 )
30 à 60 mg 4x/j
4 à 6 heures
Tramadol (palier 2)
50 à 100 mg 4xx/j
6 heures
Motphine (palier 3)
Aucune limite de dose mais augmenter les doses progressivement
Morphine à libération immédiate (LI)
Dose de départ 2.5 à 5 mg toutes les 4 heures
4 heures
Morphine à libération prolongé (LP)
10 à 20 mg toutes les 12 heures
12 heures
Prescrire de la morphine : La morphine est un analgésique puissant, efficace lorsqu’il est utilisé correctement. Préparations : La morphine se présente sous deux formes :
Morphine à libération immédiate (LI). Elle se présente sous forme de cachet ou de solution préparée à une certaine dose, par exemple 5 mg/5 ml ou 10 mg/5 ml. Prescrivez toujours la dose en mg et non en ml. La morphine LI commence à faire effet après environ 20 minutes et l’effet analgésique dure quatre heures.
Morphine à libération prolongée (LP). Il s’agit de cachets de morphine conçus pour un effet analgésique plus long. La forme la plus courante a un effet de 12 heures et doit être prise deux fois par jour à exactement 12 heures d’intervalles, par exemple à 6h00 et à 18h00 ou à 8h00 et à 20h00.
Dose : Morphine LI : Commencez par des doses de 2,5 à 5 mg toutes les 4 heures. Si le patient prend régulièrement de la codéine, vous pouvez commencer à des doses de 5 à 10 mg toutes les 4 heures. Une dose double peut être prise au moment du coucher pour éviter une dose au milieu de la nuit. Le patient peut également prendre des interdoses (doses de secours), de même quantité, à tout moment si la douleur n’est pas contrôlée par les doses régulières.
Morphine LP : Si possible, commencez toujours par administrer de la morphine LI toutes les 4 heures à un patient. Une fois que vous savez la quantité de morphine dont le patient a besoin, vous pouvez passer à de la morphine LP toutes les 12 heures. Pour calculer la dose de morphine LP ajoutez toutes les doses de morphine LI prises au cours des dernières 24 heures (il s’agit de la dose quotidienne totale de morphine), puis divisez-la par deux pour obtenir la dose de morphine LP à donner toutes les 12 heures.
Augmentation de la dose : si le patient souffre encore après 24 heures et qu’il ne présente aucun signe de toxicité ; augmentez la dose de morphine de 50 %. Continuez à augmenter la dose de 30 à 50 % par paliers de quelques jours jusqu’à ce que le patient ne souffre plus.
Arrêt de la morphine : Si un patient prend de la morphine depuis plusieurs semaines, il ne faut généralement pas arrêter soudainement car cela peut créer des symptômes de sevrage (transpiration, agitation et nausée). La dose doit être réduite sur plusieurs jours puis arrêtée.
Effets secondaires des opioïdes :
Constipation, c’est pourquoi elle doit être toujours prescrite avec un laxatif tel que (Microlax®)
Nausée : lorsqu’ils commencent à prendre de la morphine, ils devront donc prendre un antiémétique tel que metoclopramide (Primperan®) pendant les premiers jours.
Somnolence. Il est courant d’être somnolent au début de la prise de morphine
Transpiration et démangeaisons.
Toxicité et overdose : Les points suivants peuvent être des signes que la dose de morphine est trop forte et que le patient présente des signes de toxicité : somnolence, confusion, hallucinations, myoclonies (contractions brusques des membres), dépression respiratoire (respiration ralentie), déshydratation s’ils souffrent d’insuffisance rénale.
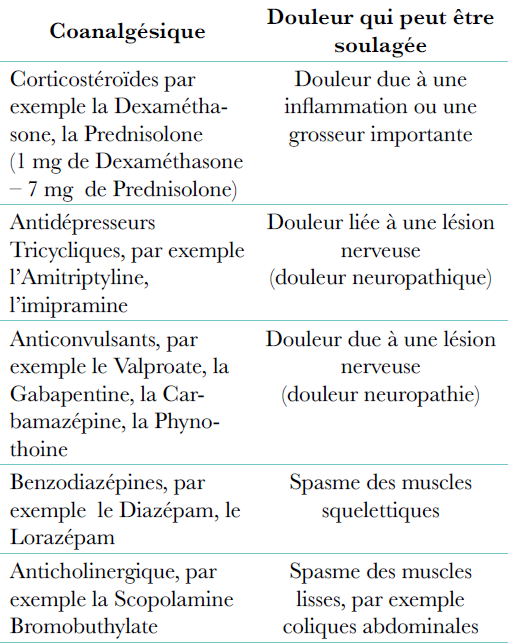
Co-analgésiques : Il s’agit de médicaments qui ne sont pas conçus pour être des analgésiques mais qui peuvent aider à soulager certaines douleurs en complément des analgésiques standards.
Douleurs qui peuvent être soulagées par des co-analgésiques :
Douleur due à une inflammation ou une grosseur importante,
Douleur due à une lésion nerveuse (douleur neuropathique).
1. La douleur due à des lésions nerveuses peut être soulagée avec les co-analgésiques suivants :
Antidépresseurs tricycliques : Ils sont utilisés à des doses inférieures au traitement de la dépression. Le médicament le plus couramment utilisé est l’amitriptyline à la posologie de 12,5 à 25 mg au coucher (cette dose peut être augmentée à 50-75 mg si le patient le tolère).
Anticonvulsivants : La dose de départ doit être faible puis augmentée graduellement, sur quelques semaines jusqu’à ce que la douleur s’amenuise. Des exemples d’anticonvulsivants:
Valproate 200 mg 2x/j (passer à 600 mg 2x/j si nécessaire), Gabapentine 300 mg 3x/j (passer à 900 mg 3x/j si nécessaire) ; Carbamazépine. 100 mg 2x/j (passer à 400 mg 2x/j si nécessaire) Phénytoïne. 100mg 2x/j (passer à 200 mg 2x/j si nécessaire).
Corticostéroïdes à forte dose : Ils peuvent soulager en cas d’inflammations ou de gonflements importants à proximité des nerfs.
2. Asthénie, immobilité :
Asthénie : Comprend des mesures visant à ménager les forces du patient tout en préservant ses capacités d’autonomie et de participation, des recherches de médicaments pris par le patient susceptibles de majorer une asthénie. Un traitement par corticoïdes en cures courtes ou plus rarement par amphétamines (adrafinil, méthylphénidate) peut être proposé.
Immobilité : Il est recommandé d’évaluer précocement le retentissement physique de la diminution de la mobilité chez le patient. La réponse aux besoins doit faire l’objet d’une concertation entre les professionnels de santé (médecin, infirmier(e), aide-soignant(e), kinésithérapeute, ergothérapeute, psychomotricien(ne), psychologue, etc.), le patient, l’entourage et éventuellement les services sociaux. Une aide technique pour l’aménagement de l’environnement (barres d’appui, fauteuil roulant, lit électrique, etc.) est envisagée.
2. Anxiété, dépression, troubles du sommeil :
Anxiété : Le recours au psychologue ou psychiatre peut être nécessaire. Des techniques corporelles (relaxation, massage, etc.) peuvent soulager le patient, un traitement par benzodiazépines (à demi-vie courte) peut être indiqué pour soulager surtout les aspects somatiques de l’anxiété.
Dépression et risque suicidaire : Il est essentiel d’écouter les plaintes et la souffrance du patient et de distinguer la tristesse, émotion naturellement ressentie chez un patient atteint d’une maladie grave évolutive (perte d’autonomie, modifications de l’image et de l’estime de soi, etc.) d’un réel syndrome dépressif. Les antidépresseurs sont proposés en test thérapeutique au moins 3 semaines.
Troubles du sommeil : Ils peuvent être un signe d’alerte d’une anxiété, d’une dépression ou d’un syndrome confusionnel. Le traitement doit être discuté avec le patient et son entourage et dépend du type d’insomnie : zolpidem et zopiclone en cas d’insomnie d’endormissement, benzodiazépine en cas d’anxiété associée, anti-dépresseur sédatif en cas de réveil précoce avec signes dépressifs.
3. Syndromes confusionnels :
Peuvent annoncer la phase terminale peuvent entraîner une déshydratation et arrêt de l’alimentation. La prise en charge dépend de l’étiologie, rechercher en premier lieu une étiologie médicamenteuse ou toxique, en second lieu une cause métabolique (surtout en cas de déshydratation) ou une cause mécanique (globe urinaire, fécalome). Les molécules indiquées sont :
En première intention les neuroleptiques, soit neuroleptiques « classiques » (halopéridol ou phénothiazines), soit en cas d’intolérance, les neuroleptiques atypiques (rispéridone, olanzapine) ;
En cas d’anxiété (en particulier dans les cas de sevrage), les benzodiazépines per os ou par voie parentérale (lorazepam, midazolam), ou le méprobamate, associés si besoin aux neuroleptiques.
4. Sécheresse de la bouche et ulcérations buccales :
Entraînent un réel inconfort voire une douleur chez le patient au niveau des lèvres, de la voix, de la salivation, de la déglutition et de la cavité buccale. Les prothèses dentaires sont nettoyées comme les dents après chaque repas et déposées la nuit. L’humidité de la bouche peut être maintenue exemple : boire de l’eau, sucer ou mastiquer des cubes d’ananas, de la gomme à mâcher, des glaçons, appliquer des compresses humides sur les lèvres.
Diminution de la pression en évitant les appuis prolongés (mobilisation, mise au fauteuil, verticalisation et reprise éventuelle de la marche) et en planifiant des changements de position toutes les 2 à 3 heures, voire à une fréquence plus élevée (les phénomènes de cisaillement et de frottement doivent être évités par une installation et une manutention adéquates du patient, le décubitus latéral oblique à 30° par rapport au plan du lit
Utilisation des supports (matelas, sur matelas, coussins de siège).
Le traitement de l’escarre repose sur la lutte contre la contamination des plaies (notamment avec les urines et les selles), l’utilisation d’un pansement adapté à l’état de la plaie.
Plaies malodorantes : des pansements absorbants au charbon activé peuvent diminuer les odeurs.
Œdèmes : Surveiller l’apparition d’une surinfection et la traiter par une antibiothérapie probabiliste anti-streptococcique. Les œdèmes diffus ou déclives peuvent être soulagés :
Par une contention légère à modérée (de classe 1 ou 2) par bandages ou dispositifs adaptés, à condition qu’il n’existe pas d’insuffisance artérielle
Par une surélévation du ou des membres atteints ;
Par des massages deux fois par jour. Le lymphœdème des membres peut parfois être réduit par une contention modérée à forte, par bandages ou dispositifs adaptés, à condition qu’il n’existe pas de thrombose veineuse.
Prurit : les soins de peau constituent le premier temps du traitement symptomatique d’un prurit. Les bains trop chauds, les agents asséchants (savons trop détergents) ou irritants (lessives, vêtements en laine) sont à éviter et l’usage fréquent d’une crème hydratante est recommandé, il peut être proposé:
En cas de prurit localisé : les corticoïdes locaux ;
En cas de prurit généralisé : les antihistaminiques, les corticoïdes par voie générale.
2. Dyspnée :
il est recommandé de rechercher des signes cliniques de gravité (fréquence respiratoire élevée et cyanose). Lorsqu’un saturomètre est disponible, une mesure de la saturation en oxygène du sang artériel peut être proposée pour confirmer une hypoxie. Pour améliorer la respiration (position demi-assise), en cas de composante obstructive, les broncho-dilatateurs β2 stimulants peuvent être utilisés. S’il existe des sécrétions bronchiques très abondantes, un anti cholinergique est proposé par voie sous-cutanée (scopolamine bromhydrate en première intention ou à défaut atropine). L’oxygénothérapie est recommandée en cas d’hypoxie prouvée (saturométrie ou gaz du sang) ou suspectée. En cas de dyspnées résistantes aux traitements précédents, l’utilisation des opioïdes est proposée.
3. Toux, hoquet rebelle :
Toux : Le traitement symptomatique débute par, l’humidification de l’air inspiré ou des aérosols hydratants (aérosols de sérum physiologique ou d’eau faiblement minéralisée). En cas de toux productive, la kinésithérapie respiratoire si elle n’est pas douloureuse peut être proposée, associée à l’humidification. En cas de sécrétions bronchiques abondantes, on peut proposer la scopolamine (bromhydrate) en sous-cutané ou à défaut l’atropine.
Hoquet rebelle : A pour principale cause une distension ou une irritation gastro-duodénale pour lesquelles on peut proposer des pansements digestifs à base de charbon ou des antiacides à base d’hydroxyde d’aluminium et de magnésium. En cas d’échec, un antiémétique stimulant la motricité gastro-duodénale (métoclopramide, dompéridone) est recommandé.
4. Dysphagie :
En cas de dysphagie, il est recommandé de rechercher une cause médicamenteuse pouvant assécher la muqueuse buccale et exacerber une dysphagie (opioïdes, neuroleptiques, anticholinergiques, métoclopramide, dompéridone, etc.) et d’adapter le traitement.
En cas de trouble de la déglutition, on peut proposer pour éviter les fausses-routes : une texture pâteuse ou gélifiée des aliments, une alimentation froide, la prise des repas en position assise. La douleur liée à la déglutition peut être soulagée par des anti-inflammatoires non stéroïdiens. En cas d’obstruction, les corticoïdes à fortes doses sont proposés. En cas d’échec, une dilatation endoscopique ou la pose d’une endoprothèse œsophagienne ou une désobstruction au laser.
Nausées, vomissements : Il est recommandé :
De lister les médicaments susceptibles de provoquer ou d’aggraver des nausées et des vomissements
De supprimer, dans la mesure du possible, les stimuli susceptibles d’aggraver les symptômes (odeurs, etc.) ;
De fractionner les repas et de proposer de petites collations. Il est recommandé d’évaluer l’état d’hydratation du patient, surtout s’il existe une diarrhée associée : en cas de déshydratation rapide en 24-36 heures, une réhydratation parentérale est à discuter. Les antiémétiques prokinétiques (metoclopramide ou dompéridone) sont recommandés.
6. Anorexie, cachexie :
En cas d’anorexie, il est proposé de commencer par favoriser l’appétit grâce à une présentation attrayante des repas (même s’ils doivent ensuite être mixés), grâce à de petites portions, à des boissons fraîches. Des préparations hyperprotidiques et hypercaloriques peuvent être essayées. Les médicaments orexigènes peuvent être prescrits si ces mesures simples ne suffisent pas : les corticoïdes et les progestatifs (acétate de megestrol, acétate de medroxyprogestérone) augmentent l’appétit et probablement le bien-être du patient.
En cas de cachexie, les mesures diététiques pour lutter contre l’anorexie sont recommandées.
7. Déshydratation :
En l’absence de cause curable, et particulièrement en phase ultime, si la réhydratation orale n’est pas possible, il n’est pas recommandé une réhydratation parentérale systématique.
Troubles du transit : constipation, occlusion, diarrhée
Constipation : Il est proposé de prévenir la constipation :
En encourageant, dans la mesure du possible, une activité physique régulière, ou à défaut en proposant des massages du cadre colique,
En maintenant la consommation de fibres alimentaires grâce à une alimentation variée et agréable pour le patient et une hydratation orale adéquate.
il est proposé de lutter contre la douleur, de traiter les nausées et les vomissements, de discuter une aspiration gastrique, d’arrêter éventuellement un traitement laxatif, maintenir une hydratation et une alimentation adaptées à l’état du patient. Pour lutter contre la douleur, l’association opioïdesantispasmodiques est préconisée. Comme antispasmodique, la scopolamine butylbromure (ou à défaut la scopolamine bromhydrate).
Diarrhée : Est à traiter rapidement car elle retentit vite sur l’hydratation et sur l’état physique et psychologique du patient. Le traitement symptomatique repose sur :
Une adaptation du régime alimentaire, selon le contexte clinique (éviction momentanée des laitages en cas de diarrhée infectieuse, arrêt des compléments nutritifs liquides, réhydratation en privilégiant la voie orale) ;
Des soins d’hygiène;
L’utilisation d’anti-diarrhéiques, après élimination d’une fausse diarrhée liée à un fécalome. Le lopéramide est proposé en première intention…
2. Troubles urinaires :
En cas de troubles urinaires, la prise en charge est d’abord étiologique.
En cas d’infection urinaire, il est rappelé que chez un patient porteur d’une sonde vésicale à demeure, une bactériurie apyrétique ne nécessite pas de traitement antibiotique.
En cas d’hématurie macroscopique persistante, il est recommandé d’essayer de supprimer la cause du saignement (embolisation artérielle en cas de saignement d’origine rénale, électrocoagulation en cas de saignement vésical). Il est proposé la pose d’une sonde vésicale à double courant permettant des lavages répétés, et/ou une irrigation continue.
Phase terminale et agonie :
La phase terminale peut durer plusieurs jours alors que l’agonie est souvent définie par les 48 à 72 heures qui précèdent la mort. L’agonie peut être identifiée par le caractère rapidement évolutif de l’état général, et l’apparition éventuelle :
De troubles de conscience (somnolence et parfois comas entrecoupés de périodes de lucidité) ;
De troubles respiratoires (râles agoniques) ;
De troubles de la déglutition ;
De troubles circulatoires (hypotension artérielle, cyanose des extrémités, entraînant troubles trophiques au niveau des points de pression) ;
D’un syndrome confusionnel ;
De myoclonies, dyskinésies, contractures, voire convulsions ;
D’une élévation de la température corporelle et d’une asthénie extrême.
Conclusions :
Développer le contrôle efficace des symptômes, le respect des souhaits et des besoins propres du patient et pour la famille constituent les pierres angulaires des soins palliatifs qui sont à prendre en compte, non seulement les symptômes physiques ou psychologiques présentés par le patient, mais également ses besoins spirituels, familiaux et sociaux. Une collaboration étroite entre les différents intervenants dans la prise en charge du cancéreux pendant cette phase délicate de son parcours de la maladie du début jusqu’à la phase finale est indispensable. Tous les centres destinés à la prise en charge de ces malades doivent être équipés pour faire face à tous les problèmes qui peuvent survenir durant cette période de soins palliatifs. La participation du médecin généraliste est indispensable pour la prise en charge des effets secondaires des traitement et les symptômes qui suivent.
Références
Recommandations ANAES « Modalités de prise en charge de l’adulte nécessi- tant des soins palliatifs, 12/03/02 (www.has-sante.fr)
Rapport annuel du comité national de suivi du développement des soins pal- liatifs et de l’accompagnement. 2008 [www.sante.gouv.fr]
Programme de développement des soins palliatifs 2008-2012 [www.sante.gouv.fr]
Amery J (2009). Children’s Palliative Care in Africa. London: Oxford Uni- versity Press.
APCA (2009). Competency framework for palliative care. Kampala: African Palliative Care Association.
APCA (2009). Standards for palliative care in Africa. Kampala: African Pal- liative Care Association.
Gwyther L, Merriman A, Mapanga Sebuyria L and Schietinger H (2006). A Clinical Guide to Supportive and Palliative Care for HIV and AIDS in Sub- Saharan Africa. Kampala: APCA.
Hospice Africa Uganda (2006). Palliative Medicine: pain and symptom control in the cancer and/or AIDS patient in Uganda and other African Countries (4th Edition). Uganda: Hospice Africa Uganda.
International Network for Cancer Treatment and Research (2008). INCTR Palliative Care Handbook. INCTR Publication No 3.
Kopf A and Patel NB (eds) (2009). Guide to Pain Management in Low Re- source Settings. United States: IASP Press.
Lavy V, Bond C and Wooldridge R (2007). Palliative Care Toolkit: Improving care from the roots up in resource-limited settings. London: Help the Hospices and WPCA.
Meiring M (2009). Guidelines for managing pain in children. South Africa: Adcock Ingram.
Mwangi-Powell FN, Downing J, Ddungu H, Kiyange F, Powell RA and Bagu- ma A (2010). ‘Palliative care in Africa’ in Ferrell BR and Coyle N (eds) Oxford Textbook of Palliative Nursing, 3rd Edition (March 2010). New York: Oxford University Press.
World Health Organization (2004). Palliative Care: Symptom management and end-of-life care. Integrated management of adolescent and adult illness. Geneva: World Health Organization.
World Health Organization (2007). WHO Model List of Essential Medicines– 15th List. Geneva: World Health Organization.
www.palliativedrugs.com, accessed March 2010. ‘Palliative drugs: Essential information for palliative and hospice care’.
Guide des bonnes pratiques de soins en EPHAD, Guide des bonnes pratiques d’une démarche palliative en établissements, Guide des lits identifiés sur le site du ministère chargé de la Santé [www.sante.gouv.fr] rubrique « Soins palliatifs» dans dossier « A à Z ».
Répertoire national des ressources de la Société française d’accompagnement et de soins palliatifs (SFAP) [http://www.sfap.org] ou le portail soins palliatifs [www.portail-soins-paliatifs.org]. Vous pourrez également y trouver des rensei- gnements sur les formations possibles.
Occlusion intestinale : la chirurgie palliative est à dis- cuter en première intention. Si elle n’est pas possible,
S. MOSTEFAI, Service de Rhumatologie. CHU Issaad Hassani, Beni Messous, Alger
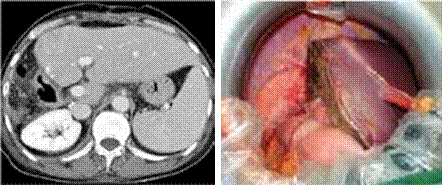
Abstract : Many studies in rheumatoid arthritis (RA) have shown an increase in cardiovascular risk such as myocardial infarction. Data are rarer for venous thromboembolic events whose risk is multiplied by 2. It is important to iden- tify risk factors for thromboembolism related to RA and treatments. A 50-year-old woman with a history of early menopause, undergoing hormone replacement therapy, hypothyroidism, osteoporosis treated with risdronate, followed for 5 years for a seropositive RA, with little defor- ming. The patient was put under methyl prednisolone 8mg/d and methotrexate 15mg /week. This treatment resulted in a marked improvement with a reduction in corticosteroids at 2 mg /day. 4 years later, the patient pre- sents a thrust of her RA and she consults for a pain in the left calf. The doppler ultrasound shows a thrombophle- bitis of the left posterior tibial vein. Two months later, the patient has a pulmonary embolism. Several mechanisms may explain thromboembolic events in RA. Inflammation is probably the link between RA and thrombosis because it is the major determining factor of lesions and dysfunc- tion of the endothelial wall of vessels. Treatments such as high dose corticosteroid therapy, methotrexate also have a deleterious effect and favor thrombosis. Conclusion: A strict control of the activity of the disease with an adapted treatment makes it possible to reduce this type of accident which increases the morbidity and the mortality in RA.
Résumé : De nombreux travaux au cours de la polyarthrite rhumatoïde (PR) ont démontré une augmentation du risque cardio vascu- laire tel que l’infarctus du myocarde. Les données sont plus rares en ce qui concerne les accidents veineux thromboembo- liques dont le risque est multiplié par 2. Il est important d’iden- tifier les facteurs de risque de thromboembolie liés à la PR et aux traitements.
Patiente âgée de 50 ans, aux antécédents de ménopause pré- coce, mise sous traitement hormonal substitutif, une hypothyroï- die, une ostéoporose traitée par risédronate, suivie pendant 5 ans pour une PR séro positive, peu déformante. La malade a été mise sous méthyl prednisolone 8mg/j et méthotrexate 15mg/semaine. Ce traitement a entraîné une nette amélioration avec une dimi- nution de la corticothérapie à 2mg/j. 4 ans plus tard, la patiente présente une poussée de sa PR et elle consulte pour une douleur du mollet gauche. L’écho doppler objective une thrombophlébite de la veine tibiale postérieure gauche. Deux mois après, la patiente présente un tableau d’embolie pulmonaire.
Plusieurs mécanismes peuvent expliquer les accidents thrombo emboliques dans la PR. L’inflammation est probablement le lien entre la PR et la thrombose, car c’est le principal facteur déterminant des lésions et du dysfonctionnement de la paroi endothéliale des vaisseaux. Les traitements comme la cortico- thérapie à forte dose, le méthotrexate ont également un effet délétère et favorisent la thrombose. Un contrôle strict de l’acti- vité de la maladie avec un traitement adapté permet de réduire ce type d’accident qui augmente la morbidité et la mortalité dans la PR.
Au cours de la polyarthrite rhumatoïde (PR), il existe denombreux travaux qui ont démontré une augmentation du risque cardiovasculaire tel que l’infarctus du myo- carde. Cependant, les données sont plus rares en ce qui concerne les accidents thromboemboliques. En effet, la thromboembolie veineuse (TEV) est une manifestation possible de la PR et elle constitue une importante cause de mortalité et de morbidité. Plusieurs facteurs dont l’inflammation pourraient jouer un rôle dans l’augmen- tation du risque thromboembolique. L’identification de ces facteurs de risque est cliniquement utile pour déter- miner les patients prédisposés à ces risques, car des tra- vaux ont montré une augmentation de l’ordre de 1,5 à 6 des accidents thromboemboliques au cours de la PR[1-7].
Observation :
Madame A. Chafia, âgée de 50 ans, G6P3 avec trois avortements et dans ses antécédents une ménopause précoce à l’âge de 30 ans ayant nécessité la mise sous traitement hormonal substitutif pendant 4 ans, une os- téoporose sous risédronate 35mg par semaine, une hy- pothyroïdie sous lévothyrox, un surpoids avec un BMI de 29, 75 et une PR évoluant pendant 5 ans.
Il s’agit d’une polyarthrite rhumatoïde séropositive, peu déformante, très active avec un DAS28 à 7,61. Biolo- giquement, une VS accélérée à 97 mm la 1ère heure et une CRP à 55 mg/l. Le facteur rhumatoïde et les anti CCP sont positifs, les FAN et les AC anticardioli- pines sont négatifs. La radiographie standard des mains et poignets objective un pincement de l’interligne radio carpien et des érosions au niveau du 5ème rayon des deux mains. La patiente a été mise sous 8 mg de méthylpred- nisolone par jour puis dégression jusqu’à 2 mg par jour et 15 mg de méthotrexate par semaine. L’évolution a été favorable sous ce traitement avec un DAS28 à 2,09. 4 ans plus tard, la patiente présente une poussée de sa PR avec un DAS28 à 5,25. Elle consulte pour une douleur aiguë du mollet gauche avec chaleur et aug- mentation du volume. L’écho doppler faite confirme le diagnostic de thrombophlébite de la veine tibiale postérieure gauche ayant nécessité un traitement hépa- rinique (HBPM) et l’arrêt du méthotrexate. Après 30 jours, bonne évolution de la thrombophlébite, on réin- troduit le méthotrexate à la dose de 10mg par semaine et le maintien du méthylprednisolone à raison de 4mg par jour.
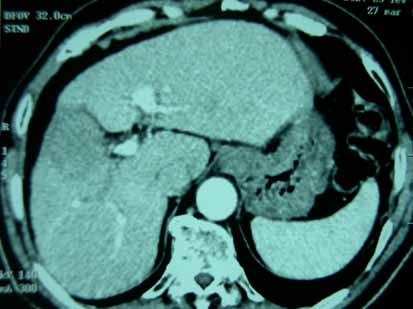
40 jours après l’épisode de thrombophlébite, la patiente consulte à nouveau pour une douleur thoracique, une toux et une dyspnée stade III de NYHA. On décide d’arrêter le méthotrexate. L’examen cardio vasculaire, l’ECG et l’échocardiographie sont sans anomalie. La patiente est adressée aux urgences de pneumologie. La tomodensitométrie thoracique permet de poser le dia- gnostic d’embolie pulmonaire de la branche droite de l’artère pulmonaire dans sa partie proximale et éten- due aux branches segmentaires. La patiente a été mise sous héparinothérapie puis relai par l’acénocoumarol
(Sintrom®). Le bilan étiologique de la thromboembo- lie veineuse retrouve un syndrome inflammatoire avec une VS accélérée à 63mm la 1ère heure, une CRP à 30mg/l, cholestérol à 2,37, HDL, LDL, triglycérides sans anomalie. Le bilan immunologique comportant les FAN, les anti DNA, l’anticoagulant lupique circulant, les anticardiolipines et les anti ß2 GP1 sont tous néga- tifs. Le bilan de thrombophilie comportant TP, TCK, TCA, fibrinémie, antithrombine, protéine C et S sont sans particularité.
Après deux ans, on décide d’arrêter le Sintrom® avec une très bonne évolution de la PR sous 2mg de méthyl- prednisolone par jour.
Discussion :
La relation entre PR et TEV a été rarement étudiée. Mais des études ont mis en évidence un lien entre la mortalité et la morbidité cardiovasculaire et les facteurs de sévérité de la PR [1,8-10]. En effet, le risque cardio- vasculaire est plus important dans les PR sévères qui s’accompagnent d’arthrites, de manifestations extra-ar- ticulaires, de VS et CRP élevées, de facteur rhumatoïde et d’anti CCP positifs et à la radiographie des érosions osseuses [11-14]. L’incidence des TEV est accrue dans la PR avec un taux d’incidence pour 1.000 patients-an- née de 6,1 (5,4-6,9) versus 2,5 (2,3-2,8) chez les sujets contrôles sans PR soit un risque relatif (RR) de 2,4 (2,1- 2,8)[15]. Pour l’embolie pulmonaire, le RR est de 2,23 (IC à 95% : 1,75 à 2,88) et pour la thrombose veineuse profonde, le RR est 2,20 (IC à 95% : 1,78 à 2,71). Le risque de TEV est plus grand au cours de la première année de suivi de la PR, car à ce stade de début de la maladie, l’activité inflammatoire n’est pas assez contrô- lée et le bénéfice du traitement anti rhumatismal n’est pas encore atteint. Ce risque diminue avec le temps.
Un certain nombre d’études a montré que le risque de TEV semble augmenté dans les maladies auto immunes comme la PR[16]. Plusieurs mécanismes sont suscep- tibles d’expliquer l’augmentation du risque de TEV dans la PR. Les facteurs de risque classiquement connus comme le tabagisme, l’hyperlipidémie, l’hyperhomocys- téinémie, l’hypertension artérielle, le diabète et l’immo- bilité peuvent être importants dans la PR. Toutefois, il semble exister d’autres facteurs en rapport direct avec l’inflammation systémique de la PR. L’inflammation peut moduler les réponses thrombotiques par l’augmen- tation de la régulation de la procoagulation, la diminu- tion de la régulation de l’anticoagulation, la suppression de la fibrinolyse[17].
Plusieurs facteurs prothrombotiques ont été identifiés chez les patients ayant une PR.
1. La dysfonction endothéliale :
Elle conduit à l’altération de la perméabilité endothé- liale avec augmentation du nombre de leucocytes et l’adhésion des plaquettes. Les plaquettes sont impli- quées dans l’inflammation, l’athérosclérose et la throm- bose. La dysfonction endothéliale est liée à l’inflamma- tion[18], au HLA-DR1[19], au génotype inhabituel[20] et elle s’améliore avec l’infliximab[21,22]. L’adhésion des monocytes est favorisée par l’augmentation de l’expres- sion des molécules d’adhésion (VCAM-1 ; ICAM1) durant l’inflammation[23]. Cette expression est induite par des cytokines pro inflammatoires comme IL1 Beta, TNFɑ et CRP[22]. Comme tous ces facteurs sont pré- sents dans la circulation systémique au cours de la PR, il est fort possible qu’ils pourraient être considérés comme des facteurs de risque cardiovasculaires. En outre, les facteurs de la coagulation comme l’augmentation du facteur de Von Willebrand et le plasminogène activa- teur inhibiteur-1[24] sont importants et peuvent jouer un rôle significatif aussi bien dans la PR que dans la thrombose[25].
2. L’hypercoagulabilité :
L’hypercoagulabilité est induite par une inflammation systémique active et par la production de cytokines comme IL-1, IL-6, IL-8 et le TNFɑ qui sont respon- sables de la dysfonction endothéliale, de la diminution de la régulation de la protéine C et l’inhibition de la fibrinolyse. Ces cytokines pourraient moduler les ré- ponses thrombotiques en activant les voies de la coagu- lation [26-28]. La poussée inflammatoire de la PR réduit la mobilité du patient, ce qui favorise la stase veineuse et augmente la coagulabilité du sang.
Il est a été prouvé que les processus de la coagulation sont actifs dans la PR à la fois en intra et extravasculaire. L’hypercoagulabilité fait suite à une activation excessive des enzymes de la coagulation et du facteur tissulaire. Il a également été démontré que le dépôt de fibrine est un facteur potentiel de l’inflammation synoviale et joue un rôle dans l’activation de la cascade de la coagulation qui est étroitement liée à l’inflammation synoviale [29,30]. Le blocage de la voie de la coagulation peut réduire le pro- cessus inflammatoire dans les maladies chroniques [31]. L’hypercoagulabilité peut être secondaire à la présence d’anticorps anticardiolipines et au profil lipidique anor- mal. 22% des PR ont des anticorps anticardiolipines. La positivité de ces anticorps chez les patients avec des maladies auto-immunes est fortement corrélée avec le
risque de l’apparition concomitante de complications thromboemboliques veineuses et artérielles. Les anti phospholipides peuvent induire un état pro inflam- matoire, pro adhésif et pro coagulant dans les cellules endothéliales, les plaquettes et les monocytes [32-34]. Un grand nombre de patients avec PR ont également une dyslipidémie qui constitue un facteur de risque impor- tant des événements thromboemboliques [35]. La lipo- protéine a (Lp a) est augmentée dans la PR et sa concen- tration plasmatique est influencée par plusieurs facteurs acquis comme certains médicaments et l’inflammation. Elle inhibe également la lyse du caillot de fibrine. La Lp a est un facteur de risque pour TEV [36,37].
3. Les conditions prothrombotiques :
Les conditions prothrombotiques sont définies par une augmentation des facteurs de risque de thrombose et une diminution des facteurs anti coagulants [38]. Les activités prothrombotiques peuvent être attribuées à un effet toxique direct sur l’endothélium qui manque de dérivé NO qui pourrait conduire à une hyperactivation des plaquettes [39,40]. Un autre mécanisme prothrombo- tique possible peut être lié à une diminution de l’expres- sion de la protéine anti coagulante, la thrombomodu- line qui est essentielle à l’activation de la protéine C [41]. L’hyperhomocystéinémie est un facteur prothrombo- tique observé dans la PR. La carence en folate, vitamine B12 et vitamine B6 peut engendrer une hyperhomocys- téinémie. L’hyperhomocystéinémie même modérée est considérée par la plupart des auteurs comme un facteur de risque vasculaire artériel et veineux. Elle aurait une toxicité directe sur les cellules endothéliales et induit la synthèse de cytokines pro inflammatoires. L’hyperho- mocystéinémie modérée coïncide avec des taux sériques bas d’acide folique et des concentrations plus élevées des marqueurs d’activateur immunitaire comme TNF- R75 et la néoptirine. Une étude australienne a montré une proportion significative de patients ayant une PR qui ont des taux élevés d’homocystéine par rapport aux témoins. Elle est due à un polymorphisme génétique et/ ou des facteurs carentiels comme la carence en folate, vitamine B12 et B6 et elle peut être également due à une cause iatrogène comme le méthotrexate et les corti- coïdes ou à une altération de la fonction rénale. L’inter- férence du méthotrexate avec le métabolisme des folates est responsable de sa toxicité [42]. Les microparticules constituent un autre mécanisme qui pourrait être impliqué dans la détermination d’une condition prothrombotique dans la PR. Ce sont des vésicules membranaires qui circulent dans le sang surtout dans les plaquettes et qui sont des médiateurs de l’inflammation et de la thrombose [40].
4. Les marqueurs de l’inflammation :
Dans la PR, l’état inflammatoire augmente le risque thrombotique de plusieurs manières, par l’augmenta- tion du nombre de plaquettes et leur activité, par l’aug- mentation de la cascade de la coagulation ce qui altère les activités anticoagulantes et fibrinolytiques condui- sant ainsi à l’hyperhomocystéinémie. La concentration plasmatique de plusieurs marqueurs de l’inflammation comme le fibrinogène, les cytokines, la CRP et les D-Di- mères est associée à un risque thrombotique. Il a été dé- montré que l’augmentation de la CRP est responsable de lésions cardiaques par activation du complément et l’inflammation et peut ainsi avoir un effet pro coagulant. En outre, le TNFɑ joue un rôle crucial dans l’inflamma- tion de la PR et peut provoquer des lésions endothéliales favorisant la coagulation et par conséquent pourrait être impliqué dans les complications cardiovasculaires de la PR. Les anti TNF diminuent le risque cardiovasculaire en améliorant l’inflammation [43-47].
5. Les complications après chirurgie orthopédique :
La TEV est une complication connue après la chirurgie pour prothèse totale de hanche ou de genou. Les données suggèrent que le risque de développer une TEV dans la PR pourrait être inférieur à celui des patients arthrosiques, car dans la PR, les patients ont tendance à être plus jeunes, avec un poids plus léger et une utilisation plus fréquente des AINS classiques qui ont une activité anti plaquettaire avec inhibition de la thromboxane A2. Cet effet n’est pas retrouvé avec les AINS sélectifs (anti COX2), ce qui peut augmenter le risque des événements thrombotiques chez des patients prédisposés [17].
6. La thérapeutique et le risque thromboembolique :
Aujourd’hui, la plupart des patients avec PR sont traités avec une variété de médicaments comportant des AINS, des corticoïdes, des DMARDS et des agents biologiques pour soulager les symptômes et arrêter la progression de la maladie.
Les AINS sont connus pour leur augmentation du risque de l’hypertension artérielle (HTA) et l’infarctus du myocarde. Certaines études ont montré qu’il pour- rait exister un risque de TEV avec les AINS. Bien qu’il existe des résultats contradictoires pour les corticoïdes et les TEV, de nombreuses études ont montré des effets défavorables des glucocorticoïdes à fortes doses, ceci par augmentation des facteurs de risque comme le diabète, l’HTA, le profil lipidique, l’obésité et les protéines de la coagulation. Cependant, pour les faibles doses de corti- coïdes, il n’y a pas de preuve de risque cardiovasculaire. L’explication pourrait être que les effets anti inflam- matoires des corticoïdes sur l’inflammation lors de la poussée balance les effets indésirables sur la coagulation et la fibrinolyse. Les données sur les risques cardiovas- culaires et de TEV avec l’utilisation des DMARDS dans la PR sont rares, excepté pour le méthotrexate et l’hydroxychloroquine. L’hydroxychloroquine dimi- nue le risque cardiovasculaire en diminuant le taux de cholestérol, du LDL et de l’IL-6. Le méthotrexate est un anti folique, responsable d’une carence en folate et d’une hyperhomocystéinémie, augmente le risque de TEV. La supplémentation en folate doit être systéma- tique pour diminuer les accidents de TEV au cours de la PR. Le léflunomide et la ciclosporine sont respon- sables d’une HTA[16]. Les anti TNFɑ et les inhibiteurs de l’IL-6 ont un effet bénéfique sur la PR et diminuent le risque thromboembolique. Une étude a montré une amélioration des paramètres cliniques, biologiques ainsi qu’une réduction de l’activation de la coagulation et de la dysfonction endothéliale chez les patients atteints de PR traités avec les anticorps monoclonaux chimériques (Infliximab). Une autre étude randomisée (MEASURE) a montré que le blocage des récepteurs de l’IL-6 réduit le fibrinogène et les D-Dimères de plus de 40% par rap- port au placebo [16,48].
Chez notre patiente, plusieurs facteurs de risque per- mettent d’expliquer la survenue de l’accident throm- boembolique, à savoir le surpoids avec un BMI de 29,75, une dyslipidémie, une ménopause précoce trai- tée par THS. À noter que cet accident a coïncidé avec une poussée de la PR qui est une cause de TEV. Sur le plan thérapeutique, la corticothérapie a été utilisée à faible dose, mais le méthotrexate n’a pas été supplé- menté par l’acide folique, ce qui a peut-être engendré une hyperhomocystéinémie, cause de TEV.
Conclusion :
La PR peut être considérée comme un état pro throm- botique avec un risque de TEV et de maladie cardio- vasculaire. L’inflammation est probablement le lien entre ces deux maladies. Cependant, il est important d’identifier les facteurs de risque d’embolie pulmonaire et de thrombose veineuse liés à la PR et aux différents traitements. Il faut augmenter de vigilance dans la surveillance et le suivi des complications thromboembo- liques. Le meilleur contrôle de l’activité inflammatoire de la PR permet de réduire la mortalité cardiovascu- laire. La PR ne figure pas parmi les facteurs de risque de TEV dans un guide de pratique clinique pour une prophylaxie anti thrombotique. Certains auteurs re- commandent l’utilisation de l’aspirine en absence de contre-indication chez les patients à risque. La prophy- laxie par héparine dans la TEV est limitée aux périodes de poussées nécessitant une immobilisation et lors d’une intervention chirurgicale.
Références
Solomon DH, Karlson EW, Rimm EB et al. Cardiovascular mor- bidity and mortality in women diagnosed with rheumatoid arthritis. Circulation 2003;107:1303-7
Goldhaber SZ, Visani L, De RM. Acute pulmonary embolism : clinical outcomes in the international Cooperative Pulmonary Embo- lism Registry (COPER). Lancet 1999;353:1386-9
WatsonDJ, Rhodes T, Guess HA. All causes mortality and vascu- lar events among patients with rheumatoid arthritis, osteoarthritis or non-arthritis in the UK General Practice Research Database. J Rheu- matol 2003;30: 1196-202
Wolf F, Freundlich B, Straus WL. Increase in cardiovascular and cerebrovascular disease prevalence in rheumatoid arthritis. J Rheum 2003;30: 36-40
Liang KP, Liang KV, Matterson EL, Mc-Clelland RL, Christianson TJ, Turesson C. Incidence of non-cardiac vascular disease in rheuma- toid arthritis and relationship to extra articular disease manifestations. Arthritis Rheum 2006; 54: 642-8
Alikhan R, Cohen AT, Combe S et al. MEDENOX study: risk fac- tors for venous thromboembolism in hospitalized patients with acute medical illness: analysis of the MEDENOX study. Arch Intern Med 2004;164:963-8
Kim SG et al. The risk of venous thromboembolism in patients with rheumatoid arthritis. Arthritis Care Res 2013; 65: 1600-7
Soubier M, Dougados M. Atherosclerosis and rheumatoid arthritis. Rev Med Interne 2006; 27:125-36
Avina-Zubieta JA, Choi HK, Sadatsafavi M et al. Risk of cardio- vascular mortality in patients with rheumatoid arthritis: a meta-analy- sis of observational studies. Arthritis Rheum 2008;59:1690-7
Van Halm VP, Peters MJ, Voskuyl AE et al. Rheumatoid arthritis versus diabetes as risk factors for cardiovascular disease, across sectio- nal study. The CARRE investigation. Ann Rheum Dis 2009;68:1395-400
Maradit-Kremers H, Cynthia P.J., CrowsonS et al. Cardiovascu- lar death in rheumatoid arthritis. Arthritis Rheum 2005;52: 722-32
Naz SM, Farragher TM, Bunn DK et al. The influence of age at symptom onset and length of followup on mortality in patients with recent-onset inflammatory polyarthritis. Arthritis Rheu 2008; 58:985-9
Turesson C, Mc Clelland RL, Christianson TJH et al. Severe extra articular disease manifestations are associated with increases risk of first even cardiovascular events in patients with rheumatoid arthritis. Ann Rheum Dis 2007; 66: 70-5
Lopez-Longo FJ, Olivier Minarro D, de la Torre I et al. Associa- tion between anti cyclic citrullinated peptide antibodies and ischemic heart disease in patients with rheuamatoid arthritis. Arthritis Rheum 2009;61: 419-24
Choi HK, Rho YH, Zhu Y, Cea-Soriano L, Avina-Zubieta JA, Zhang Y. The risk of pulmonary embolism and deep vein thrombo- sis in rheumatoid arthritis: a UK population based outpatient cohort study. Ann Rheum Dis 2013; 72: 1182-1187
I.A.M Van den Oeveret coll. Thromboembolic and cardiovascu- lar risk in rheumatoid arthritis: role of the homenostatic system. Ann Rheum Dis 2014: 73: 954-57
Mameli A, Barcellona D, Marongiu F. Review. Rheumatoid arth- ritis and thrombosis. ClinExpRheumatolo 2009; 27: 846-55
Valudo G, Marchesi S, Gerli R et al. Endothelial dysfunction in young patients with rheumatoid arthritis and lox disease activity. Ann Rheum Dis 2004; 63: 31-5
Gonzalez- Juannatey C, Testa A, Garcia- Castelo A et al. HLA- DRB1 status affects endothelial function in treated patients with rheu- matoid arthritis. Am J med 2003; 114: 647-52
Arlestig L, WallbergJonsson S, Stegmayr B, Rantapa A, Dahlqvist S. Polymorphism of genes related to cardiovascular disease in patients with rheumatoid arthritis. ClinExpRheumatol 2007; 25: 866-71
Komai N, Morita Y, Sakuta T, Kuwabara A, Kashishara N. Anti- tumor necrosis factor therapy increases serum adiponectin levels with the improvement of endothelial function in patient with rheumatoid arthritis. Mod Rheumatol 2007; 17: 385-90
Booth AD, Jaine DR, Kharbando RK et al. Infliximab improves endothelial dysfunction in systemic vasculitis: a model of vascular inflammation. Circulation 2004; 109: 1718-23
Veale DJ, Maple C, Kirk G, Mc Laren M, Belch JJ. Soluble cell adhesion molecule- P-seleclinand ICAM-1 and diseaseactivity in pa- tients receiving sulphasalazine for active rheumatoid arthritis. Scand J Rheumatol 1998; 27:296-99
Koenig W. Haemostatic risk factor for vascular disease. Eur.Heart J 1998;19 (suppl): 93
Walleberg-Jonsson S, Cvetkovic JT, Sundqvist KG, Lefvert AK, Rantapa A, Dahiqvist S. Activation of the immune system and in- flammatory activity in relation to markers of atherothrombotic di- sease and atherosclerosis in rheumatoid arthritis. J Rheumatol 2002; 29: 875-82
Fox EA, Kahn SR. The relationship between inflammation and venous thrombosis. A systematic review of clinical studies. Thromb- Haemost 2005; 94:362-365 [PubMed:16 11 38 20]
Vander PollT, Büller HR, Ten Cate H et al. Activation of coagula- tion after administration of tumor necrosis factor to normal subjects. N Engl J Med 1990;322: 1622-1627 [Pub Med 2188129]
Zöller B, Li X, Sundquist Jet al. Autoimmune diseases and venous thromboembolism : a review of the literature. Am J Cardiovasc Dis. 2012;2171-183 [Pub Med 22937487]
Zacharski LR, Brown FE, Memoli Va et al. Pathways of coagula- tion activation in situ in rheumatoid synovial tissue. ClinImmunoIm- munopathol 1992;63:155-62
Bokarewa MI, Morrissey JH, Tarkowski M. Tissue factors was a pro inflammatory agent. Arthritis Res 2002; 4:190-5
Thompson NP, Wakefield AJ, Pounder RE. Inherited disorders of coagulation appear to protect against inflammatory bowel disease. Gastroenterology 1995; 108: 1011-5
Olech E, Merrill JT. The prevalence and clinical significance of antiphospholipid antibodies in rheumatoid arthritis. CurrRheumatol Rep 2006 Apr; 8: 100-8
Runchey SS, Folsom AR, Tsai MY, Cushman M, McGovern PD. Anticardiolipin antibodies as a risk factor for venous thrombo embo- lism in a population –based prospective study. Br J Haematol 2002; 119:1005-10
Naess IA, Christiansen SC, Cannegiet –ErSc, Rosendael FR, Hammerstroem J. A prospective study of anticardiolipin antibodies as a risk factor for venous thrombosis in a general population (the HUNT study). ThrombHaemost 2006; 4:44-9
Lazarevic MB, Vitic J, Mladenovic V, Myones BL, Skosey JL, Swedler W. Dyslipoproteinemia in the course of active rheumatoid arthritis. Semin Arthritis Rheum 1992; 22:172-80
Wallberg-Jonsson S, Ohman M, Rantapa A, Dahliqvist S. Which factors are related to the presence of atherosclerosis in rheumatoid arthritis ? Scand J Rheumatol 2004; 33:373-9
Park YB, Lee SK, Lee WK. Lipid profiles in intreated patients with rheumatoid arthritis. J Rheumatol 1999; 26:1701-14
Den Heijer M, Koster T, Blom H et al. Hyperhomocysteinemia as a risk factor for deep vein thrombosis. N engl J Med 1996; 334: 759-62
Davi D, Di MG, Coppola A et al. Oxidative stress and platelet acti- vation in homozygous homocystinuria. Circulation 2001; 104:1124-8
Wolf P. The nature and significance of platelet product in human plasma. Br Haematol 1967; 13:269-88
El Mangad FEZ, Ghazi M, Qacif H, Zyani M, Niamane R. PR et thrombose veineuse profonde : rôle de l’hyperhomocystéinémie. Rev Mar Rhum 2013 ; 25 : 45-7
Mc Entegart A, Chapell HA, Creran D et al. Cardiovascular risk factor including thrombotic variables in a population with rheuma- toid arthritis. Rheumatology 2001;40: 640-4
Ferraccioli G, Gremese E. Thrombogenicity of TNFɑ in rheu- matoid arthritis defined through biological probes: TNF ɑ blockers. Autoimmun Rev 2004; 3: 261-6
Siren AL, Mc Carron R, Wang L et al. Pro inflammatory cytokine expression contributes to brain injury provoked by chronic monocyte activation. Mol Med 2001; 7: 219-29
Grignani G, Maiolo A. Cytokines and haemostasis. Haematolo- gica 2000; 85: 967-72
Davies R, Gallaway JB et al. Venous thrombotic events are not increased in patients with rheumatoid arthritis treated with anti TNF therapy. Results from the British Society for Rheumatology Biologic Register. Ann Rheum Dis 2011; 70: 1831-34
A.F. BOUTALEB(1), H. SAOULA(1)M. AISSAOUI(1), D. HAMIDOUCHE(1),R. OSMANE(1), H. MAHIOU(1),Y. AISSAT(1), A. MITICHE(1),Y. ZMIRI(1), F. HAMCHAOUI(2),M. NAKMOUCHE (1) Service d’Hépato-gastro-entérologie, Service d’Epidémiologie, CHU Lamine Debaghine, Bab El Oued
Abstract : Coeliac disease is an autoimmune reaction to gluten. The clinical manifestations are variable and not specific. The objective of this study was to determine typical, atypical and silent cases according to presenting symptoms and to evaluate their clinical and biochemical parameters. We also aimed to determine diagnosis delay of coeliac disease (interval between first symptoms and diagnosis).
317 adults patients with coeliac disease in our department between January 2009 and January 2014 were diagnosed. The coeliac diagnosis was made by serological and histological examinations. Patients were divided according to their symptoms into typical, atypical or silent form of coeliac disease.
Patients presenting atypical form of coeliac disease were predominant (78,8%). Those with typical signs were present in 21,2%. Silent form was absent in our cohort. The typical form of coeliac disease was more severe than the atypical form. In fact, patients with typical symptoms had severe clinical impact of the disease. The average of BMI (body mass index) in typical form was 19,44 versus 21,15 for the atypical form (p=0,0005). Nutrional deficiencies were also more severe. Extra-digestive clinical features were similarly observed in the two forms. In 54,2%, diagnosis delay was between 1 and 10 years. Prolonged diagnosis delay (>10 years) was seen in 15,6% of cases. Atypical form of coeliac disease was predominant in our study. It was less severe than the typical form. Diagnosis delay was between 1 and 10 years in the majority of patients. The diagnosis was considered delayed in 15,6% of cases.
Résumé : La maladie cœliaque est une entéropathie auto-immune secondaire à la consommation de gluten. Les manifestations cliniques sont très variées et non spécifiques. L’objectif de ce travail est de déterminer la fréquence des formes cliniques de la maladie cœliaque (typique, atypique et silencieuse) au sein d’une population de malades cœliaques adultes et d’étudier leurs caractéristiques cliniques et biologiques ainsi que d’établir le délai diagnostique de la maladie en fonction de la forme clinique.
Le recrutement des patients cœliaques s’est effectué de Janvier 2009 à Janvier 2014. Le diagnostic de la maladie cœliaque chez nos patients tous âgés de plus de 16 ans, repose sur les critères cliniques, sérologiques, histologiques de la maladie cœliaque. Les patients sont classés selon la symptomatologie révélatrice en formes typique, atypique ou silencieuse de maladie cœliaque (classification d’Oslo). Les différentes caractéristiques cliniques et biologiques sont analysées. Le délai diagnostique est déterminé grâce à l’interrogatoire, il est défini par la période qui sépare le début des symptômes à la date du diagnostic.
Nous avons diagnostiqué 317 patients cœliaques. 78,8% ont une forme atypique et 21,2% ont une forme typique. Aucune forme silencieuse n’a été retrouvée. La forme typique est plus sévère que la forme atypique. En effet, elle est caractérisée par un retentissement important sur le plan clinique (moyenne du BMI=19,44 versus 21,15 pour la forme atypique, p=0,0005) et un syndrome carentiel plus sévère. Les manifestations extra-digestives sont observées dans les deux formes cliniques sans différence statistiquement significative. Le délai diagnostique de la plupart de nos patients (54,2%) est compris entre 1 et 10 ans. Un diagnostic tardif (au-delà de 10 ans) est posé chez 15,6% des malades.
La forme atypique de la maladie cœliaque est la forme clinique prédominante dans notre population de malades cœliaques. Il s’agit d’une forme clinique moins sévère que la forme typique. Le délai diagnostique de la maladie cœliaque est le plus souvent compris entre 1 et 10 ans. Un diagnostic tardif est fait chez 15,6% des patients.
Mots-clés : Maladie ccoeliaque, formes cliniques, delai diagnostique.
Introduction :
La maladie cœliaque est une entéropathie auto-immune qui survient chez des sujets prédisposés génétiquement (HLA QQ2 ou DQ8) et consommant du gluten[1].
Sa prévalence est de 1% environ dans les pays d’Europe et d’Amérique du nord[2,3]. La prévalence dans les pays du Maghreb semble tout aussi élevée. En effet, une étude récente ayant fait l’objet d’une thèse réalisée dans une population d’écoliers de la wilaya d’Alger, a retrouvé un taux de 0,59% de cœliaques[4].
La maladie cœliaque est une maladie protéiforme. Les manifestations cliniques sont très variées. Les symptômes digestifs peuvent être frustes, voire absents[5]. Les signes extra-digestifs peuvent être prédominants ; le diagnostic n’est alors pas évoqué précocement, engendrant ainsi un retard diagnostique[7].
En Algérie, il existe très peu d’études qui concernent la maladie cœliaque. La plupart ont fait l’objet de thèse mais n’ont pas encore été publiées.
L’objectif de ce travail est de déterminer la fréquence des formes cliniques de maladie cœliaque (typique, atypique et silencieuse) au sein d’une population de malades cœliaques adultes et d’étudier leurs caractéristiques cliniques et biologiques ainsi que d’établir le délai diagnostique de la maladie en fonction de la forme clinique.
Matériels et méthodes :
Notre étude est transversale, observationnelle et prospective. Le recrutement des patients cœliaques s’est effectué de Janvier 2009 à Janvier 2014. Il est essentiellement réalisé au niveau de la consultation du service de gastro-entérologie du CHU Lamine Debaghine, de Bab El Oued. Certains patients proviennent des autres services du CHU Lamine Debaghine de Bab El Oued avec lesquels nous avons collaboré ; ce sont les services de médecine interne, de rhumatologie, d’endocrinologie et de pédiatrie (individus de plus de 16 ans). De rares patients proviennent des autres CHU (Mustapha Bacha, Hôpital Central de l’Armée).
Le diagnostic de maladie cœliaque chez nos patients tous âgés de plus de 16 ans, repose sur les critères diagnostiques de l’ESPGHAN (European Society for PediatricGastroenterology,HepatologyandNutrition), à savoir, des symptômes cliniques évocateurs, une sérologie positive, une histologie typique et une réponse au traitement.
Les patients sont classés en forme typique, lorsque la diarrhée chronique et le syndrome carentiel sont les manifestations révélatrices de la maladie cœliaque. Les patients qui ont des symptômes digestifs banals comme une constipation, un ballonnement, des douleurs abdominales ou un reflux gastro-œsophagien, sont considérés comme ayant une forme atypique. Des manifestations extra-digestives peuvent exister, elles peuvent dominer le tableau clinique, la maladie cœliaque est dite aussi atypique. Les patients peuvent être asymptomatiques et sont diagnostiqués grâce au dépistage sérologique. On parle alors de maladie cœliaque silencieuse.
Une fois le diagnostic posé, les renseignements concernant le patient sont reportés sur une fiche préétablie comprenant : les données démographiques, le délai diagnostique, l’examen physique avec le calcul du BMI (body mass index) selon les données du national « heart, lung and blood institute », la recherche de signes carentiels tels que la pâleur cutanéo-muqueuse, les troubles des phanères, le retard staturo-pondéral et/ou pubertaire et les résultats des examens biologiques. Ceux-ci comprennent : la formule de numération sanguine, la ferritinémie, le bilan lipidique, l’albuminémie, le bilan phosphocalcique, la vitamine D, le taux de prothrombine, la glycémie, le bilan hépatique et le bilan thyroïdien.En cas de suspicion d’une hépatopathie, un bilan étiologique est pratiqué.Une ostéodensitométrie osseuse est réalisée chez tous les patients au moment du diagnostic.
Analyse statistique :
L’analyse des données est faite sur le logiciel EPI info et le logiciel SPSS. Les tests statistiques utilisés sont le test de khi deux (variables qualitatives) pour la comparaison de pourcentages et le test de Student(variables quantitatives) pour la comparaison de deux moyennes.
Résultats :
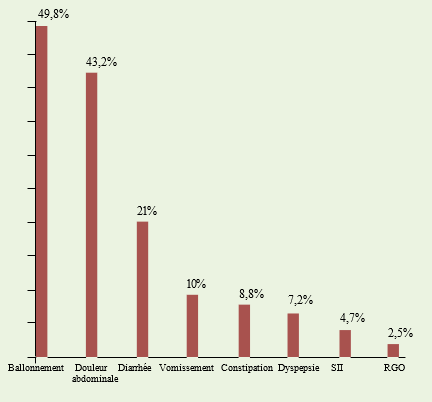
Le nombre de patients cœliaques retenus pour cette étude est de 317 patients dont la grande majorité est féminine (258 femmes pour 59 hommes). Le sex-ratio (H/F) est de 0,23. L’âge moyen de la population est de 31,64 ans avec des extrêmes de 17 à 72 ans. Les patients qui ont 50 ans et plus sont au nombre de 22, ce qui représente 7% de la population cœliaque. Le délai diagnostique a pu être déterminé chez 153 patients soit dans 48,3% des cas. Ainsi, dans 7,2% des cas le diagnostic est précoce (moins de 6 mois), dans 23% il est compris entre 6 et 11 mois ; 54,2% ont un délai diagnostique compris entre 1 et 10 ans, enfin 15,6% ont un diagnostic tardif c’est-à-dire au-delà de 10 ans Le symptôme digestif le plus fréquent est le ballonnement abdominal (50%), les douleurs abdominales sont présentes dans 43,2%, la diarrhée chronique dans 21%, les vomissements chroniques dans 10%, la constipation chronique dans 8,8%, la dyspepsie dans 7,2%, et enfin le reflux gastro-œsophagien dans 2,5%.
4,7% des patients ont des symptômes entrant dans le cadre du syndrome de l’intestin irritable selon les critères de Rome III (dans sa forme diarrhée-constipation). Voir l’histogramme représentant les symptômes digestifs.
Nos patients ont ainsi été classés : 21,2% ont une forme typique de maladie cœliaque et la grande majorité des patients, soit 78,8% ont une forme atypique. Aucun de nos patients n’a de forme silencieuse de maladie cœliaque.
Le BMI (body mass index) moyen de toute la population cœliaque est normal (20,78 +/-3,6). La moyenne du BMI des patients ayant une forme atypique (21,15+/-3,62) est supérieure à celle de la forme typique (19,44 + /-3,19) de façon statistiquement significative (p=0,0005). À l’examen physique, les signes carentiels (pâleur cutanéo-muqueuse, troubles des phanères et de la peau, retard staturo-pondéral et/ou pubertaire) ne sont présents que dans 25% des cas. Ils sont plus fréquents chez les patients ayant une forme typique mais de façon non significative (p=0,09). Le tableau 1 reporte les différents signes carentiels selon la forme clinique.
Signes carentiels
Total
Forme atypique
Forme typique
Pâleur
72 (22,7%)
50 (20%)
22 (32,8%)
Troubles de la peau et des
59 (18,6%)
35 (14%)
24 (35,8%)
phanères
Retard staturo-pondéral
19 (6%)
11 (4,4%)
8 (12%)
et/ou pubertaire
Tableau 1 : Les signes cliniques carentiels selon la forme clinique
Sur le plan biologique, l’anémie est la carence la plus fréquente (83%), elle est retrouvée le plus souvent dans la forme typique (p=0,03). Dans les deux formes cliniques, l’anémie est ferriprive et microcytaire hypochrome dans la quasi-totalité des cas. Le tableau 2 représente les taux d’anémie et ses caractéristiques selon la forme clinique. Les autres paramètres du syndrome carentiel sont significativement plus présents dans la forme typique, sauf pour la vitamine D (le p n’est pas significatif). Dans le tableau 3 sont rapportés les différents paramètres biologiques du syndrome carentiel selon la forme clinique.
Anémie et ses caractères
Total
Forme atypique
Formetypique
Anémie absente
54 (17%)
49 (19,6%)
5 (7,4%)
Anémie présente
263(83%)
201 (80,4%)
62 (92,5%)
p=0,03
Microcytaire hypochrome
261 (99,2%)
200 (80%)
61 (91%)
Mégaloblastique
2 (0,8%)
1 (0,4%)
1 (1,5%)
Fer sérique normal
30 (9,6%)
28 (11,4%)
2 (3%)
Fer sérique bas
283 (90,4%)
218 (88,6%)
65 (97%)
Ferritinémie normale
10 (7%)
9 (8,1%)
1 (3%)
Hypoferritinémie
134 (93%)
102 (91,9%)
32 (97%)
p=0,28
Tableau 2 : L’anémie et ses caractéristiques selon la forme clinique
Paramètres
Total
Forme atypique
Formetypique
Calcémie normale
231 (74,5%)
198 (81,2%)
33 (50%)
Calcémie basse
79 (25,5%)
46 (18,8%)
33 (50%)
p=0,000001
Vitamine D normale
50 (30%)
42 (31,4%)
8 (24,2%)
Vitamine D
Basse
117 (70%)
92 (68,6%)
25 (75,8%)
p=0,42
Lipides
Normaux
230 (74%)
196 (80,3%)
34 (50,7%)
Lipides bas
81(26%)
48 (19,7%)
33 (49,2%)
p=0,000001
Albuminémie normale
258 (82,4%)
222 (89,9%)
36 (54,5%)
Albuminémie basse
55 (17,6%)
25 (10,1%)
30 (45,5%)
p=0,000001
TP normal
300 (94,6%)
243 (97,2%)
57 (85%)
TP bas corrigé
par la vit K
10 (3,2%)
4 (1,6%)
6 (9%)
TP bas non
corrigé/ Vit K
7 (2,2%)
3 (1,2%)
4 (6%)
p=0,006
Tableau 3 : Les autres signes biologiques carentiels selon la forme clinique
De façon générale, les signes extra-digestifs sont présents aussi bien dans la forme atypique que dans la forme typique.
L’anémie récidivante est le symptôme extra-digestif le plus fréquemment révélateur de la maladie cœliaque. En effet, 86,7% des patients ont eu une anémie.
Le taux d’anémie chute à 83% au moment du diagnostic en raison du traitement martial administré aux patients avant le diagnostic de la maladie. Les manifestations osseuses sont présentes dans 42,3%.
Les autres manifestations articulaires (9,1%), buccales (6,6%), hépatiques (6,3%) et neurologiques (4,7%) sont moins fréquentes. Le tableau 4 représente les différentes manifestations extra-digestives selon la forme clinique.
Signesextra-digestifs
Total
Formeatypique
Formetypique
Anémie
275 (86,7%)
210 (84%)
65 (97%)
Manifestations hépatiques
20 (6,3%)
15 (6%)
5 (7,5%)
Arthralgies
29 (9,1%)
24 (9,6%)
5 (7,4%)
Manifestations neurologiques
15 (4,7%)
14 (5,6%)
1 (1,5%)
Aphtose buccale récidivante
21 (6,6%)
18 (7,2%)
3 (4,5%)
Manifestations osseuses
134 (42,3%)
107 (42,8%)
27 (40,3%)
p=0,55
Tableau 4 : Les symptômes extra-digestifs selon la forme clinique
Les manifestations osseuses sont représentées essentiellement par l’ostéopénie (67,4%), l’ostéoporose (26,8%) et l’ostéomalacie (5,8%).
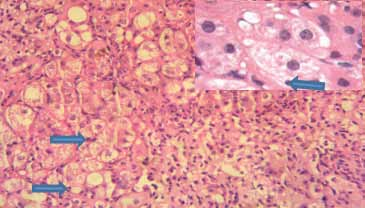
Les hépatopathies associées à la maladie cœliaque sont représentées par les cytolyses cryptogénétiques bénignes (56%) et les hépatopathies plus sévères dysimmunitaires ou non dysimmunitaires. Dans cette étude, ces dernières sont révélatrices de la maladie cœliaque dans 100% des cas. Il s’agit de cirrhoses dysimmunitaires (15,8%), de cirrhoses cryptogénétiques (7%), d’hypertensions portales sans cirrhose (17,6%), de syndrome de Budd Chiari (1,8%) et de maladie de Wilson (1,8%).
Les manifestations neurologiques sont rares. Elles sont représentées par l’épilepsie idiopathique (5 cas), la neuropathie périphérique (4 cas dont 1 cas de névrite optique), le syndrome dépressif (5 cas) et l’ataxie au gluten (1 cas).
Étant donné la grande majorité féminine de la population cœliaque, les anomalies gynéco-obstétricales sont décrites essentiellement chez la femme. Chez les hommes, un seul cas de stérilité, un cas d’anomalie du spermogramme ayant nécessité un traitement et un autre cas de baisse de la libido chez un sujet de 30 ans, ont été retrouvés.
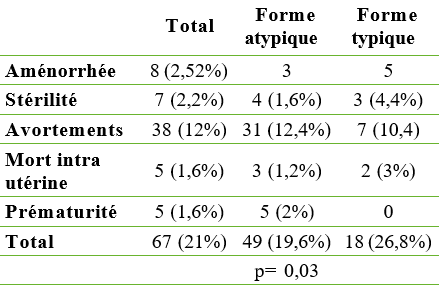
L’avortement répété représente l’anomalie gynéco-obstétricales la plus fréquente (12%). Les autres anomalies sont plus rares : stérilité dans 2,2%, mort in utero et prématurité dans 1,6%, aménorrhée primaire ou secondaire dans 0,37%. Globalement, ces anomalies gynéco-obstétricales sont plus fréquentes dans la population de patientes ayant une forme typique de maladie cœliaque, la différence étant statistiquement significative (p=0,03). Les troubles gynéco-obstétricaux selon la forme clinique sont illustrés dans le tableau 5.
Tableau 5 : Les symptômes extra-digestifs selon la forme clinique
Discussion :
La maladie cœliaque atteint l’adulte à n’importe quel âge avec cependant un pic de fréquence entre 30 et 40 ans [7]. Ces données concordent avec celles de notre étude, la moyenne d’âge de notre population de malades cœliaques étant de 31,6 ans.
Le diagnostic à un âge tardif est possible : 7% dans notre population, 6,5 à 9,3% dans la littérature [8,9]. Comme pour toute maladie auto-immune, la maladie cœliaque prédomine chez la femme (sex ratio de 0,50 à 0,25) [10,11]. Dans notre série, il est de 0,23.
La maladie cœliaque est une pathologie sous-diagnostiquée même dans les pays développés. En effet, aux USA pour chaque malade diagnostiqué, il en existe 7 à 8 non diagnostiqués [12]. En dépit d’une meilleure connaissance de la maladie, le délai diagnostique, défini par le temps écoulé entre le début des symptômes et le diagnostic, reste élevé. Dans l’étude de Fuchs, 32% des patients ont un délai diagnostique de plus de 10 ans [13]. Ceci est probablement dû au caractère très varié et non spécifique des symptômes liés à la maladie cœliaque. Dans notre étude, le diagnostic tardif (au-delà de 10 ans) représente 15,6% des malades et la majorité des patients (54,2%) ont un délai diagnostic entre 1 et 10 ans.
La maladie cœliaque peut se manifester par des symptômes variés ; aucun signe digestif n’est prédictif de la maladie même lorsque les symptômes sont associés [14]. Dans la littérature, les manifestations digestives décrites dans la maladie cœliaque sont variées et leur fréquence diffère d’une étude à l’autre. Ainsi, dans une étude multicentrique (Italie, Iran et Roumanie) [15], les symptômes digestifs varient d’un centre à un autre. Les symptômes prédominants sont la douleur abdominale et la diarrhée. La constipation, la dyspepsie et le ballonnement sont moins fréquents [15]. Devant ce grand polymorphisme symptomatique, le clinicien a du mal à sélectionner les patients susceptibles d’avoir une maladie cœliaque. Des algorithmes décisionnels ont été proposés pour aider le praticien à déterminer les patients à dépister. Certains de ces algorithmes informatisés semblent avoir une bonne sensibilité et spécificité [16], encore faut-il qu’ils soient disponibles et faciles à utiliser pour générer une meilleure adhésion des médecins. Dans notre série, les signes digestifs les plus fréquents sont le ballonnement abdominal 49,8%, les douleurs abdominales 43,2% et la diarrhée chronique 21,2%. Moins fréquemment, sont observés les vomissements 10,1%, la dyspepsie 7,2% et le reflux gastro-œsophagien 2,5%. Dans notre population cœliaque, 4,7% ont des symptômes qui correspondent aux critères de Rome III du syndrome de l’intestin irritable dans sa forme alternance diarrhée-constipation. Pour cette catégorie de patients, le traitement à base d’antispasmodiques et de médicaments anti-gaz était inefficace. En dépit du caractère réfractaire des symptômes, aucun dépistage sérologique n’a été réalisé. La fibroscopie digestive est effectuée devant l’apparition d’une anémie ferriprive permettant ainsi de faire le diagnostic de maladie cœliaque.
Dans la littérature, le taux du syndrome de l’intestin irritable dans la maladie cœliaque est très variable d’une étude à l’autre. Cette variabilité dépend des critères utilisés pour le diagnostic du syndrome de l’intestin irritable et de la population étudiée. Ainsi, lorsque les critères de Rome II sont utilisés pour identifier les patients ayant un syndrome de l’intestin irritable, 3,23% sont cœliaques dans une étude jordanienne [17] et 12% dans une étude iranienne [18]. Tandis que 36% des cœliaques ont des symptômes du syndrome de l’intestin irritable dans l’étude de Green et al [19]. Dans une étude plus récente, les critères de Rome III ont été utilisés, le taux de 0,8% de maladie cœliaque confirmée histologiquement a été rapporté [20]. Cela est probablement dû au recours à des critères diagnostiques plus sélectifs dans la classification de Rome III pour le diagnostic du syndrome de l’intestin irritable.
Dans notre population cœliaque, la forme atypique est prédominante ; elle représente 78,8% de la population cœliaque avec un rapport forme atypique/forme typique=3,7.
Ces données sont concordantes avec celles de la littérature. En effet, les formes atypiques sont les plus fréquentes, en particulier au cours de ces dix dernières années. Elles représentent 80% dans l’étude de Farrell [21]. Dans une série Italienne ce taux est de 66% [22]. Dans les rares études du Maghreb et du Moyen Orient les résultats sont très variables selon les régions.
Notre étude ne retrouve aucune forme silencieuse, cela s’explique :
d’une part, par notre respect strict de la définition d’Oslo [23] concernant la forme silencieuse (en effet, nous avons considéré qu’une maladie cœliaque révélée par une anémie isolée ou une ostéopénie isolée est une forme atypique et non silencieuse),
d’autre part, par la rareté de l’utilisation du dépistage sérologique dans notre étude.
Dans notre cohorte, les symptômes cliniques sont souvent non accompagnés de signes carentiels qui orientent vers une pathologie organique, tels que la pâleur intense observée dans seulement 22,7%, les anomalies de la peau et des phanères (18,6%), le retard staturo-pondéral (6%). Ces anomalies secondaires à des carences sévères et prolongées sont peu fréquentes dans la population cœliaque témoignant de la rareté des formes sévères. Elles sont plus observées dans les formes typiques. Cependant, la différence sur le plan statistique n’est pas significative (p=0,09).
Le signe le plus objectif lors de l’examen physique qui renseigne sur le retentissement de la maladie cœliaque est le BMI (body mass index). La moyenne du BMI de notre population de malades est normale (20,78). Toutefois, celle de la population atypique est statistiquement supérieure à celle de la population typique (21,15 vs 19,44). Cela est probablement dû au fait que la maladie soit plus sévère dans la forme typique. Ces résultats nous permettent d’attester qu’un BMI diminué n’est pas un bon indicateur de maladie cœliaque, puisque la majorité des patients ont un BMI normal. Ceci est d’ailleurs rapporté dans les études internationales [24].
Dans la littérature, l’anémie est présente lors du diagnostic de maladie cœliaque dans 12 à 69% [25]. Elle représente le symptôme extra-digestif le plus fréquent [25]. L’anémie est le plus souvent ferriprive, récidivante ou réfractaire au traitement [26]. Des carences en vitamines B12 ou acide folique peuvent être associées [25, 26]. Dans notre étude, l’anémie est le signe carentiel le plus fréquent (83%) dans les deux formes cliniques. Cette fréquence élevée (dans les deux formes cliniques) est probablement en rapport avec le fait que le fer soit absorbé dans le duodénum, qui est atteint de façon quasi constante dans la maladie cœliaque. Il est à noter que le taux d’anémie est plus important de façon significative dans la forme typique. Ce taux important observé dans notre population de malades, peut être expliqué par des carences d’apport probablement associées à la malabsorption. L’anémie est le plus souvent ferriprive (90,4%) et microcytaire hypochrome (99,2%).
Les autres carences sont beaucoup moins fréquentes que l’anémie. Elles témoignent d’une malabsorption plus sévère. En effet, l’hypocalcémie est présente dans 25,5%, l’hypolipidémie dans 26%, l’hypo albuminémie dans 17,6% et la carence en vitamine K dans 3,2%. Ces carences sont, de façon très significative, plus souvent observées dans la forme typique, ce qui témoigne de la plus grande sévérité de cette forme par rapport à la forme atypique.
Dans la littérature, les carences en albumine, électrolytes et lipides sont rares [27]. Elles sont observées surtout dans les formes typiques de maladie cœliaque comme il apparaît dans notre étude.
Les manifestations extra-digestives sont présentes aussi bien dans les formes atypiques que typiques. Comme précédemment décrit, l’anémie est le signe extra-digestif le plus fréquent (86,7%). En deuxième position viennent les manifestations osseuses (42,3%). Elles sont souvent asymptomatiques et doivent être recherchées systématiquement dès que le diagnostic de maladie cœliaque est posé. Dans la littérature, près des 2/3 des patients ont une ostéodensitométrie pathologique (ostéopénie et ostéoporose) [28]. Ces anomalies osseuses sont présentes dans les deux formes de maladie cœliaque sans différence significative, elles sont dues essentiellement à la malabsorption du calcium et de la vitamine D [29]. L’absorption du calcium se fait au niveau du duodénum constamment atteint quelle que soit la forme clinique [30].
Dans la littérature, l’hypertransaminasémie idiopathique ou « hépatite » cœliaque est présente chez près de la moitié des cœliaques non traités [31]. Dans notre cohorte, elle n’est retrouvée que chez 32 patients soit dans 10% des cas. L’augmentation de la perméabilité intestinale semble jouer un rôle non négligeable dans la survenue de ces « hépatites » [32], plus fréquentes lorsque l’atteinte intestinale est sévère. Le taux faible « d’hépatite » cœliaque observé dans notre étude pourrait être expliqué par le fait que les formes sévères soient peu fréquentes dans notre cohorte.
Dans notre série, les atteintes hépatiques sévères sont retrouvées dans 8,2%. L’étiologie dysimmunitaire des cirrhoses constitue la cause la plus fréquente (9 cirrhoses dysimmunitaires sur 13 cirrhoses). Ces données sont concordantes avec celles de la littérature. En effet, la cholangite biliaire primitive et l’hépatite auto-immune sont fréquemment associées à la maladie cœliaque [33,34,35]. La même prédisposition génétique est probablement à l’origine de cette association [36]. Le dépistage de la maladie cœliaque est recommandé au cours de ces maladies auto-immunes [37].
Dans près de la moitié des cas (14 malades sur 26), le diagnostic étiologique de l’hépatopathie n’a pas pu être établi. Il s’agit des quatre cirrhoses cryptogénétiques et des dix HTP intra-hépatiques idiopathiques. Certains auteurs ont décrit l’existence de cirrhoses cryptogénétiques [38] ou d’HTP intra-hépatique [39] associées à la maladie cœliaque. La relation entre ces atteintes hépatiques (non étiquetées) et la maladie cœliaque a été évoquée devant l’absence d’étiologie et l’amélioration sous régime sans gluten.
Les manifestations gynéco-obstétricales telles que l’aménorrhée, la stérilité ou les avortements répétés ont été décrites, associées à la maladie cœliaque dans de nombreux écrits. Elles sont dues à la malabsorption de l’acide folique, du zinc et du sélénium [40,41,42], ainsi qu’à une déficience en ghreline et leptine [43], mais aussi à une dysrégulation du système immunitaire [44]. Dans notre cohorte de patientes cœliaques, ces anomalies gynéco-obstétricales sont significativement plus fréquentes dans la forme typique de la maladie (26,8% versus 19,6% ; p=0,03), témoignant de la sévérité des carences dans cette forme clinique de maladie cœliaque.
Pour conclure : Il est important que le praticien sache évoquer la maladie cœliaque devant des symptômes digestifs banals tels que des ballonnements, des douleurs abdominales atypiques ou un syndrome de l’intestin irritable réfractaire au traitement. La connaissance des manifestations extra-digestives de la maladie cœliaque est importante. En effet, la maladie peut se manifester par une simple anémie chronique et réfractaire, une ostéopénie ou des manifestations hépatiques. De même, la forme historique avec diarrhée chronique et syndrome carentiel est certes plus sévère, mais elle ne doit plus être considérée comme la forme prédominante chez nous.
Histogramme représentant les symptômes digestifs en pourcentage
SII : syndrome de l’intestin irritable RGO : reflux gastro-œsophagien
Bibliographie :
European Society for Pediatric, Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease (Husby S et al). J Pediatr Gastroenterol Nutr. 2012; 54(1): 136-60.
West J, Logan RFA, Hill PG et al. Seroprevalence correlates and characteristics of undetected celiac disease in England, Gut 2003; 52:960-965.
Cook HB, Burt MJ, Collett JA et al. Adult coeliac disease: Pre- valence and clinical significance. J Gastroenterol Hepatol. 2000; 15 :1032-36.
Thèse de doctorat en médicine (spécialité pédiatrie) du Docteur Manoubia Bensmina soutenue en 2015 : Détermination de la Pré- valence de la maladie cœliaque chez les enfants d’âge scolaire de la wilaya d’Alger.
Jones S, D’Souza C et Haboubi N. Patterns of clinical presentation of adults coeliac disease in a rural setting. Nutr J. 2006; 5(24): 8 pages.
Fernandez A, Gonzalez L et De la Fuete J. Coeliac disease: Clini- cal features in adult populations. Rev Esp Enferm Dig. 2010; 202(8): 466-471.
Snyder CL, Young DO, Green PHR, et al. Celiac disease. 2008, in Pagon RA, Adam MP, Ardinger HH, et al editors. Gene Reviews. University of Washington, Seatle; 1993-2015.
Singh P, Sherghill S, Makharia GK, et al. Celiac disease in older adults. J Gastrointestin Liver Dis. 2013; 22: 359-60.
Zingone F, West J, Auricchio R, et al. Incidence and distribution of coeliac disease in Campania (Italy): 2011-2013. UEGJ. 2015; 3(2): 182-89.
Bardella MT, Fredalla C, Saladino V, et al. Gluten intolerance: Gender and age related differences in symptoms. Scand J Gastroen- terol. 2005; 40: 15-9.
Liorente-Alonso MJ, Fernandez-Acenero MJ et Sebastien M. Glu- ten intolerance: Sex-age-related features. Can J gastroenterol. 2006 ; 20(11) : 719-722
Mooney PD, Hadjivassiliou M, Sanders DS. Coeliac disease. BMJ. 2014; 318: g1561.
Fuchs V, Kurppa K, Huhtala H, et al. Factors associated with long diagnostic delay in celiac disease. Scand J Gastroenterol. 2014 ; 49(11) : 1304-10.
Nejad RM, Pourhosseingholi MA, Mojarad EN, et al. Atypical presentation is dominant and typical in celiac disease. 2009 ; 18(3) : 285-91.
Ehsani-Ardakani MJ, Rostami-Nejad M, Villanacci V, et al. Gas- trointestinal and non-gastrointestinal presentation in patients with celiac disease. Arch Iran Med. 2013; 16(2): 78-82
Ludvingsson JF, Pathak KJ, Murphy S, et al. Use of computerized algorithme to identify individuals in need of testing for celiac disease. J Am Med Inform Assoc. 2013; 20: e306-e310.
Jadallah KA et Khader YS. Celiac disease in patients with pre- sumed irritable bowel syndrome: A case-finding study. WJGE. 2009; 15(42) : 5321-5325
Masjedijadeh R, Hajjani E, Hashemi J, et al. Celiac disease in South-West Iran. WJGE. 2006 ; 12 : 4416-19.
Green PHR, Stavropoulos SN, Panagi SG et al. Characteristics of adult celiac disease in the USA: results of a national survey. Am J Gastroenterol 2001; 96(1) :126-31.
Sharma H, Verma AK, Das P, Gupta SD, Ahuja V et Makharia GK. Prevalence of celiac disease in Idian patients with irritable bowel syndrome and uninvestigated dyspepsia. J Dis Dig. 2015.16(8): 443-48
Volta U, Caio G, Sanghellini V et al. The changing clinical profile of celiac disease: a 15-year experience (1998-2012) in an Italian refe- ral center. Gastroenterology. 2014 ; 14:194-8)
Ludvingsson JF, Leffler DA, Bai JC, et al. The Oslo definitions for celiac disease and related terms. Gut. 2013 ; 62 : 43-52
Van Der Pals M, Myleus A, Norstrom F, et al. Body mass index is not a reliable tool in predicting celiac disease in children. BMC Pediatr. 2014; 14: 165.
Hershko C et Patz J. Ironing out mechanisms of anemia in celiac disease. Haematologica. 2008; 93(12):1761—65.
Murray JA, Van Dyke C, Plevak MF et al. Trends in the iden- tification and clinical feature of celiac disease in a North American community, 1950-2001. Clin gastroenterol Hepatol. 2003; 1(1): 19-27
Fouda MA, Khan AA, Sultan M, et al. Evaluation and manage- ment of skeletal health in celiac disease: position statement. Can J Gastroenterol. 2012; 26(11): 819-29.
Molteni N, Bardella MT, Vezzoli G, et al. Intestinal calcium ab- sorption as shown by stable strontium test in celiac disease before and after gluten-free diet. Am J Gastroenterol. 1995; 90: 2025-28.
Kurpa-Kozak U. Pathologic bone alterations in celiac disease: Etiology, epidemiology, and treatment. Nutrition. 2014; 30: 16-24.
Volta U. Liver dysfunction in celiac disease, Minerva Med 2008;99:619-29
Volta U, De Franceshi L, Lari F, et al. Coeliac disease hidden by cryptogenetic hypertransaminasaemia. Lancet. 1998; 352: 26-9
Floerani A et al, Prevalence of celiac disease in primary biliary cirrhosis and antimitochondrial antibodies in adults celiac patients in Italy, Dig Liv Dis 2002; 34:258-261
Van Gerven NM, Bakker SF, DE Boer YS, et al. Seroprevalence of celiac disease in patients with autoimmune hepatitis. Eur J Gastroen- terol Hepatol. 2014 ; 26(10) : 1104-7.
Villalta D, Girolami D, Bidoli E, et al. High prevalence of celiac disease in autoimmune hepatitis detected by anti-tissue transglutami- nases autoantibodies. J Clin Lab Anal. 2005; 19(1): 6-10.
Ludvingsson JF, Elfstrom P, Broom U, et al. Celiac disease and risk of liver disease: a general population-based study. Clin Gastroenterol Hepatol. 2007; 5(1): 63-9 e1.
National Institute for health and clinical excellence (NICE). Coe- liac disease: recognition and assessment of coeliac disease. 2009.
Wakim-Fleming J, Pagadala MR, McCullough AJ, et al. Preva- lence of celiac disease in cirrhosis and outcome of cirrhosis on a glu- ten-free diet: a prospective study. J Hepatol. 2014; 61(3): 558-63.
Maiwall R, Pulimood AB, Babji S, et al. Investigation into celiac disease in Indian population patients with portal hypertention. Ind J Gastroenterol. 2014; 33(6): 517-23.
Hirson C. Coeliac infertility-folic acid therapy. Lancet. 1970; 1(7643): 412.
Bedwal RSBA. Zinc, Copper, and Selenium in reproduction. Experimentia. 1994; 50: 626-40.
Singhal N, Alam S, Sherwani R, et al. Serum zinc levels in celiac disease. Indian Pediatr. 2008; 45: 319-21.
Martin JR, Lieber SB, McGrath J, et al. Maternal ghrelin defi- ciency compromises reproduction in femal progery through altered uterine developmental programming. Endocrinology. 2011; 152: 2060-66.
Anjum N, Baker PN, Robinson NJ, et al. Maternel celiac disease autoantibodies from celiac patients are responsible for trophoblast damage via apoptosis in vitro. Am J Gastroenterol. 2010.
A.BALAMANE(1), B.BENHABYLES(2), N. SMAIL(2) ,Service de Gastroentérologie, CHU Isaad Hassani, Beni Messous, Alger. Service d’Épidémiologie, CHU Mustapha Bacha, Alger.
Abstract : In the world, regional distribution of Inflammatory Bowel Disease (IBD) mainly ulcerative colitis (UC) and Crohn’s Disease (CD) is ubiquitous though unequal. Algeria is considered as a low incidence country of IBD. In this work, we have studied the epidemiologic evolution of that disease from 1981 to 2006 in Algiers department through the comparison of 3 epidemiologic studies using the same methodologic approach done between 1981-1985, 1994-1998, 2003-2006.
Some conclusion can be drawn from the results of this work. 1. IBD are not rare in Algiers; 2. Incidence of UC remains stable during the 25 years study period; 1.24.105/y to 1.38.105/y CD incidence showed a different evolution with firstly an important increase from 0.79 105/y to 1.64 105/y then a stabilization of the incidence rate (1.49 105/y); 3. Disease was as frequent in men than in women; 4. 70% of the patients were young adults: the first peak of incidence was comprised between 20 and 29 years but a second peak was observed after 50 y; 5. Anatomoclinical aspects did not changed during the whole period study and our data are close to those of the literature.
Résumé : Représentées essentiellement par la Rectocolite Hémorragique (RCH) et la Maladie de Crohn (MC), les Maladies Inflammatoires chroniques de l’Intestin (MICI) sont des affections de cause inconnue à répartition ubiquitaire mais très inégale selon les régions. L’Algérie est considérée comme une zone de faible prévalence de ces maladies. Notre travail a consisté essentiellement en l’étude de l’évolution épidémiologique des MICI dans la Wilaya d’Alger entre 1981 et 2006 à travers la comparaison de 3 études successives utilisant une méthodologie sensiblement identique : 1981-1985, 1994-1998, 2003-2006. Quelles conclusions peut-on tirer de ce travail ? 1. Les MICI ne sont pas des maladies rares à Alger ; 2. L’incidence de la RCH est restée relativement stable en 25 ans passant de 1.24.105/h/an à 1.54.105/h/an et 1.38.105/h/an. La MC a connu une évolution différente après une nette augmentation entre 1981 et 1998 (0.79.105/h/an à 1.04.105/h/an) correspondant à un accroissement de 107%, l’incidence se stabilise à 1.49.105/h/an. Cette évolution correspond à l’évolution générale des MICI dans le monde ; 3. Hommes et femmes sont touchés de façon sensiblement égale ; 4. L’affection touche essentiellement l’adulte jeune (70% des patients sont âgés de 20 à 39 ans) mais il existe un 2ème pic d’incidence entre 50 et 59 ans ; 5. Le profil anatomo-clinique de l’affection n’a pas connu d’évolution majeure en 25 ans et ses caractéristiques, appréciées sur les données de la dernière enquête sont tout à fait classiques et ne différent pas de celles de la littérature.
Mots-clés : Maladies Inflammatoires Chroniques de l’Intestin, Épidémiologie, Profil anatomo-clinique
Introduction :
Les Maladies Inflammatoires Chroniques de l’Intestin (MICI) sont caractérisées par un état inflammatoire du tube digestif d’évolution chronique.
Cette entité anatomique regroupe principalement deux affections, la Rectocolite Hémorragique (RCH) et la Maladie de Crohn (MC), reconnues et distinguées l’une de l’autre sur la base d’un faisceau d’arguments cliniques, endoscopique, radiologique et anatomopathologiques. Plus rarement (10% des cas en moyenne), lorsque devant un état inflammatoire colique chronique il n’est pas possible de le rattacher à une RCH ou à une MC, on parle de Colite Inclassable (CI). Lorsque la difficulté diagnostique est telle que la distinction ne peut pas se faire même sur la pièce opératoire, il s’agit d’une colite indéterminée (C Ind.).
La pathogénie de ces affections est très incomplètement comprise. Elle semble être multifactorielle et implique une prédisposition génétique, des facteurs environnementaux qui engendrent un dysfonctionnement immunitaire intestinal à l’origine d’une inflammation chronique.
La RCH est une condition inflammatoire chronique de la muqueuse qui touche constamment le rectum et s’étend de façon continue, symétrique et variable sur le reste du colon. Cette affection est guérie par la colectomie.
En revanche, la MC, bien que prédominante au niveau de l’intestin, peut intéresser la totalité du tube digestif « de la bouche à l’anus ». Cette affection inflammatoire hétérogène à distribution asymétrique, discontinue et polymorphe se manifeste habituellement par une diarrhée rarement sanglante, des accidents obstructifs digestifs intermittents qui traduisent bien la nature volontiers sténosante et fistulisante des lésions intestinales ; des lésions anales particulières sont fréquentes ; histologiquement se trouve réalisée dans les cas typiques, une inflammation granulomateuse intestinale transmurale. La chirurgie ne guérit pas définitivement la maladie et l’exérèse chirurgicale est souvent suivie de récidive.
Dans les deux affections, aux manifestations intestinales s’associent de façon inconstante des manifestations extra intestinales (articulaires, cutanées, oculaires, hépatobiliaires, vasculaires …) de gravité et d’évolution variable, certaines d’entre elles étant susceptibles d’évoluer de façon autonome pour leur propre compte.
L’évolution des MICI se fait le plus souvent de façon intermittente par poussées entrecoupées de périodes de rémissions ; les formes chroniques évoluant d’un seul tenant sont plus rares et parfois plus sévères. Les poussées sont d’intensité variable ; elles peuvent être graves ou compliquées mettant en jeu le pronostic vital du patient. En l’absence d’une pathogénie bien définie, le traitement médical des MICI demeure symptomatique.
Il fait appel habituellement à la corticothérapie ou aux aminosalicylates, les immunosuppresseurs sont réservés aux formes corticodépendantes et les thérapeutiques biologiques d’introduction plus récente (anticorps anti Tumor Necrosis Factor) sont réservées notamment aux formes réfractaires de la maladie.
L’étude de l’épidémiologie des MICI et en particulier celle de son évolution dans le temps peut avoir une importance considérable non seulement sur un plan purement scientifique mais aussi sur le plan de la santé publique.
Elle peut permettre :
D’une part, d’initier ou de tester des hypothèses pathogéniques en identifiant notamment des facteurs de risque liés à l’environnement en se basant sur d’éventuelles différences géographiques quant à l’incidence et aux données démographiques,
D’établir des prévisions épidémiologiques en fonction de l’évolution connue de la maladie dans le temps et des modifications locales de l’environnement en se fondant sur les données recueillies au niveau des pays ayant une longue histoire de MICI,
D’autre part, de guider l’action des services de santé, d’organiser la prise en charge des patients et de mettre en place des programmes adéquats.
De nombreuses études épidémiologiques ont été consacrées aux MICI au cours de ces 5 dernières décennies. Les études sont de valeur très inégale et présentent des différences méthodologiques parfois importantes, ce qui rend leur comparaison délicate en particulier entre les études anciennes ou provenant de zones géographiques différentes.
Cependant, les études épidémiologiques exposent à de multiples difficultés méthodologiques.
Les difficultés d’interprétation d’une étude épidémiologique sont nombreuses et se situent à différents niveaux des enquêtes.
En premier lieu, il existe, en l’absence d’étiologie des MICI, des difficultés à identifier de façon certaine et définitive ces états pathologiques et les séparer d’autres états inflammatoires.
Le diagnostic de MICI n’est pas toujours évident.
Le polymorphisme clinique (mode de début, tableaux atypiques pseudo infectieux) impose bien souvent, dans les formes inaugurales un diagnostic différentiel laborieux basé sur les résultats d’explorations adéquates et de valeur indiscutable,
Il peut exister une variation inter observateur ; ceci est surtout valable pour l’interprétation des examens complémentaires (endoscopie, histologie),
La classification « étiologique » initiale peut être erronée et dans 5 à 10% des cas, le diagnostic initial peut connaître des changements dans le temps,
Le délai entre le début clinique réel de la maladie et le moment du diagnostic peut, lorsqu’il est important, conduire un biais de recrutement,La disparité entre les systèmes de santé influe sur le recrutement et l’enregistrement des patients,
La sélection de la population à prendre en compte est capitale et peut être responsable de biais de recrutement (séries de patients hospitalisés Vs séries issues de la population générale).
Les séries hospitalières conduisaient à une sous-estimation certaine de l’incidence et de la prévalence de la maladie (les patients les moins atteints n’étant pas comptabilisés), alors que la sévérité, la morbidité et la mortalité étaient surévaluées. Les études de population, plus récemment introduites estiment de façon plus sûre l’incidence et la prévalence de la maladie dans une zone géographique déterminée. Dans les pays disposant d’un registre national des MICI, les résultats sont encore plus proches de la réalité épidémiologique de la maladie (Suède-Danemark).
La méthodologie de collecte des données peut également être à l’origine d’insuffisances ; les études les plus fiables sont les études prospectives qui permettent de recueillir des informations de haute qualité.
Il existe également un manque d’uniformité dans les possibilités et moyens de diagnostic disponibles selon les pays.
Dans le monde, la répartition des MICI est ubiquitaire mais se fait de façon très inégale. C’est ainsi que les MICI prédominent dans les pays occidentaux bénéficiant d’un niveau de vie élevé (USA, Pays Nordiques, Grande Bretagne) ; elles sont moins fréquentes dans les pays d’Europe du Sud (en Espagne, Italie, Grèce) et encore moins dans les pays en développement encore que les écarts, en termes d’incidence et de prévalence, aient tendance à se resserrer.
En Algérie comme dans le reste du Maghreb, les MICI étaient classiquement considérées comme des maladies rares voire exceptionnelles.
En 1979 et en 1980, deux travaux effectués à Alger battaient en brèche cette assertion et avaient attiré l’attention sur la relative fréquence des MICI sous nos climats. Néanmoins, ces deux études rétrospectives ne pouvaient, en aucun cas, permettre des conclusions définitives.
Il nous avait paru intéressant et essentiel de compléter ces premiers travaux par des enquêtes épidémiologiques prospectives.
Une première étude effectuée à Alger entre le 1er janvier 1981 et le 31 décembre 1985 devait révéler que la RCH et la MC n’étaient pas des affections rares au niveau de la Wilaya d’Alger.
Une deuxième étude, reposant sur les mêmes critères de diagnostic et intéressant la population de la même région a été réalisée entre le 1er janvier 1985 et le 31 décembre 1998.
Enfin, une troisième enquête devait être réalisée dans les mêmes conditions entre 2003 et 2006.
Données épidémiologiques :
Dans le monde, la plupart des études épidémiologiques réalisées dans la même région et au cours de la même période, concluent que la RCH représente la MICI la plus fréquente.
En Europe, le ratio RCH/MC a été évalué à 1,44/1 à 5,28/1 ; il est de 1,1/1 à1,43/1 aux USA et 2,41/1 en Asie (Corée). En revanche, la MC a été trouvée plus fréquente que la RCH en Ecosse (0,45/1), en France (0,73/1) en Grande Bretagne (0,69/1), au Canada (0,85/1).
Dans le monde, il existe d’importantes variations géographiques de l’incidence de la MC ; rapportée à 100.000 habitants/an, elle est de 6,9 aux USA, de 14,6 à 15,8 au Canada, de 2,3 à 6,3 en Europe, de 0 ,5.105 / an à 1,6.105 /an en Afrique.
La prévalence des MICI a été évaluée récemment. Celle de la RCH a été estimée de 4,9 à 505.105 en Europe, de 4,9 à 168,3.105 en Asie et au Moyen Orient et de 37,5 à 248.105 en Amérique du Nord. La prévalence de la MC est de 0,6 à 322.105 en Europe, de 16,7 à 318,5.105 en Amérique du Nord.
Ces différences s’expliquent, au moins en grande partie, par l’existence d’un gradient Nord-Sud qui a été mis en évidence aussi bien en Europe qu’aux USA.
Ainsi, l’étude Européenne (rapportée par Shivanada) concernant une période allant du 1/10/91 au 30/9/93 a intéressé 20 pays (8 Centres en Europe du Nord et 12 en Europe du Sud).
L’incidence de la MC a été trouvée supérieure de 40% dans les pays du Nord, comparée à celle des pays du Sud.
Aux USA, 2 études réalisées par Sonnenberg portant d’une part sur les hospitalisations pour MC dans les Hôpitaux des Vétérans de l’Armée et d’autre part au niveau des archives de la Sécurité Sociale (MEDICARE), ont montré que les taux de fréquence de prise en charge étaient plus élevés dans les Etats du Nord des USA.
L’incidence des MICI en général et de la MC en particulier a subi d’importantes modifications dans le temps. Nous disposons de plusieurs études privilégiées qui ont comporté un suivi allant de 10 à 60 ans. Dans les pays à forte incidence de MICI, la RCH, après augmentation rapide de son incidence entre 1940 et 1970 connait aujourd’hui une stabilisation. La MC, historiquement, a subi un accroissement plus long de sa fréquence mais connaitrait actuellement une stabilisation de son incidence.
Cependant, dans les pays initialement à faible incidence (pays du Sud par exemple), la MC est en constante augmentation (Italie, Grèce, Espagne, Asie).
En Algérie :
À Alger, l’évolution à long terme de l’incidence de la MC a connu une évolution en 2 temps ; d’abord une augmentation franche entre 1981 et 1998 (de 0,79.105
/an à 1,64.105 /an), puis une stabilisation entre 1998 et 2006 (1,49.105/an).
La répartition des cas de MICI en fonction de sexe montre des résultats variables selon les pays et la nature de la maladie. On décrit habituellement une légère pré-dominance féminine dans la MC et masculine dans la RCH. Il en a été ainsi dans notre dernière étude (2003- 2006) à Alger (RCH : SR H/F=1,04/1, MC : SRF/H :1,05/1.
Les MICI débutent chez l’adulte jeune dans la majorité des pays. Le maximum de fréquence de l’âge de début se situe habituellement entre 20 et 40 ans (> 60% des cas). Dans la majorité des cas, la courbe de répartition des cas de MC en fonction de l’âge est unimodale avec un pic de fréquence qui se situe entre 25 et 35 ans mais il peut être décalé vers 35-40 ans. Dans quelques études, la répartition est bimodale avec un deuxième pic tardif et beaucoup moins important, au-delà de 60 ans.
On notera que ce type de répartition est en général commun à l’homme et à la femme.
L’évolution démographique à long terme a été peu étudiée ; concernant la MC le sex ratio F/H est en général resté constant dans le temps. Pour notre part, nous avons noté certaines différences entre les courbes de répartition des incidences par tranches d’âge chez l’homme et chez la femme avec un pic de fréquence de 20 à 29 ans chez l’homme et 30-39 ans chez la femme ; avec une augmentation sensible des cas féminins, le sex ratio H/F passant de 1,89/1 (1980) à 0,94/1 (2006).
Mais le fait marquant qui émerge de nos études est l’augmentation importante (X2) de la MC en 25 ans.
De ce fait, compte tenu de sa prévalence, de sa gravité potentielle et du coût élevé de sa prise en charge, la MC est en passe de devenir un véritable problème de santé publique dans notre pays.
Quelles conclusions peut-on tirer de ce travail ?
Les MICI ne sont pas des maladies rares à Alger ;
L’incidence de la RCH est restée relativement stable en 25 ans passant de 1.24.105/h/an à 1.54.105/h/ an et 1.38.105/h/an. La MC a connu une évolution différente après une nette augmentation entre 1981 et 1998 (0.79.105/h/an à 1.04.105/h/an) correspondant à un accroissement de 107%, l’incidence se stabilise à 1.49.105/h/an. Cette évolution correspond à l’évolution générale des MICI dans le monde ;
Hommes et femmes sont touchés de façon sensiblement égale ;
L’affection touche essentiellement l’adulte jeune (70% des patients sont âgés de 20 à 39 ans) mais il existe un 2ème pic d’incidence entre 50 et 59 ans ;
Le profil anatomoclinique de l’affection n’a pas connu d’évolution majeure en 25 ans et ses caractéristiques, appréciées sur les données de la dernière enquête sont tout à fait classiques et ne différent pas de celles de la littérature.
Référence bibliographiques
Boucekkine T. Colite ulcéreuse et Maladie de Crohn. Rapport de la Société de Médecine d’Alger au IXème Congrès Médical Maghrébin- Rabat 12-14 mai 1980
Boucekkine T., Khedis A, Ziari A, Asselah F, Asselah H et Illoul G. Aspects anatomopathologiques de la Colite Ulcéreuse à Alger. Ann. Gas- troentérologie Hépatologie 1982, 18,5 ; 351-359
Boucekkine T., Kermouni M, Mehdi F, Illoul G. Epidemiologic evolution of Ulcerative Colitis in Algiers (1981-1985). Digestion 1998, 5 (Suppl 3), 152, Fol, M1357
Boucekkine T. Baiod N, Nakmouche M, Illoul G. Epidemiologic evolution of Inflammatory Bowel Disease (IBD) in Algiers (1981-1991
Sedlack RE, Whistant J, Everback LR. Incidence of Crohn’s di- sease in Olmested Country, Minnesota (1935-1975) Ann. J. Epidemio- logy 1980,112, 759-769.
Loftus EV Jr. Clinical epidemiology of IBD: incidence, prevalence and environmental influences Gastroenterology. 2004, 126, 1504- 1517.
Ekbom A, Helmick C, Zack M, Adami HO. The epidemiology of inflammatory Bowel Disease: a large population based study in Swi- den. Gastroenterology 1991,100, 350-358
Munkolm P, Langholz E, Haagen Nielsen O, Kreiner S, Binder U. Incidence and Prevalence of Crohn’s Disease in the county of Copen- hagen. (1962-1987). Scand J Gastroenterology 1992,27, 609-614.
Balamane A. Maladies Inflammatoires Cryptogéniques de l’intes- tin à Alger- Evolution épidémiologique et profils anatomocliniques et évolutifs. Thèse DEMS-Alger, 2010.
Martinez Salmeron JF, Rodrigo M, Deteresa J, Nagueras F, Garcia Montero M, De Sola. 10. Epidemiology of inflammatory Bowel Disease in province of Granada, Spain: a retrospective study from 1979-1988 Gut 1993, 34, 1207-1209.
Vucelic B, Kovac B, Sentic M, Milicic D, Hadzic N, Juresa V, Bozikov J, Rotkvic I, Baljehac. Epidemiology of Crohn’s Disease in Zaghreb, Yuguslavia a ten-year prospective study Int. J. Epidemiology 1991, 20,216-220.
Tsianos EV, Masalas CN, Merkourpoulous M, Dakelos GN, Lo- gan RFA. Incidence of inflammatory Bowel Disease in North Greece: rarity of Crohn’s disease in an area where Ulcerative Colitis is com- mon Gut 1994, 35; 3693-3702.
Shivananda, Lennard Jones J, Logan, R, Fear N, Price A, Carpen- tal L, Blankeinstein Van M. Incidence of Inflammatory Bowel
Disease across Europe, is there a difference between north and south? Results of the European Collaborative Study on Inflammatory Bowel Disease. Gut 1996; 39:690-697
Pagenault M, Trou I, Alexandre JL, Cruchant E, Dabadie A, Cha- pern J, Robaszkiewicz M, Incidence des Maladies inflammatoires du Tube Digestif en Bretagne (1998-1995). Gastroentero Clin Biol 1997, 21, 483-490.
Thomas GAO, Millard Jones D, Rhodes J, Roberts GM. Incidence of Crohn’s Disease in Cardiff over 60 Years (1986-1990) an uptade. European journal of Gastroenterology and Hepatology 1995, 7,401- 405.
Sonnenberg A, Mc Carthy DJ, Jacobsen SG. Geographic varia- tion of Inflammatory Bowel Disease within the United States Gas- troenterology 1991, 100,143-149.
Sonnenberg A, Wasserman JH. Epidemiology of Inflammatory Bowel Disease among US Milatary Veterans. Gastroenterology 1991, 101,122-130.
Gollop JH, Phillips SF, Melton III U, Zinsmeister AR. Epide- miologic aspects of Crohn’s Disease: a population based study in Olmested County, Minnesota 1947-1982. Gut 1988,29,45,56.
Loftus EV, Silverstein MD, Sandborn WJ, Tremaine J, Harmesea WS, Zinsmeister AR. Crohn’s Disease in Olmsted County 1940-1993, incidence, prevalence and Survival. 19. Gastroenterology1998, 114, 1161-1168
Lee FI, Costello FT. Crohn’s Disease in Blackpool: Incidence and prevalence 1968-1980 Gut 1985, 26,274-278.
K. BELHOCINE, Service de Gastro-Entérologie, CHU Mustapha Bacha, Alger.
Abstract : Gastroesophageal reflux disease (GERD) is one of the most common digestive disorders over the world. It is a chronic illness with a significant impact on patients’ quality of life and may lead to complications and esophageal cancer. Diagnosis and management of GERD are based on clinical analysis. However, in some cases, testing is needed (i.e.: endoscopy, pH and pH-impedance monitoring, esophageal manometry). GERD is traditionally and empirically managed with anti-secretory medication; surgery may be required in selected patients. In the future, the better understanding of the disease’s pathophysiology will likely offer new therapeutic strategies.
Résumé : Le reflux gastro-œsophagien est une pathologie fréquente devenue ubiquitaire. Motif fréquent de consultation en soins primaires, secondaires et tertiaires, il s’agit d’une entité qui expose à certaines complications qui peuvent grever le pronostic vital. Sa prise en charge repose sur l’analyse clinique et le traitement médical dans les situations les plus simples et typiques, mais nécessite aussi le recours à des explorations spécialisées et d’autres alternatives thérapeutiques, dans les situations plus complexes de résistance thérapeutique ou de manifestions extra-digestives dominantes. Des mécanismes étiopathogéniques multiples et intriqués, une meilleure maitrise thérapeutique et la satisfaction des patients passeront certainement par le contrôle de ces différentes cibles pathogéniques.
Mots-clés : Reflux gastro-œsophagien, traitement du RGO, reflux.
Introduction – Définition :
Le reflux gastro-œsophagien (RGO) est une entité pathologique liée à la remontée involontaire du contenu gastrique vers l’œsophage due à l’incompétence du système anti-reflux, qui expose le patient à une altération de sa qualité de vie et à des complications évolutives pouvant compromettre son pronostic vital à court, moyen et long terme.
Le reflux gastro-œsophagien est une pathologie très fréquente, dont la prévalence croissante affecte plus de 30% des adultes dans les pays occidentaux(1). Il s’agit d’un véritable problème de santé publique et un motif fréquent de consultation, non seulement du fait de son impact sur le quotidien du patient mais aussi du fait de sa gravité potentielle liée aux complications possibles (hémorragies et sténoses) mais surtout au risque de dégénérescence cancéreuse sur endobrachyœsophage(2). Ces complications sont la traduction de l’histoire naturelle des lésions œsophagiennes anatomiques non traitées(3).
La physiopathologie(4) du RGO, complexe et multifactorielle, est imparfaitement élucidée. Si l’acidité et les perturbations mécaniques de l’œsophage, de l’estomac et de la jonction œsogastrique interviennent grandement dans la pathogénie du RGO et de ses complications, les caractéristiques de l’atmosphère physico-chimique endoœsophagienne participent certainement à la survenue des lésions macroscopiques et microscopiques observées ainsi qu’à la perception des symptômes par le patient(5,6).
Diagnostic – Explorations :
Le reflux gastro-œsophagien est largement prédominant dans les pays occidentaux ; cependant, la modernisation de la vie, les modifications hygiéno-diététiques et l’accroissement de la pandémie d’obésité en font une pathologie devenue ubiquitaire et croissant dans de multiples zones géographiques. Dans la population occidentale, 30% à 45% des personnes rapportent un épisode mensuel de reflux, 5% à 25% un épisode hebdomadaire et 5% à10% un épisode quotidien(7).
Du point de vue clinique, le RGO se décline sous divers aspects symptomatiques se regroupant sous un large spectre clinique intégrant des manifestations digestives et extradigestives telles que définies par la classification de Montréal(8). On distingue ainsi, la forme classique au décours de laquelle les manifestations habituelles s’articulent autour d’une symptomatologie digestive, de la forme atypique se traduisant par des symptômes extra-digestifs.
Le RGO se manifeste essentiellement par des symptômes digestifs typiques cardinaux que sont le pyrosis et les régurgitations, souvent déclenchés par l’alimentation et/ou le syndrome postural. Le pyrosis se définit comme une sensation de brûlure rétro-sternale prenant origine au niveau de l’épigastre et remontant le long du sternum ; le pyrosis a une bonne spécificité (78%) mais une faible sensibilité (60%) pour le diagnostic de RGO en utilisant la pH-métrie comme test de référence. Quant aux régurgitations, elles consistent en la remontée du contenu gastrique au niveau œsopharyngien voire de la bouche pouvant être à nouveau dégluties, leur spécifici- té et leur sensibilité sont assez médiocres(9). Les vomissements, les douleurs épigastriques, la dysphagie peuvent être présents au cours du RGO dans le cadre du syndrome œsophagien symptomatique. Les aspects du « syndrome œsophagien lésionnel » de la classification de Montréal sont représentés par les œsophagites peptiques gradées selon la classification de Los Angeles(10), la sténose, l’endobrachyœsophage et plus rarement l’adénocarcinome œsophagien.
Quant aux manifestations extradigestives, elles peuvent être isolées ou associées aux symptômes digestifs. Ce sont essentiellement des manifestations pulmonaires (toux, bronchites, manifestations asthmatiformes …), ORL (laryngites, dysphonie …), pseudo-cardiaques (douleurs angineuses …), dentaires (érosions …). Elles posent principalement la question de l’imputabilité de ces manifestions isolées à un véritable RGO, souvent absent par ailleurs, et imposent ainsi la réalisation d’explorations exhaustives souvent spécialisées(11,12).
Ainsi, le diagnostic de RGO repose essentiellement sur l’approche clinique (13) mais des explorations spécifiques, sont parfois requises. Ces investigations peuvent se concevoir dans quatre situations particulières : pour asseoir un diagnostic notamment dans le cadre de manifestations atypiques isolées, pour expliquer et palier un défaut de réponse thérapeutique, pour évaluer une indication de chirurgie anti-reflux et dans le cadre de recherches physiopathologiques, permettant ainsi d’étudier les mécanismes pathogéniques et d’évaluer voire valider de nouvelles armes thérapeutiques.
De façon pragmatique, devant un patient présentant des symptômes évocateurs de RGO, l’analyse minutieuse et détaillée des plaintes est capitale et peut souvent suffire à retenir le diagnostic en se basant sur la définition de Montréal(8) ; en présence de symptômes typiques isolés ou associés à des manifestations atypiques, il est licite de proposer une stratégie coût-efficacité optimale basée sur un traitement empirique par les inhibiteurs de la pompe à protons avec dans une grande majorité des cas une réponse satisfaisante(14).
Des explorations sont souhaitables, et en premier lieu l’endoscopie digestive haute(15), en l’absence de réponse au traitement anti-sécrétoire optimisé, mais aussi d’emblée lorsqu’il existe des signes d’alarme (dysphagie, anémie, hémorragie, amaigrissement), chez un patient de plus de 50 ans, devant une évolution des symptômes de plus de 5 ans, devant une récidive précoce à l’arrêt du traitement, devant des symptômes atypiques isolés, pour contrôler le traitement d’une œsophagite sévère ou pour surveiller un endobrachyœsophage(16,17,18).
Cette endoscopie pourra évoquer avec une forte probabilité (bonne spécificité, médiocre sensibilité) le diagnostic de RGO, écarter des diagnostics différentiels, rechercher des signes de complications (œsophagites, endobrachyœsophage, dégénérescence néoplasique) ou une œsophagite à éosinophiles parfois associée ou mimant un RGO grâce aux biopsies effectuées(19,20,21). Des explorations plus spécialisées disponibles dans des laboratoires d’explorations fonctionnelles digestives (pH-métrie, pH-Impédancemétrie, manométrie œsophagienne) deviennent nécessaires si le patient demeure symptomatique malgré un traitement optimal, en l’absence de lésion endoscopique et devant des symptômes atypiques isolés(22,23,24,25). La pH-métrie et la pH-Impédancemétrie permettront avec l’aide de l’analyse d’association symptômes-reflux de rattacher les plaintes du patient, qu’il s’agisse de plaintes digestives ou extra-digestives, à un RGO acide, non-acide ou faiblement acide, à un œsophage hypersensible ou à un diagnostic de pyrosis fonctionnel voire d’écarter la réalité d’un véritable reflux gastro-œsophagien. Ces explorations sont aussi indispensables si l’on propose une chirurgie anti-reflux au patient afin de confirmer le diagnostic avant un geste invasif, écarter toute contre-indication à type de troubles sévères de la motricité (achalasie du cardia) et représente également un examen de référence permettant d’analyser une éventuelle symptomatologie postopératoire(26,27,28,29).
Prise en charge thérapeutique :
Les buts du traitement, au cours du reflux gastro-œsophagien, sont de soulager efficacement les symptômes, de cicatriser et maintenir en rémission les œsophagites, de prévenir les complications, et de rétablir une qualité de vie du patient, satisfaisante (30).
Le traitement médical, primordial, reste basé essentiellement sur la prescription d’anti-sécrétoires. Une meilleure compréhension des mécanismes physiopathologiques de la maladie a permis de proposer de nouvelles cibles thérapeutiques et de nouvelles molécules agissant directement sur ces mécanismes étiopathogéniques. À côté de l’approche médicale, des techniques endoscopiques ont été tentées mais en l’absence d’efficacité avérée, elles ont été abandonnées pour la plupart ; quant à la chirurgie, elle trouve une place certaine chez des patients sélectionnés.
Ainsi, l’approche mécanistique du RGO a pour objectif de renforcer la barrière anti-reflux par la correction du defect anatomique d’une hernie hiatale, de neutraliser le contenu agressif gastrique par divers traitements : antiacides, anti sécrétoires (inhibiteurs de la pompe à protons – IPP, antagonistes des récepteurs H2 – antiH2), molécules anti-reflux (agonistes des récepteurs Gaba – Baclofène, antagonistes des récepteurs m5Glutamate), protecteurs muqueux (prostaglandines, alginates), régulateurs de la clearance œsogastrique (prokinétiques) et modulateurs des signaux nociceptifs (antidépresseurs à
faible dose : Citalopram, Amitriptyline, ou médecine alternative : hypnose, acupuncture…) (31).
En pratique, les règles hygiéno-diététiques (repas léger le soir, proscrire tabac et alcool, éviter de se coucher juste après les repas, position surélevée de la partie haute du corps pendant le sommeil), sont largement utilisées ; elles ne reposent, cependant, sur aucune recommandation formelle en « Evidence Base Médecine » ; par contre, il est impératif et d’efficacité avérée de lutter contre l’obésité (32).
Le traitement médical a été bouleversé par l’avènement des anti-sécrétoires et notamment des inhibiteurs de la pompe à protons dans les années 90. La méta-analyse de Chiba montrant une supériorité des IPP versus Anti- H2, portait sur plus de 7.000 patients atteints d’œsophagite modérée ou sévère et rapportait des taux de cicatrisation de 84 ± 11 % avec les IPP de première génération (oméprazole, lansoprazole ou pantoprazole) contre 52 ± 17 % pour les anti-H2 ; les proportions de sujets asymptomatiques à l’issue de ce traitement initial étaient de 77 ± 10 % versus 48 ± 15 % pour les IPP et les anti-H2, respectivement (33,34).
L’efficacité globale des IPP est très satisfaisante en termes de cicatrisation lésionnelle œsophagienne (85%) et de contrôle symptomatique (80%), un traitement d’entretien ou à la demande est souvent nécessaire ; cette réponse est cependant moindre en cas de RGO sans lésion endoscopique (Non Erosive Reflux Disease – NERD). Les résultats obtenus avec les IPP de nouvelle génération (rabéprazole et ésoméprazole) semblent légèrement supérieurs (35,36).
L’échec au traitement se voit dans un tiers des cas. Les causes d’échec sont multiples et doivent être recherchées, notamment par les explorations spécialisées qui montrent dans plus d’un tiers ces cas de non-réponse aux IPP, l’absence de relation entre les plaintes du patient et un diagnostic de RGO.
Dans un premier temps, il convient de vérifier la compliance au traitement et d’optimiser le traitement médical (durée, dose, horaire). Dans un second temps il faudra rechercher la notion de prise d’anti-inflammatoires non stéroïdiens, éliminer un éventuel syndrome de Zollinger-Ellison ; pour finir par réaliser les examens spécialisés qui vérifieront la réalité du RGO, sa nature chimique, l’efficacité du traitement prescrit et qui permettront ainsi de guider une stratégie thérapeutique efficiente.
Les IPP sont aussi proposés en cas de manifestations atypiques avec de moins bons résultats (50% de résolution globalement) mais la problématique première est de rattacher formellement les symptômes extradigestifs à un RGO (12).
La chirurgie demeure une alternative thérapeutique efficace chez des patients rigoureusement sélectionnés et doit donc s’accompagner d’un bilan exhaustif. Elle sera plus facilement proposée aux patients jeunes avec symptômes typiques et anciens de RGO, bons répondeurs aux IPP et désireux d’arrêter leur traitement médical avec une exposition acide œsophagienne pathologique en pH-métrie. (37). Les recommandations de la Société Américaine de Gastro-entérologie(25,38) retenaient comme indication à la chirurgie :
Une alternative au traitement au long cours des patients reflueurs notamment ceux désirant interrompre les IPP,
La présence d’effets secondaires sous traitement médical,
La présence d’une volumineuse hernie hiatale,
Une œsophagite réfractaire ou des symptômes persistants documentés liés au RGO.
La fundoplicature de type Nissen par voie laparoscopique représente actuellement la procédure de référence avec des résultats très satisfaisants dans les centres experts (39).
À côté de ces traitements de référence que constituent les IPP et la chirurgie et face aux situations d’échec de ces deux options, de nouvelles approches ont été envisagées (40) : des traitements endoscopiques par radiofréquence tels que le dispositif Stretta* (41,42), une alternative « médicale » à la fundoplicature chirurgicale soit un montage anti-reflux réalisé par voie endoscopique
« l’EsophyX, EndoGastric Solutions, Inc » (43). De nouveaux dispositifs comme l’anneau magnétique « Linx Reflux Management System, Torax Medical, Inc » (44) et des techniques nouvelles de stimulation du sphincter inférieur de l’œsophage par système de neurostimulation implantable « EndoStim » (45) présentant des résultats encourageants sont aussi en cours d’étude.
Conclusion :
Le reflux gastro-oesophagien est une entité pathologique, au spectre clinique étendu, représentant un motif fréquent de consultation en unité de soins primaires, secondaires et tertiaires. Le diagnostic essentiellement clinique nécessite aussi, parfois, des explorations spécifiques endoscopiques mais aussi hautement spécialisées que sont les explorations fonctionnelles œsophagiennes (pH-métrie, pH-impédancemétrie-manométrie œsophagienne). Les complications du reflux gastro-œsophagien, bien que devenues plus rares, peuvent être de gravité variable et représenter indéniablement une source de morbidité lourde et un important fardeau pour le patient et le système de santé. Leur prévention passe donc par une prise en charge optimale du reflux et par son diagnostic précoce. Le traitement actuel est essentiellement médical reposant sur les anti-sécrétoires et notamment les inhibiteurs de la pompe à protons, la chirurgie demeure réservée à des patients bien sélectionnés. Bien qu’imparfaitement élucidés, les mécanismes physiopathologiques à l’origine de cette entité représentent la cible privilégiée de stratégies thérapeutiques nouvelles afin de contrôler et de maitriser définitivement les symptômes, les lésions anatomiques et l’évolution vers les complications.
Références
Locke GR 3rd, Talley NJ, Fett SL, Zinsmeister AR, Melton LJ 3rd . Prevalence and clinical spectrum of gastroesophageal reflux: a popu- lation-based study in Olmsted County, Minnesota. Gastroenterology 1997;112:1448-56.
Moayyedi P, Axon AT. Review article: gastro-esophageal reflux disease- the extent of the problem. Aliment Pharmacol Ther. 2005; 22:11-19
Savarino E, de Bortoli N, De Cassan C, Della Coletta M, Bartolo O, Furnari M, Ottonello A, Marabotto E, Bodini G, Savarino V. The natural history of gastro-esophageal reflux disease: a comprehensive review. Dis Esophagus. 2017;30:1-9
Fass R and Ofman JJ. Gastroesophageal reflux disease–should we adopt a new conceptual framework? Am J. Gastroenterol. 2002 ;97:1901-9
Mikami DJ, Murayama KM. Physiology and pathogenesis of gas- troesophageal reflux disease. Surg Clin North Am. 2015;95:515-25.
Lee YY, McColl KE. Pathophysiology of gastroesophageal reflux disease. Best Pract Res Clin Gastroenterol. 2013 ;27:339-51
Ronkainen J. Agréus L. Epidemiology of reflux symptoms and GORD. Best Pract Res Clin Gastroenterol.2013;27:325-37
Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R. The Montreal definition and classification of gastroesophageal reflux disease: a glo- bal evidence-based consensus. Am J Gastroenterol 2006; 101:1900-20
Klauser AG, Schindlbeck NE, Müller-Lissner SA. Symptoms in gastro-oesophageal reflux disease. Lancet. 1990;335:205-8
Lundell LR, Dent J, Bennett JR, et al. Endoscopic assessment of oesophagitis: clinical and functional correlates and further validation of the Los Angeles classification. Gut. 1999;45:172–180
Oh TH. Atypical manifestation of gastroesophageal reflux di- sease: a disease with a thousand faces. J Neurogastroenterol Motil. 2014 ;20:1-
Sidhwa F, Moore A, Alligood E, Fisichella PM Diagnosis and Treatment of the Extraesophageal Manifestations of Gastroesopha- geal Reflux Disease. Ann Surg 2017;265:63–67
Kuo P, Holloway RH. A pragmatic symptom-based approach. Best Practice & Research Clinical Gastroenterology 2010;24: 765–773
Scarpignato C, Pelosini I, Di Mario F. Review. Acid suppression therapy: where do we go from here? Dig Dis. 2006;24:11-46.
Hatlebakk JG. Endoscopy in gastro-oesophageal reflux disease. Best Pract Res Clin Gastroenterol. 2010;24:775-86
Shaheen NJ, Weinberg DS, Denberg TD, Chou R, Qaseem A,Shekelle P; Clinical Guidelines committee of the American College of Physicians. Upper endoscopy for gastroesophageal reflux disease: best practice advice from the clinical guidelines committee of the American College of Physicians. Ann Intern Med. 2012;157:808-16
Kahrilas PJ, Shaheen NJ, Vaezi MF, Hiltz SW, Black E, Modlin IM, Johnson SP, Allen J, Brill JV. American Gastroenterological Asso- ciation. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383-1391
Hirano I., Richter JE; Practice Parameters Committee of the American College of Gastroenterology. ACG practice guidelines: eso- phageal reflux testing. Am J Gastroenterol. 2007;102:668-85
Molina-Infante J, Ferrando-Lamana L, Ripoll C et al. Esophageal eosinophilic infiltration responds to proton pump inhibition in most adults. Clin Gastroenterol Hepatol 2011; 9 :110 -7
Molina-Infante J., Katzka DA. Tissue biomarkers distinguishing EoE from GERD: Concerns about the control group. Am J Gastroen- terol 2013;108:452–3
Hirano I. Should Patients with Suspected eosinophilic Esophagitis Undergo a Therapeutic Trial of Proton Pump Inhibition? Am J Gas- troenterol 2013; 108:373 – 375
Vaezi MF. Review article: the role of pH monitoring in extraoeso- phageal gastro-oesophageal reflux disease. Aliment Pharmacol Ther. 2006 ;23 :40-9
Sifrim D, Zerbib F. Diagnosis and management of patients with reflux symptoms refractory to proton pump inhibitors. Gut. 2012;61:1340-54
Sifrim D, Dupont L, Blondeau K, Zhang X, Tack J, Janssens J. Weakly acidic reflux in patients with chronic unexplained cough during 24-hour pressure, pH, and impedance monitoring. Gut. 2005;54:449-54
Mainie I, Tutuian R, Shay S, Vela M, Zhang X, Sifrim D, Castell DO. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: a multicentre study using combined ambulatory impedance-pH monitoring. Gut 2006;55: 1398-402.
Katz PO, Gerson LB, Vela MF. Guidelines for the Diagnosis and Management of Gastroesophageal Reflux Disease. Am J Gastroente- rol. 2013; 108:308 – 328
Tutuian R, Mainie I, Agrawal A, et al. Nonacid reflux in patients with chronic cough on acid-suppressive therapy. Chest 2006; 130: 386–391
Kessing BF, Bredenoord AJ, Smout AJ. Erroneous diagnosis of gastroesophageal reflux disease in achalasia. Clin Gastroenterol He- patol. 2011;9:1020-4
Stoikes N, Drapekin J, Kushnir V, Shaker A, Brunt LM, Gyawali CP. The value of multiple rapid swallows during preoperative esopha- geal manometry before laparoscopic antireflux surgery. Surg Endosc. 2012;26:3401-7.
Scarpellini E, Ang D, Pauwels A, De Santis A, Vanuytsel T, Tack J. Management of refractory typical GERD symptoms. Nat Rev Gas- troenterol Hepatol. 2016 May;13(5):281-949.
Wang YK, Hsu WH, Wang SS, Lu CY, Kuo FC, Su YC, Yang SF, Chen CY, Wu DC, Kuo CH. Current pharmacological management of gastroesophageal reflux disease. Gastroenterol Res Pract. 2013; 2013: 98365.
Dalbir S. Sandhu and Ronnie Fass. Current Trends in the Mana- gement of Gastroesophageal Reflux Disease. Gut Liver. 2017 : 24
Chiba N, De Gara CJ, Wilkinson JM, Hunt RH. Speed of healing and symptom relief in grade II to IV gastroesophageal reflux disease: a meta-analysis. Gastroenterology. 1997; 112: 1798-810.
Moayyedi P, Santana J, Khan M, Preston C, Donnellan C. WIT- HDRAWN : Medical treatments in the short term management of reflux oesophagitis. Cochrane Data base Syst Rev. 2011 16
Sigterman KE, Van Pinxteren B, Bonis P, Lau J, Numans ME. Short-term treatment with proton pump inhibitors, H2- receptor antagonists and prokinetics for gastro-oesophageal reflux disease-like symptoms and endoscopy negative reflux disease. Cochrane Data base Syst Rev 2013; 5
Savarino V, Di Mario F, Scarpignato C. Proton pump inhibitors in GORD An overview of their pharmacology, efficacy and safety. Pharmacol Res. 2009;59:135-53
Mainie I, Tutuian R, Agrawal A, et al. Combined multichannel intraluminal impedance-pH monitoring to select patients with per- sistent gastro-oesophageal reflux for laparoscopic Nissen fundoplica- tion. Br J Surg. 2006;93:1483-7
Wilshire CL, Watson TJ. Surgical management of gastroesopha- geal reflux disease. Gastroenterol Clin North Am. 2013; 42:119-31
Broeders JA, Rijnhart-de Jong HG, Draaisma WA, Bredenoord AJ, Smout AJ, Gooszen HG. Ten-year outcome of laparoscopic and conventional nissen fundoplication: randomized clinical trial. Ann Surg. 2009; 250:698-706
Kethman W, Hawn M.New Approaches to Gastroesophageal Reflux Disease.J Gastrointest Surg. 2017 Jun 16
Coron E, Sebille V, Cadiot G, Zerbib F, Ducrotte P, Ducrot F, Pouderoux P, Arts J, Le Rhun M, Piche T, Bruley des Varannes S, Gal- miche JP; Consortium de Recherche Indépendant sur le Traitement et L’exploration du Reflux Gastro-oesophagien et de L’endobrachyœ- sophage (CRITERE). Clinical trial: radiofrequency energy delivery in proton pump inhibitor-dependent gastro-oesophageal reflux disease patients. Aliment Pharmacol Ther. 2008; 28:1147-58
Yew KC, Chuah SK. Antireflux endoluminal therapies: past and present. Gastroenterol Res Pract. 2013;2013:48141
Bell RC, Hufford RJ, Fearon J, Freeman KD. Revision of failed traditional fundoplication using EsophyX transoral fundoplication. Surg Endosc. 2013; 27:761-7.
Ganz RA, Peters JH, Horgan S, et al. Esophageal sphincter device for gastro¬esophageal reflux disease. N Engl J Med. 2013;368:719-72.
Rodríguez L, Rodriguez P, Gómez B, Ayala JC, Oksenberg D, Perez-Castilla A, Netto MG, Soffer E, Crowell MD. Long-term re- sults of electrical stimulation of the lower esophageal sphincter for the treatment of gastroesophageal reflux disease. Endoscopy 2013; 45: 595-604.
N. DEBZI(1), S.A. FARAOUN(2), K. BENTABAK(3), N. AFREDJ(1), N. GUESSAB(1), H. AIT KACI(4), A. NANI (1),T. BENATHMANE(6), I. OULED CHEIKH(1), R. KERBOUCHE(1), S. YAHIATÈNE(5), D. BENMOUSSA(5), L. BACHIRI(5), D. OURRAD(5), Z. BEKKOUCHE (5), M. AMROUN(5), F. REZOUG(5), M. LAKEHAL(9),j.M. BODIN(9), N. TERKI(4), M. REGHIS(7), O. CHAFA(7), C. AMAOUz(7), S. ADJALI(7),Y. KINANE(7),D. HANOUN(8), T. BOUCEKKINE(1), A. GRABA(3), B. GRIÈNE(5) , S.E. BENDIB(2), K. BOUDjEMA(9).Service d’Hépatologie, CHU Mustapha Bacha, Alger.Service d’imagerie médicale, centre Pierre et Marie Curie, Alger.Service de Chirurgie générale et oncologique, centre Pierre et Marie Curie, Alger.Service d’Anatomie Pathologique, centre Pierre et Marie Curie, Alger.Service d’Anesthésie Réanimation,centre Pierre et Marie Curie, Alger. (6) Service de Psychiatrie, CHU Mustapha Bacha.(7) Service d’Hémobiologie et de transfusion, CHU Mustapha Bacha. (8) Institut National de Santé Publique, Alger.(9) Service de Chirurgie Digestive et de Transplantation, CHU Pontchaillou, Rennes, France.
Résumé :
En Février 2003, la première transplantation hépatique à donneur vivant apparenté (THDV) a été réalisée en Algérie. Le développement de la greffe hépatique à donneur cadavérique (THDC) se heurte à des obstacles logistiques et organisationnels.L’objectif principal de ce travail est d’évaluer rétrospectivement, les résultats et le suivi à long terme des patients transplantés.
Du 1/3/2002 au 31/3/2012, nous avons colligé les dossiers de 39 receveurs : THDV (n=34), THDC (n =5 : 3 TH, 2 greffes combinées foie rein). L’âge moyen était de 38 ans (16-58), il s’agissait de 23 hommes et 16 femmes avec un sex-ratio H/F de 1.43. Le score moyen de Child- Pugh était de 10(6-14). L’étiologie de la cirrhose était : virale (n=12 : 7VHC, 5VHB), syndrome de Budd-Chiari (SBC, n=6) , auto -immune (HAI, n=5) , cryptogénique (n=4), biliaire secondaire (CBS, n= 3) , alcoolique (n= 3) , CBP (n=2) , stéatohépatite non alcoolique (n=1) , CHC sur cirrhose cryptogénétique (n=1) , maladie de Wilson (n=1) et hyperoxa- lurie primitive (n=1). L’évaluation du donneur vivant comporte 3 phases: psycho-sociale, clinique et morphologique. Pour les indications de la greffe, nous avons utilisé la classification de Child-Pugh. Chez le receveur, nous avons étudié la morbi-mortalité précoce et tardive. Chez le donneur vivant, les complications des hépatectomies ont été analysées selon la classification de Dindo-Clavien. L’immunosuppression comprenait la ciclosporine (CSA) associée au mycophénolate mofétil (MMF) ou le tacrolimus associé au MMF. Pour le suivi à long terme, nous avons étudié : le rejet, les complications de l’immunosuppression, les infections, les complications biliaires, la récidive de la maladie initiale et la survenue de cancers.
Aucun décès n’est survenu chez les donneurs. Nous rapportons 16% (n=7) de complications majeures chez 5 donneurs. Le taux de survie des receveurs à 3 ans était de 61% (n=26) avec un suivi médian de 58 mois, 43% des receveurs sont décédés (n=13), 9 décès sont survenus dans la période péri-opératoire. L’infection constitue la principale cause de décès (n=6) soit 46%. 10 de nos patients soit 33% ont eu une récidive de la maladie initiale.
Ces résultats préliminaires encourageants, incitent à développer la transplantation hépatique dans notre pays.
Abstract : In February 2003, the first living donor liver transplantation (LDLT) was performed in Algeria, up to now cadaveric donor is impossible to perform because of logistical reasons.
The main objective of this study was to evaluate our results and long-term follow up of transplanted patients.
9 recipients (LDLT n=34, CD n=5: 3 LT, 2 Liver-Kidney transplantation) were followed from 12/1/2002 to 3/31/2012, the mean age was 38 years (16-58) they were 23 men and 16 females with sex ratio M/F =1.43. The mean Child-Pugh score was 10 (6-14). The etiology of the cirrhosis was: viral (n=12: 7HCV, 4HBV), Budd Chiari syndrome (BCS, n=6), auto-immune (AIH, n=5), cryptogenic (n=4), secondary biliary (n=3), alcoholic (n=3) , PBC (n=2), NASH (n=1), hepatocellular carcinoma (n=1), Wilson disease (n=1), and hyperoxaluria (n=1). The evaluation of living donor featured three phases: psychological, clinical and imaging. The indications for transplantation was made according to Child- Pugh score. In recipients, we have studied early and late morbidity mortality. Among the Living donor, complications of hepatectomy were analyzed according to Dindo– Clavien system. The immunosuppression regimen included cyclosporine (CSA) combined with mycophenolate mofetil (MMF) or tacrolimus with MMF. For Long term follow up we have studied: the rejection, complications of immunosuppression, infections, biliary complications, recurrence of the initial disease, and occurrence of cancers. No donor mortality occurred; 16% (n=7) of major hepatectomy complications were recognized in 5 donors. The recipient survival rate at 3 years was 61% (n=26) with median follow up of 58 months. 43% of recipients died (n=13), all in LDLT group, 9 deaths occurred in perioperative period, the main cause of death is infection (46%). 33 % (n=10) recipients presented recurrence of the initial disease.These preliminary results encourage the development of liver transplantation in our country.
Key-words :Transplantation, living donor, cirrhosis.
Introduction :
L’a transplantation hépatique constitue la solution thérapeutique ultime, chez les patients en insuffisance hépatique terminale, particulièrement au cours des cirrhoses décompensées ou des hépatites fulminantes(1).
Des programmes cliniques de transplantation hépatique à donneur vivant (THDV) ont été développés entre 1989 et 1991, en Asie pour des raisons culturelles, aux Etats-Unis et en Europe pour des raisons de pénurie de greffons pédiatriques responsable d’une mortalité élevée sur liste d’attente (30-50% des cas). La THDV trouve également sa justification dans les pays où les contraintes organisationnelles et/ou légales ne permettent pas le recours au don cadavérique(1,2,5,6,7).
En Février 2003, la première transplantation hépatique à donneur vivant apparenté a été réalisée en Algérie, en étroite collaboration avec l’équipe médico-chirurgicale du service de chirurgie hépatobiliaire et de transplantation hépatique de l’hôpital Pontchaillou de Rennes (France).
À ce jour, même si les contraintes cultuelles et culturelles sont levées, aucune transplantation hépatique à donneur cadavérique (THDC) n’a été réalisée en Algérie. Des raisons organisationnelles et logistiques freinent le développement et la réalisation de la greffe hépatique à donneur cadavérique(3).
Objectifs :
L’objectif principal de ce travail est d’évaluer rétrospectivement, les résultats et le suivi à long terme des patients transplantés.
Patients et méthodes :
Du 1/3/2002 au 31/3/2012, nous avons colligé les dossiers de 39 receveurs : 34 THDV et 5 THDC. Sur l’ensemble des receveurs étudiés, l’âge moyen était de 38 ans (16-58), il s’agissait de 23 hommes et 16 femmes avec un sex ratio H/F de 1,43. Le score moyen de Child-Pugh était de 10 (extrêmes : 6 -14), les receveurs se répartissaient comme suit : 73% (n=25) des patients étaient classés C, 23% (n = 8) B et une patiente atteinte d’un carcinome hépatocellulaire (CHC) sur cirrhose cryptogénétique était A.
L’évaluation du donneur vivant comporte globalement 3 phases : psycho-sociale, clinique, et d’imagerie. L’imagerie non invasive comprend : l’écho-Doppler, le scanner multibarette, la Bili-IRM et l’invasive : l’artériographie coelio-mésentérique, que nous ne pratiquons plus depuis la 15ème transplantation, remplacée par le scanner multibarette. Dans notre protocole la biopsie du foie du donneur est devenue obligatoire depuis la 17ème THDV (5,6,7), nous estimons qu’elle renforce la sécurité du donneur et affine les critères de sélection.
Selon le lien de parenté, les donneurs se répartissaient comme suit : sœur (n=9), fille (n=8), frère (n=7), fils (n=3), neveu (n=2), père (n=2), cousin (n=1) et tante (n=1). L’âge moyen était de 28 ans (18-58).
Chez les patients ayant bénéficié d’une THDV (n = 34), l’étiologie de la cirrhose était d’origine virale dans 9 cas, cinq d’entre eux avaient le VHC, de génotype 1 (n=4) et de génotype 2 (n=1). Quatre patients avaient le VHB, parmi lesquels deux avaient une infection active (ADN-VHB > 2000 UI/ml), le premier receveur a été traité par la lamivudine et le second par l’entécavir, le troisième avait une infection inactive et a reçu un traitement préemptif par la lamivudine. Ces trois patients traités avaient une charge virale indétectable avant la greffe, le dernier receveur avait un profil d’immunité naturelle. Un SBC primitif a été noté dans 6 cas, chez deux receveurs la cause de la maladie était un syndrome myéloprolifératif latent et chez quatre d’entre eux aucune étiologie n’a été retrouvée. La cirrhose était liée à une HAI (n=5), cryptogénétique (n=4), CBS (n=3) : secondaire à une CSP (n=1), maladie de Caroli (n=1), traumatisme des voies biliaires (n=1) ; alcoolique (n=3), CBP (n=2), stéatohépatite non alcoolique (n=1), CHC (n=1) retenu sur les critères de l’UCSF2, avec un taux d’AFP à 3000 UI.
Sur les 34 THDV : 33 ont été réalisées dans notre centre et une en Jordanie. Il s’agissait d’un prélèvement de foie droit dans 31 cas, un foie gauche dans un cas et chez un donneur la procédure a été interrompue en raison du décès du receveur sur table suite à un choc hémorragique.
Cinq patients ont bénéficié d’une THDC à l’étranger. Trois en France, sur foie total pour maladie de Wilson, un split pour cirrhose VHC et une greffe combinée foie-rein pour cirrhose VHC et insuffisance rénale chronique secondaire à une malformation de la jonction urétérale. Une greffe a été réalisée en Belgique pour cirrhose VHB inactive (AC anti-HBc isolés). Une greffe combinée foie-rein a été réalisée aux Etats-Unis pour hyperoxalurie primitive.
En somme, les patients présentant une étiologie virale C, étaient 7 (THDV = 5, Split=1, greffe combinée foie-rein=1), parmi lesquels 3 ont été traités avant la greffe mais avec échec à la bithérapie interféron-ribavirine pour intolérance hématologique (n=2) et rechute (n=1).
Nous avons étudié séparément la morbi-mortalité précoce inférieure à 3 mois, et tardive au-delà de 3 mois.
Chez le donneur vivant, les complications générales et celles des hépatectomies ont été classées selon la classification de Dindo-Clavien(4).
L’immunosuppression comprenait la ciclosporine (CSA) associée au mycophénolate mofétil (MMF) ou le tacrolimus associé au MMF. Les corticostéroïdes sont main tenus six mois ; en cas de cirrhose auto-immune, nous gardons une dose minimale efficace au long terme pour prévenir la récidive de la maladie initiale.
Pour 35 de nos receveurs, le traitement immunosup presseur administré était la CSA associée au MMF et le tacrolimus, plus MMF dans 4 cas.
Durant le suivi, nous avons réalisé systématiquement une ponction biopsie hépatique post transplantation (PBHPT) à un an, à 5 ans et en cas de suspicion de rejet. Chez les receveurs VHC, la PBHPT est réalisée chaque année pour juger de l’évolutivité de la fibrose et pour poser l’indication du traitement antiviral. L’association interféron pégylé-ribavirine a été administrée en post transplantation lorsque le score histopathologique de Métavir montrait une fibrose égale ou supérieure à F1(8).
Chez les receveurs présentant une étiologie virale B, le traitement antiviral par les analogues (lamivudine ou entécavir) et les immunoglobulines spécifiques anti-Hbs ont été administrés selon le protocole de l’hôpital Paul Brousse (9). Les patients atteints de syndrome de SBC recevaient une anticoagulation avec l’acénocoumarol en pré et post-greffe. Pour la cirrhose alcoolique, le sevrage est de règle au moins 6 mois avant la transplantation.
Le suivi de la récidive du CHC comportait une échographie Doppler, un taux d’alpha fœto-protéine tous les 6 mois et une tomodensitométrie annuelle.
Pour le suivi à long terme, nous avons étudié : le rejet, les complications de l’immunosuppression (le syndrome métabolique, l’insuffisance rénale, l’HTA), la survenue d’infections, les complications biliaires, la récidive de la maladie initiale et l’apparition de cancers.
Résultats :
À ce jour, aucun décès n’est survenu chez les donneurs. Selon la classification de Dindo-Clavien, nous avons noté 26 complications chez 16 donneurs, il s’agissait d’une complication unique chez 9 donneurs, deux chez 4 donneurs et trois chez 3 donneurs. Les complications générales (n=8) figurent sur le tableau 1 et celles des hépatectomies (n=18) sur le tableau 2. En les regroupant, elles étaient mineures dans 19 cas (73%) et majeures dans 7 cas (27%). Ces complications majeures ont été retrouvées chez 5 donneurs (16%).
Classification de clavien
Grade 1
Grade 2
Grade 3a
Grade 3b
Grade 4a
Colique néphrétique
1
Pharyngite
1
Infection urinaire
1
Fièvre
2
Pneumopathie
3
Tableau 1 : Complications générales (n=8)
Classification de clavien
Grade 1
Grade 2
Grade 3a
Grade 3b
Grade 4a
Élévation transaminases
1
Collection sous-phrénique
4
Infection de paroi
3
Thrombose veine hépatique
1
Thrombose veine porte
1
Transfusion peropératoire
1
Epanchement pleural/ascites
2
Pneumothorax
1
Abcès sous-phrénique
1
Hémorragie intra-abdominale
2
1
Tableau 2 : Complications des hépatectomies (n = 18)
Le taux de survie globale (THDV-THDC) des receveurs à 3 ans était de 61% (n=26) avec un suivi médian de 58 mois, (figure 1), et 73% (n=19) étaient encore en vie à plus de 60 mois. Le taux de la survie des THDV était de 62% (n=21) (figure 2).
Parmi les receveurs THDV, 43% d’entre eux sont décédés (n=13), 9 décès sont survenus dans la période péri-opératoire (Tableau 3) et 4 à distance (Tableau 4). Aucun décès n’est survenu chez les patients ayant bénéficié d’une transplantation hépatique à donneur cadavérique.
L’infection constitue la principale cause de décès (n=6) soit 46%. Selon l’étiologie, le SBC représente la première cause de décès (n=4) soit 30%.
Le SBC constitue également la cause principale de décès à long terme (n=2), un décès dans un tableau de SBC fulminant malgré une anti-coagulation efficace et le second suite à un accident aux anticoagulants.
Un patient transplanté pour cirrhose à VHB a présenté une thrombocytopénie idiopathique au 4ème mois post greffe, traité par rituximab après échec des corticostéroïdes et des immunoglobulines standards. Un mois après, il décède dans un tableau de syndrome de détresse respiratoire aiguë (SDRA) secondaire à un sepsis pulmonaire probablement d’origine virale, les bilans bactériologique et parasitaire étaient négatifs. Un receveur transplanté pour cirrhose alcoolique est décédé au 6ème mois post greffe dans un tableau de sepsis pulmonaire.
Quant aux complications biliaires, elles sont retrouvées chez 9 receveurs soit 23%, si on les rapporte aux THDV, le taux est de 38% (n=8). Elles étaient précoces chez trois receveurs des THDV, où on note une péritonite biliaire par lâchage sur anastomose hépatico-jéjunale 28 PBHPT ont été réalisées, 27 par voie transcutanée sous contrôle échographique et une en per-opératoire. Les patients transplantés pour VHC (n=7), cinq sont vivants, deux à 24 mois, parmi eux le patient ayant bénéficié d’un split chez lequel a été découvert incidemment un CHC sur l’explant, le troisième à 42 mois, le quatrième à 78 mois et le cinquième à 88 mois.
La PBHPT faite au 9ème mois (n=1) pour suspicion de rejet a retrouvé selon la classification Metavir une hépatite A1F1, et à un an (n=4) une hépatite A1F1(n=2), A2F2 (n=1), et une hyperplasie nodulaire régénérative (HNR, n=1) chez le patient avec greffe combinée foie-rein. La PBHPT à 2 ans (n=4) a montré les mêmes résultats histopathologiques, à trois ans (n=2) des lésions stables A1F1 (n=1) une progression à A2F2 (n=1) et à 5 ans (n=2) une hépatite A2F2 (n=1) et un A1F1 (n=1).
Quatre receveurs ont bénéficié en post-greffe d’un trai- tement par interféron pégylé alpha 2a à 135ug/par semaine, associé à la ribavirine à 600 mg/jour, deux receveurs ont eu une réponse virologique soutenue, le premier de génotype 1 traité pendant 48 semaines et le second de génotype 2 traité pendant 24 semaines.
La tolérance était acceptable, nous avons utilisé l’érythropoïétine alpha pendant presque toute la durée du traitement. Nous avons arrêté le traitement chez deux patients, pour le premier à 1 mois en raison d’une neutropénie sévère, d’une thrombocytopénie et d’une anémie hémolytique, ce patient avait le même profil de tolérance qu’en traitement pré-greffe, et pour le second au 2ème mois, à cause d’une anémie hémolytique et la survenue d’une insuffisance rénale.
Concernant les patients transplantés VHB (n=5), trois d’entre eux étaient actifs avant la greffe et ont répondu aux analogues, un patient est décédé à 6 mois d’un SDRA comme cité précédemment et deux receveurs sont toujours inactifs respectivement à 5 et 7 ans, ils reçoivent des immunoglobulines anti-Hbs selon le protocole de Paul Brousse(9). La PBHPT a été faite chez deux receveurs à 1 an, et n’a pas montré d’anomalies histologiques, et l’ADN VHB fait annuellement reste indétectable.
Les patients transplantés pour cirrhose HAI (n=5), quatre d’entre eux sont encore en vie, respectivement à 40, 87, 77 et 100 mois, la PBHPT à 2 ans (n=1) a confirmé la récidive clinique chez un receveur, mis en rémission par corticostéroïdes.
La PBHPT à 5 ans (n=3) a montré une cirrhose inactive dans un cas et aucune récidive chez deux receveurs. Les transplantés pour cirrhose cryptogénétique (n=4), sont tous en vie respectivement à 48, 60, 72 et 108 mois. Un des receveurs a présenté à un an et à quatre ans, un ictère avec élévation des transaminases 8 fois la normale, sur la PBH nous avons retrouvé des anomalies histologiques compatibles avec une hépatite auto-immune de novo, sur le plan thérapeutique le MMF a été remplacé par l’azathioprine.
Cette patiente est en rémission clinique et biologique et a mené une grossesse normale à l’âge de 35 ans soit à la 9ème année post greffe. Dans ce même groupe, chez un second receveur la PBHPT faite à 5 ans a montré une hépatite chronique cirrhogène active. La PBHPT faite chez le 3ème receveur pendant la chirurgie pour sténose anastomotique biliaire comme mentionné précédemment a montré une hépatite cholestatique avec fibrose septale et la PBHPT à 5 ans chez ce même patient a retrouvé une cirrhose inactive. Pour le dernier receveur nous sommes à la quatrième année, la PBHPT est programmée à 5 ans.
Pour les transplantés pour cirrhose alcoolique (n=3), deux sont vivants respectivement à 60 et 84 mois, pour le premier receveur, il y a une récidive de son alcoolisme, la PBHPT faite à un an a montré une stéatohépatite, pour le deuxième receveur on note la survenue d’un cancer du cavum à la cinquième année post greffe, traité par radio-chimiothérapie.
Les patients transplantés pour maladie cholestatique (THDV, n=5) seulement 2 d’entre eux sont encore en vie, respectivement à 6 ans pour le receveur de la CSP et 7 ans pour celui de la CBP, la PBHPT réalisée à 5 ans a montré l’absence de récidive pour ces deux receveurs. La patiente ayant reçu la greffe combinée foie-rein pour hyperoxalurie primitive est vivante à 5 ans. Le receveur ayant la maladie de Wilson est vivant à 7 ans avec une PBHPT sans anomalies à 5 ans, deux receveurs SBC à 7 et 8 ans, et le transplanté pour CHC est indemne de récidive à 2 ans.
En somme, 33% de nos patients (n=10) ont eu une récidive de la maladie initiale, et deux patients ont présenté des lésions de novo, une HAI chez le premier et une HNR chez le second.
En ce qui concerne les complications infectieuses à long terme, un patient transplanté pour cirrhose VHB a présenté à 15 mois une aspergillose pulmonaire ayant répondu à la capsofungine, il est vivant à 4 ans.
Le deuxième patient, greffe combinée foie-rein pour cirrhose VHC a présenté la deuxième année post greffe un syndrome de Hunt Ramsay traité par acyclovir, il est vivant à 6 ans.
Le syndrome métabolique du transplanté a été observé dans 61% des cas (n=16), 34% (n=9) des receveurs sont en surpoids avec un IMC moyen à 27, 34 kg/m2, 34% ont une hypertension artérielle (n=9), 3 receveurs ont une HTA associée à une dyslipidémie, 15% ont une hyperuricémie (n=4), 19% ont un diabète (n=5) avec 3 cas de diabète post-greffe, et 23% (n=6) des receveurs ont présenté une insuffisance rénale, chez 4 d’entre eux, elle est modérée avec un de taux de créatininémie compris entre 16 et 18 mg /l et chez 2 receveurs transplantés pour VHC , elle est sévère, avec une clairance de la créatinine respectivement à 20 et 22 ml / min et une diurèse conservée, l’un d’entre eux est le patient ayant reçu la greffe combinée foie-rein, aucun des deux n’est au stade de dialyse.
Discussion :
La survie globale à trois ans des THDV est de 62%, la majorité des décès ont eu lieu dans la période post-opératoire immédiate. L’infection est la principale cause de décès. Ce pourcentage de survie est légèrement différent de celui rapporté dans la littérature (10-13), cela peut être expliqué par deux raisons : la première est relative au choix des indications, nous avons toujours privilégié les patients les plus atteints, la seconde a trait à notre expérience débutante dans ce domaine.
Dans notre expérience de THDV, l’hépatectomie a été associée à un taux significatif de complications. En dehors des complications mineures, sept complications majeures ont été retrouvées chez 5 donneurs soit 16%, de grade IIIa, IIIb et IV. Le traitement chirurgical ou par radiologie interventionnelle a été mené avec succès dans tous les cas(4, 7,14).
Le taux de complications biliaires chez les receveurs était relativement élevé, comme attendu, plus important chez les receveurs des THDV(15, 16, 17), avec un décès lié à ce type de complication. Aucun des donneurs n’a présenté de complications biliaires.
À long terme, le meilleur taux de survie a été observé chez les patients transplantés pour cirrhose auto-immune ou cryptogénétique(18,19), suivies des cirrhoses virales.
Par contre, la survie au cours des SBC est différente comparativement aux données de la littérature(20-23). Nous avons eu quatre décès, deux en période post-opératoire et deux à distance. À notre avis, deux raisons majeures expliquent ces résultats peu satisfaisants.
La première, est en rapport avec les critères de sélection, comme signalé en introduction notre politique d’indication privilégie toujours les patients les plus avancés, nos patients arrivent à la transplantation avec des facteurs prédictifs de mauvais pronostic tels que : l’infection, le syndrome hépato-rénal et la dénutrition. La seconde raison est relative à l’absence de préparation spécifique pour ce type d’affection par la radiologie interventionnelle, de tels patients avec des ascites réfractaires devaient bénéficier de mise en place de TIPS avant la greffe programmée(21,22,23).
Trente-trois pourcent (33%) de nos patients ont eu une récidive de la maladie initiale. La récidive de l’infection à VHC a été observée dans tous les cas, la plupart du temps avec des fibroses minimes à modérées, deux patients seulement ont répondu à la bithérapie interféron pégylé ribavirine(8,24,25). Chez les patients transplantés pour cirrhose cryptogénétique, nous avons relevé une progression plus rapide vers la cirrhose(26). La biopsie hépatique reste essentielle pour le suivi des transplantés (25 27). Le syndrome métabolique et les complications de l’immunosuppression comme l’insuffisance rénale, constituent le revers de la médaille, ils nécessitent un suivi rigoureux afin de préserver le greffon(28, 29).
Conclusion :
La transplantation hépatique demeure le traitement de référence pour les patients en insuffisance hépatique terminale. Les hépatectomies en THDV exposent le donneur à une morbidité importante. La majorité de nos décès sont survenus au cours de la période péri-opératoire. Un tiers de nos patients ont présenté une récidive de la maladie initiale confirmée histologiquement. Ces résultats préliminaires encourageants, incitent à développer la transplantation hépatique dans notre pays.
Conflit d’intérêt : aucun.
Références
Varma V, Mehta N, Kumaran V, Nundy S. Indications and contrain- dications for liver transplantation. Int J Hepatol. 2011;2011:121862.
Consensus conference: Indications for Liver Transplantation, Ja- nuary 19 and 20, 2005, Lyon-Palais Des Congrès: text of recommen- dations (long version). Liver Transpl. 2006 Jun;12(6):998-1011.
Bentabak K, Graba A, Boudjema K, Griène B, Debzi N, Bekkouche N, Yahiatène S, Fellah N, Benmoussa D, Faraoun SA, Bodin JM, Lakehal M, Bendib SE, Boucekkine T. Adult-to-adult living related liver transplantation: preliminary results of the Hepatic Transplan- tation Group in Algiers. Transplant Proc. 2005 Jul-Aug;37(6):2873-4
Dindo D, Demartines N, Clavien PA. Classification of surgical complications: a new proposal with evaluation in a cohort of 6336 patients and results of a survey. Ann Surg. 2004 Aug;240(2):205-13.
Trotter JF, Wachs M, Everson GT, Kam I. Adult-to-adult trans- plantation of the right hepatic lobe from a living donor. Review N Engl J Med. 2002 Apr 4;346(14):1074-82.
Broering DC, Sterneck M, Rogiers X. Living donor liver transplan- tation. Review. J Hepatol. 2003;38Suppl 1:S119-35.
Umeshita K, Fujiwara K, Kiyosawa K, Makuuchi M, Satomi S, Sugimachi K, Tanaka K, Monden M; Japanese Liver Transplantation Society. Operative morbidity of living liver donors in Japan.Lancet. 2003 Aug 30;362(9385):687-90.
Berenguer M, Palau A, Aguilera V, Rayón JM, Juan FS, Prieto M. Clinical benefits of antiviral therapy in patients with recurrent hepatitis C following liver transplantation. Am J Transplant. 2008 Mar;8(3):679-87.
Roche B, Feray C, Samuel D. Prevention and treatment of hepatitis B virus infection after liver transplantation. Gastroenterol Clin Biol. 2002 Mar;26(3):271-83.
Muñoz LE, Nañez H, Rositas F, Pérez E, Razo S, Cordero P, Torres L, Zapata H, Hernández MA, Escobedo MM. Long-term complications and survival of patients after orthotopic liver transplan- tation. – Transplant Proc. 2010 Jul-Aug;42(6):2381-2.
Elola-Olaso AM, Gonzalez EM, Diaz JC, Garcia García I, Segu- rola CL, Usera MA, Romero CJ, Perez-Saborido B, Suarez YF, Oliva MC. Short- and long-term outcomes after living donor liver trans- plantation. Transplant Proc. 2005 Nov;37(9):3884-6.
Detry O, De Roover A, Delwaide J, Coimbra C, Kaba A, Joris J, Damas P, Meurisse, Honoré P. Living related liver transplanta- tion in adults: first year experience at the University of Liège. Acta Chir Belg. 2004 Apr;104(2):166-71.
Azoulay D, Castaing D, Adam R, Savier E, Smail A, Veilhan LA, Samuel D, Féray C, Saliba F, Ichai P, Roche B, Duclos-Vallée JC,Bismuth H. Adult to adult living-related liver transplantation. The Paul-Brousse Hospital preliminary experience. Gastroenterol Clin Biol. 2001 Aug-Sep;25(8-9):773-80
Shin M, Song S, Kim JM, Kwon CH, Kim SJ, Lee SK, Joh JW. Donor Morbidity Including Biliary Complications in Living-Donor Liver Transplantation: Single-Center Analysis of 827 Cases Trans- plantation. 2012 Feb 20.
Yaprak O, Dayangac M, Akyildiz M, Demirbas T, Guler N, Bu- lutcu F, Bassullu N, Akun E, Yuzer Y, Tokat Y. Biliary complications after right lobe living donor liver transplantation: a single-centre expe- rience. HPB (Oxford). 2012 Jan;14(1):49-53.
Chang JH, Lee IS, Choi JY, Yoon SK, Kim DG, You YK, Chun HJ, Lee DK, Choi MG, Chung IS. Biliary Stricture after Adult Right- Lobe Living-Donor Liver Transplantation with Duct-to-Duct Anasto- mosis: Long-Term Outcome and Its Related Factors after Endoscopic Treatment. Gut Liver. 2010 Jun;4(2):226-33.
Weber A, Prinz C, Gerngross C, Ludwig L, Huber W, Neu B, Ebert MP, Meining A, Weidenbach H, Schmid RM, Schulte-Fro- hlinde E. Long-term outcome of endoscopic and/or percutaneous transhepatic therapy inpatients with biliary stricture after orthotopic liver transplantation. J Gastroenterol. 2009;44(12):1195-202
Khalaf H, Mourad W, El-Sheikh Y, Abdo A, Helmy A, Medhat Y, Al-Sofayan M, Al-Sagheir M, Al-Sebayel M. Liver transplantation for autoimmune hepatitis: a single-center experience. Transplant Proc. 2007 May;39(4):1166-70.
Álamo JM, Bernal C, Barrera L, Marín LM, Suárez G, Serra- no J, Gómez MA, Padillo FJ. Liver transplantation in patients with cryptogenic cirrhosis: long-term follow-up. Transplant Proc. 2011 Jul- Aug;43(6):2230-2.
Srinivasan P, Rela M, Prachalias A, Muiesan P, Portmann B, Mufti GJ, Pagliuca A, O’Grady J, Heaton N.Liver transplantation for Budd- Chiari syndrome Transplantation. 2002 Mar 27;73(6):973-7.
Yamada T, Tanaka K, Ogura Y, Ko S, Nakajima Y, Takada Y, Uemoto S. Surgical techniques and long-term outcomes of living do-
nor liver transplantation for Budd-Chiari syndrome. Am J Transplant. 2006 Oct;6(10):2463-9.
Ulrich F, Pratschke J, Neumann U, Pascher A, Puhl G, Fellmer P, Weiss S, Jonas S, Neuhaus P. Eighteen years of liver transplantation experience in patients with advanced Budd-Chiari syndrome. Liver Transpl. 2008 Feb;14(2):144-50.
Choi GS, Park JB, Jung GO, Chun JM, Kim JM, Moon JI, Kwon CH, Kim SJ, Joh JW, Lee SK. Living donor liver transplantation in Budd-Chiari syndrome: a single-center experience. Transplant Proc. 2010 Apr;42(3):839-42.
Yosry A, Abdel-Rahman M, Esmat G, El-Serafy M, Omar A, Doss W, Zayed N, Said M, Ismail T, Hosny A, Marawan E, El-Malt O, Kamel RR, Hatata Y, El-Taweel A, Ghali A, Sabri H, Kamel S, El- Gabaly H. Recurrence of hepatitis C virus (genotype 4) infection after living-donor liver transplant in Egyptian patients. Exp Clin Trans- plant. 2009 Sep;7(3):157-63.
Neumann UP, Berg T, Bahra M, Puhl G, Guckelberger O, Lan- grehr JM, Neuhaus P. Long-term outcome of liver transplants for chronic hepatitis C: a 10-year follow-up. Transplantation. 2004 Jan 27;77(2):226-31.
Heneghan MA, Zolfino T, Muiesan P, Portmann BC, Rela M, Heaton ND, O’gradyJG.An evaluation of long-term outcomes after liver transplantation for cryptogenic cirrhosis. LiverTranspl. 2003 Sep;9(9):921-8.
Duclos-Vallee JC, Sebagh M. Recurrence of autoimmune disease, primary sclerosing cholangitis, primary biliary cirrhosis, and autoim- mune hepatitis after liver transplantation.
Sutedja DS, Wai CT, Teoh KF, Lee HL, Da Costa M, Lee M, Kaur M, Lee YM, Lee KH, Mak K, Quak SH, Isaac J, Lim SG, Prabhakaran K. Renal impairment and diabetes mellitus after liver transplant. Transplant Proc. 2004 Oct;36(8):2324-7.
Laish I, Braun M, Mor E, Sulkes J, Harif Y, Ben Ari Z. Metabolic syndrome in liver transplant recipients: prevalence, risk factors, and asso- ciation with cardiovascular events. Liver Transpl. 2011 Jan;17(1):15-22.
SA. FARAOUN(1), N. DEBZI(2), K. BENTABAK(3), A. GRABA(3), B. Griene(4),T. BOUCEKKINE(5), N. GUESSAB(2), N. AFREDJ(2), ME. BOUDJELLA(6), N. BENIDIR(7),C. BOUZID(3), K. BOUDJEMAA(8), SE. BENDIB(9) Service d’imagerie médicale, CHU Lamine Debaghine Bab El Oued, Alger. Service d’hépatologie, CHU Mustapha Bacha, Alger. Service de chirurgie, Centre Pierre et Marie Curie, Alger. Service d’anesthésie réanimation, Centre Pierre et Marie Curie, Alger. Service de gastroentérologie, CHU Mustapha Bacha, Alger. Service de médecine interne, Hôpital Bachir Mentouri de Kouba, Alger. Service d’anatomie pathologique, HSN, Alger. Service de chirurgie hépatobiliaire et digestive, CHU de Rennes, France. Service d’imagerie médicale, Centre Pierre et Marie Curie, Alger.
Abstract : Liver transplantation is a surgery with two types of difficulty. A purely technical, in relation to the vascular and biliary variants, and the other pathophysiological related to the volume of the transplanted liver. Prevention of these difficulties is closely related to the reliability of preoperative morphological assessment of living donors. The aims of our study are to evaluate the sensitivity of the multidetector CT scan in the identification of vascular anatomical variants that may have an impact on the operative strategy, and to compare the graft volume, estimated by imaging, with the actual weight of grafts. This is a prospective unicentric study of 20 living donors for liver transplantation. The evaluation was carried using a 16 section CT scan, in multiphasic acquisition. 30% of the donors had an arterial anatomical variant. A portal anatomical variant was noted in 25% of cases. The sensitivity of the multidetector CT scan in the identification of anatomical variants was 100%. The calculation of the kappa coefficient, evaluated at 1, made it possible to note an excellent concordance with the operating data. Our study has shown that the multidetector CT scan allows the surgeon to anticipate and plan safely its operative strategy.
Key words : Hepatic transplantation, multi-detector scanner, living donor.
Résumé : La transplantation hépatique est une chirurgie présentant deux types de difficulté. Une purement technique, en relation avec les variantes vasculaires et biliaires, et l’autre d’ordre physiopathologique liée au volume du foie transplanté. L’atténuation, voire la prévention de ces difficultés est étroitement liée à la fiabilité de l’évaluation morphologique préopératoire des donneurs vivants. Les objectifs de notre étude sont d’évaluer la sensibilité du scanner multidétecteur dans l’identification des variantes anatomiques vasculaires pouvant avoir un impact sur la stratégie opératoire, et de comparer le volume des greffons, estimé par l’imagerie, au poids réel des greffons pesés après prélèvement. Il s’agit d’une étude prospective unicentrique qui a porté sur 20 donneurs vivants pour transplantation hépatique. L’évaluation a été réalisée à l’aide d’un scanner 16 coupes, en acquisition multiphasique. 30% des donneurs avaient une variante anatomique artérielle. Une variante anatomique portale a été notée dans 25% des cas. La sensibilité du scanner multidétecteur dans l’identification des variantes anatomiques était de 100%. Le calcul du coefficient kappa, évalué à 1, a permis de noter une excellente concordance avec les données opératoires. Notre étude a montré que le scanner multidétecteur permet au chirurgien d’anticiper et de planifier avec sécurité sa stratégie opératoire.
C’est en Février 2003 que le programme algérien de transplantation hépatique (TH) à donneur vivant apparenté a été lancé. En pratique, cette technique est confrontée à des difficultés liées aussi bien au donneur qu’au receveur. Celles-ci sont de deux types : des problèmes purement techniques, en relation avec les variantes vasculaires et biliaires des deux sujets, et des problèmes d’ordre physiopathologique en relation avec le volume du foie transplanté. Le greffon doit être prélevé avec les voies biliaires et les pédicules vasculaires nécessaires à son implantation chez le receveur, sans pour autant compromettre la vascularisation et le drainage biliaire du foie restant chez le donneur. D’autre part, le volume du foie transplanté doit être suffisant pour assurer un retour rapide à une fonction hépatocellulaire normale chez le receveur, alors que le volume restant au donneur doit être suffisant pour lui épargner toute complication liée à la réduction du volume fonctionnel hépatique. L’atténuation, voire la prévention de toutes ces difficultés est étroitement liée à la fiabilité de l’évaluation morphologique pré-greffe réalisée par le scanner multidétecteur chez les donneurs de foie.
Les objectifs de cette étude sont d’évaluer la sensibilité du scanner multidétecteur dans l’identification des variantes anatomiques vasculaires pouvant avoir un impact sur la stratégie opératoire, et de comparer le volume des greffons, estimé par l’imagerie, au poids réel des greffons pesés après prélèvement.
Patients et méthodes :
Il s’agit d’une étude prospective unicentrique qui a porté sur 20 donneurs vivants pour transplantation hépatique. Il y avait 9 hommes et 11 femmes, indemnes de toute maladie, dont l’âge moyen était de 27 ans (extrêmes : 18-52 ans). L’évaluation a été réalisée à l’aide d’un scanner 16 coupes, en acquisition multiphasique. La phase artérielle a été déclenchée par un tracking bolus, la phase portale à 40 secondes et la phase tardive à 60 secondes. Les variantes anatomiques vasculaires ont été notées alors que le volume hépatique total, le volume hépatique droit et gauche ont été calculés après délimitation manuelle des contours hépatiques. Le volume fonctionnel restant moyen a été évalué par le calcul du rapport entre le volume restant prévu et le volume total. Les données de l’imagerie ont été comparées aux données opératoires. Leur concordance a été évaluée par le calcul du coefficient kappa
Résultats :
30% (6/20) des donneurs avaient une variante anatomique artérielle (tableau 1, Figure 1-3), la plus fréquente étant la naissance de l’artère hépatique gauche à partir de l’artère gastrique gauche retrouvée chez 4/20 donneurs (20%). Une variante anatomique portale a été notée dans 25 % des cas (5/20) (tableau 2, Figure 4). Il s’agissait le plus souvent d’un glissement de la branche porte postérieure droite sur le tronc porte observé chez 3/20 donneurs (15%). Quant aux variantes anatomiques veineuses (tableau 3, Figure 5 et 6), celles-ci ont été observées chez 35% des donneurs (7/20), la VSH droite inférieure accessoire notée chez 7/20 d’entre eux (30%), étant la plus fréquemment notée. Par ailleurs, 7 donneurs avaient une VSH signifiante du segment VIII et/ou du segment V. La sensibilité du scanner multidétecteur dans l’identification des variantes anatomiques était de 100%. Le calcul du coefficient kappa, évalué à 1, a permis de noter une excellente concordance avec les données opératoires.
Le volume hépatique total scanographique moyen était de 1.300 cm3 (extrêmes : 1.150 cm3 à 1.800 cm3). Les valeurs respectives du volume moyen des hépatectomies virtuelles et du volume moyen des explants hépatiques étaient de 700 cm3 (extrêmes : 300 cm3-1.200 cm3) et de 643 cm3 (extrêmes : 280 cm3 -1.150 cm3). Le volume fonctionnel moyen a été évalué à 35% (extrêmes : 29 à 40 %). Une surestimation moyenne de 8.2% a été constatée dans la comparaison des volumes estimés par l’imagerie et le poids réel des greffons (Figure 7).
Tableau 1 : Prévalence des variantes anatomiques artérielles, AHG : artère hépatique gauche. AHD : artère hépatique droite.Tableau 2 : Prévalence des variantes anatomiques portales
Le prélèvement sur donneur vivant a pour principal objectif de prélever un greffon comportant tous ses pédicules vasculaires sans pour autant compromettre la vascularisation et le drainage veineux du foie restant. L’identification des variantes anatomiques chez le donneur est donc essentielle pour prévenir les difficultés opératoires et les complications ischémiques post-opératoires susceptibles de compromettre la fonction hépatocellulaire, indépendamment du volume réséqué.
La plupart des variantes anatomiques vasculaires ne contre indiquent pas un prélèvement hépatique. Théoriquement, seule une distribution étagée de la veine porte et la naissance d’une artère hépatique du segment IV à partir de l’artère hépatique droite peuvent contre indiquer un geste de résection. Au niveau des veines sus hépatique, la ligature de l’une d’entre elles n’a pas de conséquences négatives en raison de l’existence d’anastomoses intra-hépatiques qui exercent un effet protecteur sur la fonction hépatique. Toutefois, il est préférable pour les segments IV, V et VIII, qui sont généralement situés de part et d’autre du plan d’hépatectomie, d’identifier leurs veines dominantes de façon à réaliser une chirurgie conservant un retour veineux satisfaisant à partir du greffon, une fois implanté, et du foie laissé en place. Il faut également préciser la position des veines accessoires de gros calibre (de plus de 5 mm de diamètre) qui pourront faire l’objet d’une réimplantation séparée.
Notre étude confirme la faisabilité et la fiabilité du scanner multiphasique dans la réalisation d’un bilan vasculaire préopératoire complet. Elle a également démontré que cette technique permet d’estimer avec une très bonne précision le volume total du foie et le volume résiduel postopératoire après simulation du plan de résection chirurgicale, technique décrite sous le terme d’hépatectomie virtuelle. Le volume résiduel estimé en préopératoire apparaît dans plusieurs études comme un bon facteur prédictif de l’insuffisance hépatocellulaire avant résection hépatique majeure. Il n’y a pas de consensus sur le volume minimum restant nécessaire, mais il est généralement admis que, pour un foie normal, un volume restant inférieur ou égal à 25 % expose à un risque significatif de complications chez le donneur. La plus redoutée d’entre elles étant l’insuffisance hépatocellulaire.
Dans notre étude, une surestimation moyenne de 8.2 % a été constatée dans la comparaison des volumes estimés par l’imagerie et le poids réel du greffon. Cette surestimation est probablement liée au fait que l’hépatectomie chirurgicale, générant forcément des manipulations tissulaires de la glande hépatique, ne peut pas intégralement reproduire la tranche de section obtenue par l’hépatectomie virtuelle. En effet, L’hépatectomie réalisée par l’imagerie suit un plan virtuel parfait passant par la scissure porte principale, alors que l’hépatectomie chirurgicale, bien que guidée par des repères anatomiques échographiques, passe par une tranche
de section réelle en suivant la ligne de dévascularition constatée après clampage porte. D’autre part, l’hépatectomie virtuelle passe à ras du bord droit de la VSH médiane, comme tracé au crayon alors que la tranche de section réelle est irrégulière et peut sur certains endroits dépasser de quelques millimètres ce bord droit. Enfin, cette surestimation préopératoire du poids du greffon par le scanner multiphasique peut être expliquée par une réduction de son poids après prélèvement, après rinçage par les solutés de conservation.
Conclusion :
Notre étude a montré que le scanner multidetecteur est une technologie avancée et fiable permettant en un seul temps une évaluation morphologique complète de la cartographie vasculaire et le calcul des volumes hépatiques. Ainsi, elle permet au chirurgien d’anticiper et de planifier avec sécurité sa stratégie opératoire tout en tenant compte d’une éventuelle surestimation par l’imagerie du volume des greffons.
Bibliographie
1- Pascher A, Sauer IM, Walter M et al. Donor evaluation, donor risks, in transplantation. Liver Transpl 2002;8:829-37. 2- Pomfret EA, Pomposelli JJ et al. Live donor adult liver trans- plantation using right lobe grafts: donor evaluation and surgi- cal outcome. Arch Surg 2001; 136:425.
Kamel IR, Kruskal JB, Pomfret EA, Keogan MT, Warm- brand G, Raptopoulos V. Impact of Multidetector CT on Do- nor Selection and Surgical Planning Before Living Adult Right Lobe Liver Transplantation. AJR 2001;76:193-200.
Hogemann D, Stamm G, Shin H et al. Individual planning of liver surgery interventions with a virtual model of the liver and its associated structures. Radiology 2000;40:267-73.
Glombitza G, Demiris AM et al. Virtual planning of liver resections: image processing, visualization and volumetric eva- luation. Int J Med Inf 1999; 53:225-37.
Lamade W, Glombitza G, Fischer L et al. The impact of 3-dimensional reconstructions on operation planning in liver surgery. Arch Surg 2000;135:1256-61.
Kiuchi Tet all. Impact of graft size mismatching on graft pro- gnosis in liver transplantation from living donors. Transplantation 1999;67: 321-7.
Kamel IR. Accuracy of Volumetric measurements After Virtual Right Hepatectomy in Potential Donors Under- going Living Adult Liver Transplantation. Am J Roentgenol 2001;176:483-7.
Boillot O, Dawahra M, Mechet I et al. Liver transplantation using a right liver lobe from a living donor. Transplant Proc 2002;34:773
K. BENTABAK(1), SA. FARAOUN(2) Service de chirurgie oncologique « A » ,Service d’imagerie médicale, EHS, Centre Pierre et Marie Curie, Alger, Algérie.
Introduction :
Le carcinome hépatocellulaire (CHC) est le 5e cancer le plus fréquent dans le monde et la tumeur maligne primitive du foie la plus fréquente [1,2]. Il est dans 90% des cas associé à une maladie chronique du foie, le plus souvent au stade de cirrhose. Le traitement chirurgical à visée curative comporte la résection et la transplantation hépatique qui ne peuvent cependant être proposées qu’à 30% des patients [3]. La résection hépatique sur foie pathologique, a longtemps été limitée par une mortalité et une morbidité élevées ainsi que par un risque de récidive important lié, à la survenue de nodules de novo en rapport avec l’hépatopathie sous jacente [4]. Les indications et les résultats de la résection hépatique dépendent du stade tumoral au moment du diagnostic, de la réserve fonctionnelle hépatique sous jacente et de l’utilisation de techniques chirurgicales adaptées. Dans ce contexte, la résection hépatique du CHC va poser le problème du terrain souvent fragile sur lequel elle survient, des capacités de régénération hépatique diminuées par la fibrose, de l’exérèse limitée par le risque d’insuffisance hépatique postopératoire irréversible pouvant conduire au décès du patient et du risque élevé de récidive. Une sélection rigoureuse des patients comportant l’évaluation du stade tumoral et celle de la réserve fonctionnelle hépatique est alors nécessaire. La résection ne se conçoit que s’il est possible de réaliser une exérèse à visée curative avec un risque de mortalité et de morbidité acceptable.
Évaluation du stade tumoral :
L’évaluation du stade tumoral repose sur une exploration comportant une échographie abdominale, une imagerie hépatique en coupe par scanner triphasique et une imagerie par résonnance magnétique. L’imagerie doit préciser la taille de la tumeur, son siège segmentaire précis, ses connexions vasculaires et l’existence d’un envahissement veineux endoluminal, c’est-à-dire un thrombus tumoral portal ou veineux hépatique caractéristique du CHC, la présence de nodules satellites ou à distance, et comporter une étude volumétrique. La recherche de localisations secondaires, en particulier par scanner thoracique, est indispensable et rentre dans le cadre du bilan pré thérapeutique.
Indications de la résection hépatique :
Les indications d’exérèse dépendent de la taille, du nombre des lésions, de leur siège et de l’estimation du foie restant après exérèse. Les meilleurs candidats à l’exérèse sont les patients ayant une tumeur unique latéralisée permettant de conserver un parenchyme hépatique restant de plus de 50% [5,6]. Les politiques de dépistage du CHC permettent de découvrir des tumeurs à un stade précoce (<à 5 cm) répondant à ces critères. Dans ce cas, la résection est en compétition avec la transplantation hépatique et les traitements percutanés. Le siège de la tumeur est alors un paramètre important dans le choix du traitement. Lorsqu’il s’agit d’une tumeur de siège périphérique, une exérèse limitée peut être réalisée de façon curative et anatomique avec un sacrifice parenchymateux faible. Au contraire, l’existence d’une petite lésion centrale de moins de 5 cm peut nécessiter un sacrifice parenchymateux plus important avec un risque d’insuffisance hépatique postopératoire faisant orienter vers le traitement percutané ou la transplantation.
L’existence de nodules satellites témoigne souvent d’une lésion métastatique locorégionale liée à un envahissement vasculaire microscopique. Le pronostic en est plus sombre mais cela ne représente pas une contre-indication opératoire. L’existence de lésions bilobaires témoigne habituellement d’une carcinogénèse multicentrique rendant peu probable le caractère curatif de la résection. Les contre-indications à la résection du CHC sont la présence de métastases extra-hépatiques, la présence de lésions multiples bilobaires, la présence d’un envahissement de la convergence biliaire et la présence d’une thrombose tumorale extensive du tronc porte ou de la veine cave inférieure. Cependant, un grand nombre de malades continuent de se présenter à un stade tumoral avancé avec une tumeur de plus de 5 cm, dépassant la limite au-delà de laquelle la transplantation peut être proposée[7].
Évaluation de la réserve fonctionnelle hépatique :
Outre l’évaluation tumorale, l’évaluation de la fonction hépatique est essentielle à la sélection des candidats à la résection hépatique sur foie pathologique. L’hépatectomie sur foie de cirrhose est grevée d’une morbidité et d’une mortalité beaucoup plus élevées. Le risque majeur est celui du développement d’une insuffisance hépatocellulaire postopératoire puis d’une décompensation de la cirrhose. Les critères prédictifs de survenue d’une insuffisance hépatique postopératoire sont essentiellement liés à la fonction hépatique préopératoire et à l’état histologique du foie sous-jacent. L’évaluation de la fonction hépatique préopératoire est essentiellement basée sur le score de Child-Pugh[3,8,9] regroupant trois classes : la classe A où la fonction hépatique est normale, la classe B où il existe une insuffisance hépatocellulaire minime à modérée et la classe C où l’insuffisance hépatocellulaire est sévère. La plupart des auteurs s’accordent pour considérer que seuls les cirrhotiques Child-Pugh A sont candidats à une résection hépatique. Certains auteurs, en particulier asiatiques, utilisent le test au vert d’indocyanine qui consiste à injecter le colorant et à mesurer sa concentration résiduelle à 15 minutes. Ainsi, une hépatectomie majeure peut être réalisée jusqu’à 15% de rétention alors que la résection hépatique doit être limitée au-delà de 20% [10]. De même, l’évaluation du volume de parenchyme hépatique restant après exérèse est une donnée essentielle de la sélection fonctionnelle. Elle nécessite une analyse précise de l’imagerie tenant compte des volumes respectifs du foie total, du foie réséqué et de la tumeur.
Facteurs de risque :
Il a été démontré que l’existence d’une hypertension portale était corrélée à la décompensation postopératoire de la cirrhose, en particulier sous forme d’ascite[11]. Le degré d’hypertension peut être évalué par la recherche de varices œsophagiennes à l’endoscopie digestive, l’existence d’une splénomégalie et d’une circulation collatérale à la tomodensitométrie. L’existence d’une thrombopénie témoignant d’un hypersplénisme est également un stigmate d’hypertension portale. Une résection hépatique majeure est contre-indiquée en présence d’une hypertension portale. Une résection limitée peut être envisagée en cas d’hypertension portale modérée.
L’étiologie de l’hépatopathie sous-jacente et son activité doivent être considérées dans le risque d’une hépatectomie. L’activité de l’hépatopathie est évaluée sur la concentration sérique des transaminases, qui constitue un facteur de risque bien établi [12,13]. Il a été montré qu’une élévation des transaminases à plus de deux fois la normale était associée à une majoration de la mortalité [12]. En cas d’hépatopathie active, la résection peut être préparée par un traitement antiviral ou un sevrage alcoolique, selon les cas, jusqu’à normalisation des transaminases. L’état histologique du foie sous-jacent peut être évalué par la biopsie du foie non tumoral pour évaluer le degré de fibrose et une éventuelle activité. La quantification de la fibrose par le score METAVIR (F0 à F4) est utile dans la prise de décision [14]. Le risque opératoire est maximal en cas de cirrhose constituée (fibrose F4), il est élevé en cas de fibrose sévère (F3), et il est faible en cas de fibrose modérée (F2). En cas de fibrose minime F1, le risque est proche de celui d’une hépatectomie sur foie sain.
Résection hépatique :
Trois principes président à la résection du CHC sur foie pathologique : l’épargne parenchymateuse, l’exérèse anatomique et la minimisation de l’hémorragie.
L’exérèse sur foie pathologique impose une épargne parenchymateuse du fait des facultés de régénération qui sont réduites par la fibrose hépatique, surtout s’il existe une cirrhose constituée, ce qui entraîne un risque d’insuffisance hépatique postopératoire.
Si l’exérèse anatomique doit rester la référence, la préservation parenchymateuse peut devenir prioritaire par rapport à l’exérèse anatomique chez les patients ayant une fonction hépatique limite. Une hépatectomie majeure ne doit être envisagée qu’en cas de tumeur volumineuse inaccessible à un geste plus limité. Dans les tumeurs profondes de petites tailles, le principe d’épargne parenchymateuse doit conduire à des exérèses plus complexes aboutissant à ne réséquer que les segments ou secteurs intéressés par la tumeur. Lorsqu’il s’agit d’une tumeur périphérique, une exérèse limitée est indiquée, à type de segmentectomie ou sous-segmentectomie. Lorsque le foie est très dysmorphique et qu’il existe une hypertension portale, il est parfois préférable d’avoir recours à une exérèse non anatomique représentée par une tumorectomie emportant une marge de parenchyme hépatique supérieure à 1 cm.
L’hémorragie préopératoire et les transfusions sanguines influencent le pronostic des résections hépatiques pour cancer. Cette influence s’exerce sur la morbidité et la mortalité opératoires, mais également sur la survie à long terme, l’hémorragie et les transfusions sanguines étant associées à une fréquence plus élevée des récidives tumorales[15, 16]. La réduction de l’hémorragie fait intervenir l’utilisation des clampages vasculaires et la gestion du remplissage vasculaire au cours de l’anesthésie. La tolérance du foie pathologique à l’ischémie est inférieure à celle du foie sain et il est maintenant bien établi que l’utilisation du clampage pédiculaire hépatique intermittent (clampage de 15 mn, déclampage de 5 mn) est parfaitement toléré par le foie de cirrhose et permet de réduire de façon très efficace l’hémorragie [15,16,17]. Outre le clampage, le maintien d’une pression veineuse centrale basse par un remplissage vasculaire le plus limité possible en peropératoire est un élément essentiel dans la réduction de l’hémorragie.
Résultats de la résection hépatique :
Les progrès récents dans les techniques de résection hépatique ainsi qu’une meilleure connaissance de ses facteurs de risque ont permis d’améliorer les résultats de l’hépatectomie partielle pour CHC tant sur le risque opératoire que sur les résultats à long terme[18,19].
Le taux de morbidité des résections hépatiques sur foie de cirrhose est de l’ordre de 30 à 50% et comporte des complications sévères[20,21]. La principale est la survenue d’une insuffisance hépatique postopératoire dont les manifestations en sont l’ictère, la baisse des facteurs de coagulation, l’ascite et l’encéphalopathie hépatique. La morbidité comporte également les complications pulmonaires, l’infection et l’insuffisance rénale pouvant elles mêmes être secondaires à l’insuffisance hépatique ou en précipiter la survenue. L’évaluation du risque opératoire et la réduction de l’hémorragie ont permis de réduire la mortalité opératoire qui est passée de plus de 10% dans les années 1980 à moins de 5% dans la plupart des centres spécialisés actuels[5,9,15,16,18,19,22,23].
Quant aux résultats en termes de survie à long terme, ils sont variables d’une série à l’autre du fait de critères d’inclusion très disparates. Pour les malades ayant les meilleures caractéristiques, c’est-à-dire tumeur unique, Child-Pugh A et sans hypertension portale, des taux de survie de 50 à 70% sont rapportés [24,25]. Pour le groupe de Barcelone, la présence d’une hypertension portale et un taux de bilirubine supérieur à 10 mg/l constituaient les deux facteurs pronostiques les plus importants qui influençaient négativement la survie à long terme. La survie à 5 ans après hépatectomie était respectivement de 74%, 50% et 25% chez les patients qui ne présentaient aucun facteur, ou qui présentaient 1 ou 2 facteurs[26].
Récidive de la maladie :
Du fait de la persistance de l’hépatopathie sous-jacente, la récidive intra-hépatique est très fréquente : 60 à 80% à 5 ans dans la littérature [8,27]. De nombreux facteurs pronostiques influençant la survie après résection pour CHC ont été identifiés. Il s’agit principalement de la sévérité de l’hépatopathie sous-jacente, la taille et le nombre des tumeurs ainsi que l’existence ou non d’un envahissement vasculaire macroscopique ou microscopique.
Dans ce contexte, le traitement des récidives repose sur la résection itérative, la destruction percutanée et la transplantation de rattrapage [28,29,30,31]. La re-ré-section est rarement réalisée et les séries publiées sont peu nombreuses et comportent peu de malades. Cette option peut se concevoir en cas de récidive uni focale sur hépatopathie compensée. La destruction percutanée peut être envisagée en cas de récidive uni focale de petite taille. La transplantation hépatique peut se concevoir dans 2 situations distinctes : soit de sauvetage une fois la récidive diagnostiquée, soit préemptive selon les critères de récidive présents lors de l’analyse de la pièce opératoire qui sont : la marge de résection, le nombre et la taille des différents nodules de CHC, la présence de nodules satellites, le degré de différenciation, la présence d’une micro invasion vasculaire et la présence de capsule [32]. La transplantation hépatique peut être proposée en respectant les critères de Milan [7], mais elle reste limitée par la pénurie de greffon.
Conclusion :
La résection peut ainsi être considérée comme une méthode de sélection des candidats à la greffe, voire comme un traitement d’attente chez les patients inscrits en liste d’attente et ayant une tumeur périphérique aisée à réséquer. Ailleurs, elle peut être considérée comme la seule alternative à la transplantation hépatique en cas d’absence de greffon qu’il soit à partir du cadavre ou du vivant. Chez des patients sélectionnés, la résection hépatique sur foie pathologique constitue une bonne option et offre une survie similaire à la transplantation hépatique. C’est pourquoi, un suivi régulier des patients présentant une hépatopathie chronique ou une cirrhose compensée est nécessaire pour dépister le CHC à un stade utile.
Image 1 : Résection hépatique emportant la tumeurImage 2 : Résection sectorielle antérieure droite emportant les segments 5 et 8Image 3 : Scanner montrant un CHC de 5 cm du secteur antérieur droit (segments 5 et 8) sur foie de cirrhose
Références :
Bruix J, Sherman M, Llovet JM, et al. Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. European Association for the Study of the Liver. J Hepa- tol 2001 Sep;35(3):421–30.
Bruix J, Sherman M. Management of hepatocellular carcinoma. AASLD practice guideline. Hepatology 2005;42(5):1208–35.
Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lan- cet 2003; 362: 1907-17.
Bismuth H, Chiche L, Adam R, Castaing, Diamond T, Dennison liver resection versus transplantation for hepatocellular carcinoma in cirrhotic patients. Ann Surg 1993 ; 218 : 145-51.
Azoulay D, Castaing D, Krissat J, Hargreaves GM, Lemoine A, et al. Percutaneous portal vein embolization increase the feasibility and safety of major liver resection for hepatocellular carcinoma in injured liver. Ann Surg 2000 ; 232 :665-72
Farges O, Belghiti J, Kianmanesh R, Regimbeau JM, Santano R, Vilgrain V, et al. Portal vein embolization before right hepatectomy:prospective clinical trial. Ann Surg 2003; 237: 208-17.
Mazzaferro V, Regalia E, Doci R, Andreola S, Pulvirenti A, Boz- zetti F, Montalto F, Ammatun M, A Orabito A, Gennari L. Liver transplantation for the treatment of small hepatocellular carconoma in patients with cirrhosis. NEJM 1996; 334, 11: 693-99.
Bismuth H, Chiche L, Adam R, Castaing D, Diamond T, Dennison Liver resection versus transplantation for hepatocellular carcinoma in cirrhotic patients. Ann Surg 1993;218 :145-51.
Belghiti J, Regimbeau JM, Durand F, Kianmanesh AR, Dondero F, Terris B, & al. Resection of hepatocellular carcinoma: a European experience on 328 cases. Hepatogastroenterology2002;49:41-6
Imamura H, Seyama Y, Kokudo N, Maema A, Sugawara Y, Sano K, et al. One thousand fifty-six hepatectomies without mortality in 8 years. Arch Surg 2003; 232: 665-72.
Bruix J, Castells A, Bosch J, Feu F, Garcia-Pagan JC, et al. Surgical resection of hepatocellular carcinoma in cirrhotic patients: prognostic value of portal pressure. Gastroenterology 1996; 111: 1018-22.
FargesO, Malassagne B, Flejou JF, Balzan S, Sauvanet A, Belghiti
J. Risk of major liver resection in patients with underlying chronic liver disease : a reappraisal. Ann Surg 1999 ; 229 : 210-5.
Ercolani G, Grazi GL, Ravaioli M, Del Gaudio M, Gardini A, Cescon M, et al. Liver resection for hepatocellular carcinoma on cir- rhosis: univariate and multivariate analysis of risk factors for intrahe- patic recurrence. Ann Surg 2003; 237: 536-43.
The French METAVIR Cooperative Study Group. Intra obser- ver and interserver variations in liver biopsy interpretation in patients with chronic hepatitis C. Hepatology 1994; 20: 15-20.
Poon RT, Fan ST, Lo CM, Liu CL, Lam CM, Wong J. Improving survival results after resection of hepatocellular carcinoma: a prospec- tive study of 377 patients over 10 years. Ann Surg 2001; 234: 63-70.
ImamuraH, Seyama Y, Kokudo N, Maema A, Sugawara Y, Sano K, et al. One thousand fifty-six hepatectomies without mortality in 8 years. Arch Surg 2003; 232: 665-72.
Belghiti J, Noun R, Malafosse R, Jagot P, Sauvanet A, Pierangeli F et al. Continuous versus intermittent portal triad clamping for liver resection: acontroled study. Ann Surg 1999; 229: 369-75.
Llovet JM, Fuster J, Bruix J. intention-to-treat analysis of surgical treatment for early hepatocellular carcinoma: resection versus trans- lantatio. Hepatology 1999 ; 10 : S115-20.
Chouillard E, Cherqui D, Tayar C, Brunetti F, Fagniez PL. Ana- tomical bi- and trisegmentectomies as alternatives to extensive liver
resection. Ann Surg 2003; 238:29-34.
Balzan S, Belghiti J, Farges O, Sauvanet A, Delefosse D, Duranr F. The « 50-50 criteria » on postoperative day 5: an accurate predictor of liver failure and death after hepatectomy. Ann Surg 2005; 242: 824-8.
Wei AC, Tung-Ping Poon R, Fan ST, Wong J. risk factors for pe- roperative morbidity and mortality after extended hepatectomy for hepatocellular carcinoma. BR J Surg 2003; 90 : 33-41.
Bismuth H, chiche L, Adam R, Castaing D, Diamond T, Denni- son A. Liver resection versus transplantation for hepatocellular carci- noma in cirrhotic patients. Ann Surg 1993; 218: 145-51.
Cha CH, Ruo L, Fong Y, Jarnagin WR, Shia J, Blumgart LH, De Matteo RP. Resection ot hepatocellular carcinoma in patients othe- rwise eligible for transplantation. Ann Surg 2003; 238: 315-21.
Ikai I, Arii S, Kojiro M, Makkuchi M, Matsuyama Y, et al. Ree- valuation of prognostic factors for survival after liver resection in patients with hepatocellular carcinoma in a japonese nationwide sur- gery. Cancer 2004; 101 :796-802.
Esnaola NF, Mirza N, Lauwers GY, Ika I, Regimbeau JM, Belghiti J, et al. Comparaison of clinicopathological characteristics and out- comes after resection in patients with hepatocellular carcinoma treated in the United States, France en Japan. Ann Surg 2003; 238: 711-9.
Llovet JM, Bru C, Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis 1999; 19(3): 329–38.
Belghiti J, Panis Y, Farges O, Benhamou JP, Fekete F. intrahepatic recurrence after resection of hepatocellular carcinoma complicating cirhisis. Ann Surg 1991; 214 :114-7.
Minagawa M, Makuuchi M, Takayama T, Kokudo N. Selection criteria for repeat hepatectomy in patients with reccurent hepatocel- lular carcinoma. Ann Surg 2003; 238: 703-10.
Poon RT Fan ST, Lo CM, Liu CL, Wong J. Long-term survi- val and pattern of recurrence after resection of small hepatocellular carcinoma in patients with preserved liver function: implications for a strategy of salvage transplantation. Ann Surg2002 ; 235 : 373-82.
Nakajima Y, Ko S, Kanamura T, Nagao M, Kanehiro H, Hisa- naga M et al. Repeat liver resection for hepatocellular carcinoma. J Am CollSurg 2001; 192: 339-44.
Belghiti J, Cortes A, Abdalla EK, Regimboeau JM, Prakash K, Durand F & al. Resection prior to liver transplantation for hepatocel- lular carcinoma. Ann Surg 2003; 238 :885-92.
Lauwers GY et al. Pronostic histological indicators of curatively resec- ted hepatocellular carcinomas. Am J Surg Pathol 2002; 26: 25-34.
S. TAHARBOUCHT, Service de Médecine Interne, EPH El Biar, Alger.
Abstract : Non-alcoholic steatohepatitis or NASH is characterized by a hepatic steatosis exceeding 5% associated with inflammation that can progress to cirrhosis and its complications. Described in 1980 by Ludwig and al, this condition is currently one of the leading causes of chronic liver diseases in the world. Considerable advances have been made in the understanding of its pathophysiology, its non-invasive diagnosis, although liver biopsy is still essential for its confirmation, and also in its drug treatment, several of which are currently being tested.
Key-words : Non alcoholic steatohepatitis – NASH-NAFLD
Résumé : La stéatohépatite non alcoolique ou NASH (non alcoholic steatohepatitis) est caractérisée par la présence d’une stéatose hépatique dépassant les 5% associée à une inflammation qui peut évoluer vers la cirrhose et ses complications. Décrite dans l’année 1980 par Ludwig et col, cette affection est actuellement une des premières causes d’hépatopathies chroniques au monde. De considérables avancées ont été réalisées dans la compréhension de sa physiopathologie, de son diagnostic non invasif même si la biopsie hépatique est encore indispensable pour sa confirmation, et également dans son traitement médicamenteux dont plusieurs essais sont en cours de réalisation.
Mots-clés : Stéatohépatite non alcoolique – NASH-NAFLD
Introduction :
L’udwig et collaborateurs l’avaient décrite pour la première fois en 1980 en observant des lésions histologiques d’hépatite alcoolique chez une vingtaine de patients qui n’étaient pas des consommateurs d’alcool(1). Ils l’avaient baptisé stéatohépatite non alcoolique ou non alcoholic steatohepatitis (NASH). En fait, il s’agit d’une entité bien individualisée qui fait partie avec la stéatose pure, d’un groupe plus large d’affections plus connu sous l’acronyme NAFLD (Non alcoholic fatty liver disease). La NASH est caractérisée par la présence d’une stéatose hépatique supérieure à 5% associée à une inflammation qui peut évoluer vers la fibrose, la cirrhose et le carcinome hépatocellulaire (CHC). Elle met ainsi en jeu le pronostic hépatique et vital du patient NAFLD. Quant à la stéatose pure, celle-ci est caractérisée par la présence d’une stéatose sans inflammation, son pronostic est généralement moins péjoratif que celui de la NASH.
Épidémiologie :
La NAFLD est actuellement considérée comme un véritable problème de santé mondial. C’est l’une des premières causes d’hépatopathies chroniques dans le monde. Sa prévalence exacte n’est toujours pas bien précisée, elle varie selon la population d’étude et la méthode diagnostique utilisée. Selon les séries rapportées, sa prévalence dans la population générale se situe entre 10 et 50%(2). La prévalence de la NASH diffère également selon la population étudiée. Histologiquement prouvée, cette affection était retrouvée chez 43 à 55% des patients NAFLD avec cytolyse, chez 49% d’obèses morbides et chez 67% des cas d’hépatopathies chroniques incidentes(3). Dans la population générale, principalement chez les donneurs de foie, sa prévalence était estimée entre 3% et 16% en Europe, et entre 6 % et 15% aux USA(3). En Algérie, la prévalence de la NASH n’est pas connue. Dans un travail de thèse du docteur Chikhi, 1,2% des patients ayant eu un CHC, étaient liés à la NASH(4).
Physiopathologie :
La physiopathologie de la NASH n’est pas encore totalement élucidée. Néanmoins, plusieurs facteurs intervenant dans sa genèse ont été identifiés. Ces facteurs interagissent entre eux selon un modèle « multi Hit » proposé par Tilg et Moshen (5).
La prédisposition génétique et l’insulinorésistance favorisent la formation de la stéatose, définie par l’infiltration d’au moins 5% des hépatocytes par des vacuoles lipidiques constituées en majorité par les triglycérides. Cette stéatose est le point central unissant la NASH à la stéatose simple. Elle peut résulter de l’afflux important d’acides gras libres (AGL) vers le foie, d’un excès de synthèse intra hépatique (de novo), d’un défaut d’oxydation ou d’un défaut de sécrétion des very low density lipoproteins (VLDL). Cet excès d’acides gras libres et les produits issus de la lipotoxicité (diacylglycerol, céramides), l’insulinorésistance, la dysfonction du tissu adipeux périphérique, et enfin le microbiote intestinal via les endotoxines bactériennes, peuvent intervenir dans l’activation et la production de cytokines pro inflammatoires (6). C’est au cours de cette étape et avec le recrutement et l’activation de cellules immunitaires, que se passe la progression de la stéatose simple à la stéatohépatite.
L’inflammation est la principale caractéristique pathologique de la NASH. Néanmoins, environ 20% à 30% des stéatoses simples peuvent évoluer vers la NASH(5,7-10) et cette inflammation peut parfois précéder la stéatose dans certains cas(11) .
Diagnostic de la NASH :
Sur le plan clinique, le patient NAFLD est souvent asymptomatique ou peut parfois présenter des symptômes non spécifiques tels une asthénie et ou une pesanteur de l’hypochondre droit, son diagnostic est fréquemment posé fortuitement ou au décours d’une complication hépatique.
Le bilan hépatique peut être normal ou perturbé (cytolyse modérée, les phosphatases alcalines (PAL) et ou les gamma-glutamyl transférases (GGT) peuvent s’élever) aussi bien dans la NASH que dans la stéatose simple.
Le recours à la biopsie hépatique est à l’heure actuelle le seul moyen pour confirmer le diagnostic de la NASH. Cet examen invasif n’est pas dénué de désavantages : une mortalité estimée entre 0 à 3,3 décès pour 10.000 biopsies(12), des erreurs d’échantillonnage et une variabilité inter observateurs lors de son interprétation(13).
Les explorations morphologiques conventionnelles telles l’échographie, la tomodensitométrie et l’imagerie par résonnance magnétique (IRM) permettent seulement d’identifier la surcharge graisseuse (stéatose) mais sans pouvoir différencier la NASH de la stéatose simple. Ces dernières années, des méthodes non invasives ont été développées pour prédire la NASH. Il s’agit principalement de scores établis à partir de données cliniques et ou biologiques (tableau 1)(14).
Cependant, aucune de ces méthodes n’a pu remplacer la biopsie hépatique qui reste le gold standard du diagnostic.
Tableau 1 : Les principaux scores prédictifs de la NASH (14)
Il existe également des méthodes non invasives permettant le diagnostic indirect de la fibrose hépatique. Certaines de ces méthodes se basent sur des paramètres cliniques et biologiques qui sont regroupés dans le tableau 2, d’autres se basent sur un principe radiologique, celui de la vitesse de propagation des ondes de cisaillement élastiques à travers le tissu hépatique (élastographie) (Tableau 3).
Tableau 2 : Les principales investigations non invasives de la fibrose hépatique
Le fibroscan® :
Son principe repose sur l’estimation de la vitesse avec laquelle se propagent les ondes de cisaillement élastiques à travers le tissu hépatique. Il est disponible avec 3 types de sondes, Small (S) pour l’enfant, medium (M) et extra large (XL) pour l’adulte.
L’élastographie intégrée à l’échographie ou ARFI (acoustic radiation force impulse) (15)
Figure 2. Elastographe de marque siemens Acuson S2000.
L’élastographie par résonnance magnétique (ERM) Sa performance diagnostique pour la fibrose avancée est excellente (sensibilité 85 %, spécificité 92%).
Anatomopathologie :
Malgré son caractère invasif, la biopsie hépatique est à l’heure actuelle l’examen de référence pour le diagnostic de la NASH. Elle permet à la fois d’identifier les principales lésions histologiques de la NASH : une stéatose supérieure à 5% des hépatocytes à prédominance macro vacuolaire, des foyers nécrotico inflammatoires à cellules mononuclées ou à polynucléaires, une ballonisation des hépatocytes avec ou sans corps de Mallory et une fibrose peri-sinusoïdale, mais également d’évaluer la sévérité de l’atteinte. Le score SAF (Stéatosis activityfibrose) est le plus récent des scores histologiques disponibles pour la NASH. Il combine l’évaluation des trois principales lésions de la NASH, le degré de stéatose (S) , l’activité (A) selon la classification NAFLD Activity score (NAS) proposée par la NASH Clinical Researsh Network (NASH CRN), et l’inflammation et fibrose (F) selon Kleiner et al. (Tableau 3).
La combinaison d’une alimentation saine et d’une activité physique régulière constitue à l’heure actuelle la pierre angulaire de la prise en charge du patient NASH. Elle a pour objectif principal de faire baisser progressivement le poids du patient NASH en surpoids. En effet, des études ont montré qu’une modification du style de vie chez des patients NASH qui s’accompagnent d’une perte de poids modérée (entre 5 à 10%) était corrélée positivement à l’amélioration du score NAS et des lésions histologiques(15,16). Par contre, une baisse importante ou trop rapide du poids pourrait aggraver les lésions inflammatoires et la fibrose portale(17). Pour atteindre ces objectifs, Il est recommandé de suivre un régime hypocalorique (1.200 Kcal par jour initialement voire 600 à 800 Kcal par jour si les objectifs ne sont pas atteints) en réduisant l’apport quotidien en carbohydrates à moins de 100g, les lipides à moins de 10g et les protéines entre 45-100 g, et une activité physique régulière (150 à 200mn par semaine) d’intensité modérée (par exemple : marche rapide, vélo d’appartement, aérobic..)(18). Néanmoins, cette hygiène de vie doit être adaptée selon la présence d’un diabète sucré, de maladie cardiovasculaire et/ou de dyslipidémie
II. Correction des facteurs métaboliques associés à la NASH :
Le recours aux médicaments si échec des règles hygiéno-diététiques des différents facteurs métaboliques associés à la NASH (diabète, dyslipidémie et/ou de l’HTA), doit faire privilégier les moins hépatotoxiques.
III. Le traitement médicamenteux de la NASH : Il est réservé essentiellement aux formes de NASH progressive (fibrose en pont et cirrhose), les formes actives avec des lésions nécrotico inflammatoires importantes et enfin, les formes de NASH peu actives mais avec des facteurs de risque de fibrose progressive (tableau 4)(18).
Tableau 4.(NASH : non alcoholic steatohepatitis, ALAT : Alanine amino trans- ferase).
Dans le tableau 5, figurent les principaux médicaments utilisés et leurs effets sur la NASH, et dans le tableau 6 les médicaments qui sont en cours d’expérimentation. Néanmoins, aucun de ces médicaments n’a encore eu d’AMM pour le traitement de la NASH. La quête d’une molécule ou d’une combinaison de molécules efficaces à la fois sur la stéatose, l’inflammation et la fibrose, est toujours en cours. Cette difficulté est liée à la multiplicité et complexité des facteurs intervenant dans la physiopathologie de la NASH.
Option thérapeutique
Mécanisme
Effets sur la NASH
Les insulino-sensibilisateurs
Agissent contre l’insulinorésistance
– Metformine
Améliore les paramètres glucido lipidiques
mais pas d’effet sur les transaminases ni
l’histologie hépatique (20)
– Thiazolidinediones ou glitazones
Réduisent les ALAT, la stéatose, les lésions
(pioglitazone, rosiglitazone.)
nécrotico- inflammatoires mais pas la fibrose (21).
Nouveaux antidiabétiques :
– Les analogues GLP1
Incrétine intestinale améliore l’ho-
N’a pas d’AMM pour cette indication.
(Liraglutide)
méostasie glycémique
Une seule étude avec petit effectif
(52 patients) a démontré une disparition des
lésions histologiques de la NASH (22).
Clofibrate, atorvastatine
Correction des paramètres lipidiques
Pas d’effet démontré sur les lésions de
NASH
Acide ursodesoxycolique (AUDC)
Action anti TNFα, réduit l’insulinoré-
Résultats non concluants de la plupart des
sistance et possible action sur le stress
études contrôlées(18)
oxydatif
α Tocophérol (vitamine E)
Anti oxydant
Essai PIVENS : la vit E a amélioré la stéa-
tose, l’inflammation et la ballonisation dans
la NASH(21). Recommandée en cas de fibrose
avancée chez les non diabétiques(18).
Les produits amaigrissants
Perte de poids
Non approuvés par les sociétés savantes
(à base de plantes ou autres)
américaines et européennes
Tableau 5 : Tableau non exhaustif des médicaments utilisés dans la NASH(19) (NASH : non-alcoholic steatohepatitis ; ALAT : Alanine Amino Transférase).
Tableau 6 : Quelques molécules en cours d’expérimentation (23) (NASH, non-alcoholic steatohepatitis; PPAR, peroxisome proliferator-activated receptor FXR, farnesoid X receptor ; ALAT : Alanine Amino Transférase; ASBT: apical sodium dependent bile acid transporter; NAS : nafld activity score).
La chirurgie bariatrique :
Il s’agit d’une option réservée essentiellement aux patients ayant une obésité morbide après avoir épuisé toutes les autres ressources thérapeutiques en raison des complications éventuelles de cette chirurgie (sténose, ulcères, hernies, dumping syndrome, désordres nutritionnels…etc.) (24,25).
Aucun essai randomisé n’a étudié les effets de la chirurgie bariatrique sur la NAFLD.
Les données de la littérature à ce sujet sont issues principalement d’études rétrospectives et des séries de cas qui toutes, ont montré un bénéfice de la chirurgie bariatrique sur la baisse du poids, la résolution de la NASH et de la fibrose (26,27).
Dans une étude française sur 109 patients obèses avec NASH (prouvée histologiquement) ayant subi une chirurgie bariatrique entre 1999 et 2013 ; il y avait une résolution de la NASH chez 85% d’entre eux après une année post chirurgie. La fibrose hépatique était réduite chez 33,8% de ces patients (28).
L’endoscopie bariatrique :
Il s’agit de nouvelles techniques qui n’ont pas encore été bien évaluées dans la NASH.
La mise en place d’un ballon intra-gastrique chez les patients obèses, a permis une baisse du poids de 14,7 kg (méta-analyse de 15 études soit 3.608 patients)(29). Néanmoins, quelques effets secondaires ont été rapportés, des nausées-vomissements, érosions et ulcères, dégonflement et migration du ballon, perforation, déshydratation … etc.(19)
La stéatohépatite non alcoolique (NASH) fait partie du spectre large des stéatopathies métaboliques ou NAFLD (non alcoholic fatty liver disease), son principal risque est l’évolution vers la cirrhose et/ou le cancer hépato cellulaire (CHC). C’est une pathologie qui reste encore sous diagnostiquée en raison de l’absence de moyens diagnostiques non invasifs fiables qui pourraient un jour remplacer la biopsie hépatique pour confirmer son diagnostic. Le progrès enregistré dans la compréhension de sa physiopathologie multifactorielle et complexe, a permis de lancer plusieurs essais cliniques à la quête d’un traitement médicamenteux efficace.
Bibliographie
Ludwig J, Viggiano TR, McGill DB, et al. Nonalcoholic stea- tohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clinic proceedings. 1980;55(7):434-8.
Hashimoto E, Tokushige K, Ludwig J. Diagnosis and classi- fication of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis: Current concepts and remaining challenges. Hepatology research : the official journal of the Japan So- ciety of Hepatology. 2015;45(1):20-8.
Puoti C, Elmo MG, Ceccarelli D, et al. Liver steatosis: The new epidemic of the Third Millennium. Benign liver state or silent killer? European journal of internal medicine. 2017.
Chikhi y. Aspects anatomocliniques et évolutifs des carci- nomes hépatocellulaires [médecine]: Alger; 2016.
Tilg H, Moschen AR. Evolution of inflammation in nonal- coholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52(5):1836-46.
Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pa- thogenesis of non-alcoholic fatty liver disease (NAFLD). Meta- bolism: clinical and experimental. 2016.
Levene AP, Goldin RD. The epidemiology, pathogene- sis and histopathology of fatty liver disease. Histopathology. 2012;61(2):141-52.
Macaluso FS, Maida M, Petta S. Genetic background in nonalcoholic fatty liver disease: A comprehensive review. World journal of gastroenterology : WJG. 2015;21(39):11088-111.
Schattenberg JM, Schuppan D. Nonalcoholic steatohepatitis: the therapeutic challenge of a global epidemic. Current opinion in lipidology. 2011;22(6):479-88.
Zhang X-Q, Xu C-F, Yu C-H, et al. Role of endoplas- mic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World journal of gastroenterology : WJG. 2014;20(7):1768-76.
Liu W, Baker RD, Bhatia T, et al. Pathogenesis of nonal- coholic steatohepatitis. Cellular and molecular life sciences : CMLS. 2016;73(10):1969-87.<Ponction-Biopsie-Hepatique.pdf>.
Bedossa P. Current histological classification of NAFLD:strength and limitations. Hepatology international. 2013;7 Suppl 2:765-70.
Alkhouri N, Feldstein AE. Noninvasive Diagnosis of Nonal- coholic Fatty Liver Disease: Are We There Yet? Metabolism: clinical and experimental.
Promrat K, Kleiner DE, Niemeier HM, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51(1):121-9.
Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, et al. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroente- rology. 2015;149(2):367-78.e5; quiz e14-5.
Andersen T, Gluud C, Franzmann MB, et al. Hepatic effects of dietary weight loss in morbidly obese subjects. Journal of he- patology. 1991;12(2):224-9.
European Association for the Study of the Liver . Electronic address eee, European Association for the Study of D, Euro- pean Association for the Study of O. EASL-EASD-EASO Cli- nical Practice Guidelines for the management of non-alcoholic fatty liver disease. Journal of hepatology. 2016.
Issa D, Wattacheril J, Sanyal AJ. Treatment options for no- nalcoholic steatohepatitis – a safety evaluation. Expert opinion on drug safety. 2017;16(8):903-13.
Haukeland JW, Konopski Z, Eggesbo HB, et al. Metformin in patients with non-alcoholic fatty liver disease: a randomized, controlled trial. Scandinavian journal of gastroenterology. 2009;44(7):853-60.
Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. The New England journal of medicine. 2010;362(18):1675-85.
Armstrong MJ, Barton D, Gaunt P, et al. Liraglutide efficacy and action in non-alcoholic steatohepatitis (LEAN): study pro- tocol for a phase II multicentre, double-blinded, randomised, controlled trial. BMJ open. 2013;3(11):e003995.
Townsend SA, Newsome PN. Review article: new treat- ments in non-alcoholic fatty liver disease. Alimentary pharma- cology & therapeutics. 2017;46(5):494-507.
Lalor PF, Tucker ON, Szomstein S, et al. Complications after laparoscopic sleeve gastrectomy. Surgery for obesity and related diseases : official journal of the American Society for Bariatric Surgery. 2008;4(1):33-8.
Higa KD, Boone KB, Ho T. Complications of the lapa- roscopic Roux-en-Y gastric bypass: 1,040 patients–what have we learned? Obesity surgery. 2000;10(6):509-13.
Hafeez S, Ahmed MH. Bariatric surgery as potential treat- ment for nonalcoholic fatty liver disease: a future treatment by choice or by chance? Journal of obesity. 2013;2013:839275.
de Almeida SR, Rocha PR, Sanches MD, et al. Roux-en- Y gastric bypass improves the nonalcoholic steatohepatitis (NASH) of morbid obesity. Obesity surgery. 2006;16(3):270-8.
Lassailly G, Caiazzo R, Buob D, et al. Bariatric Surgery Re- duces Features of Nonalcoholic Steatohepatitis in Morbidly Obese Patients. Gastroenterology. 2015;149(2):379-88; quiz e15-6.
Imaz I, Martinez-Cervell C, Garcia-Alvarez EE, et al. Sa- fety and effectiveness of the intragastric balloon for obesity. A meta-analysis. Obesity surgery. 2008;18(7):841-6.
N. AFREDJ, Service d’Hépatologie, CHU Mustapha Bacha, Alger
Abstract : Autoimmune hepatitis (AIH) is a non-resolving disease that affects mainly women. It can be asymptomatic, discovered incidentally, or manifest as acute, fulminant or chronic hepatitis. Its diagnosis is based on the combination of elevated liver enzymes, hypergammaglobulinemia, elevated autoantibodies and interface hepatitis with resetting hepatocytes on liver biopsy. Although these hallmarks are highly suggestive, they are not specific to AIH. To overcome this issue, in 1999, the «International Autoimmune Hepatitis Group» defined a score system simplified in 2008, to facilitate the diagnosis and provide guidance in the treatment of AIH. Corticosteroids combined to azathioprine remain the standard treatment of AIH. In case of non-response or intolerance, other immunosuppressive drugs can be used. For patients with advanced cirrhosis or fulminant AIH not responding to treatment, a liver transplantation should be considered. An overall ten-year rate survival higher than 80 % can be expected.
Résumé : L’hépatite auto-immune (HAI) est une affection de cause non encore élucidée, nettement prédominante chez la femme. Sa traduction clinique est variable, pouvant être complètement asymptomatique de découverte fortuite, ou se manifester par une hépatite aiguë, fulminante ou chronique. Son diagnostic repose sur l’association d’une cytolyse hépatique, d’une hypergammaglobulinémie, d’une élévation des auto-anticorps et d’une hépatite d’interface associées parfois à un aspect en rosettes des hépatocytes à l’histologie. Tous ces éléments, bien que fortement suggestifs, ne sont pas spécifiques de l’HAI. Pour cela, un score a été élaboré par l’« International Autoimmune Hepatitis Group» en 1999, puis simplifié en 2008, afin de faciliter le diagnostic de l’HAI et de standardiser sa prise en charge. Le traitement de cette affection repose sur l’association corticoïdes et azathioprine. D’autres immunosuppresseurs peuvent être utilisés en cas de non-réponse ou d’intolérance à ce traitement. En cas de cirrhose avancée ou d’HAI fulminante ne répondant pas au traitement médical, la transplantation hépatique est à envisager. Elle permet d’obtenir un taux de survie à 10 ans supérieur à 80 %.
L’hépatite auto-immune (HAI) est une maladie inflammatoire chronique du foie, faisant partie du large spectre des hépatopathies dysimmunitaires. Elle est caractérisée par une nette prédominance féminine, une hypergammaglobulinémie, une association à des auto-anticorps (aAC), une hépatite d’interface à l’histologie et une bonne réponse au traitement immunosuppresseur[1,2,3]. L’association à l’HLA DR3 ou DR4 a été récemment intégrée aux critères de définition de l’HAI[3].
Décrite pour la première fois par Waldenström en 1951[4], cette affection a d’abord été appelée « hépatite lupoïde » en 1956 [5], du fait de son association à des anticorps antinucléaires, avant d’être distinguée du lupus érythémateux disséminé quelques années plus tard, et appelée hépatite auto-immune. Cette appellation a finalement été validée par le groupe international des HAI (AIHG) en 1993[6].
L’HAI est relativement rare. Sa prévalence est estimée entre 16 et 25 cas/100.000 habitants en Europe[3,7] et à 42,9 cas/100.000 habitants en Alaska[8]. Son incidence est de 0,8-1,9/100.000 habitants en Europe[9].
Elle atteint les deux sexes, avec une nette prédominance chez les femmes, les hommes ne représentant que 25-30 % de l’ensemble des patients[7]. Toutes les tranches d’âge peuvent être concernées. Il existe chez l’adulte deux pics d’incidence, à 30 ans et à 55 ans, notamment chez la femme ménopausée[10]. Chez les enfants, l’HAI de type I reste prédominante, 2/3 des cas, et touche surtout les enfants vers la puberté, alors que l’HAI de type 2 est plus fréquente chez le jeune enfant [11]. Les sujets âgés présentent en général une HAI de type 1, d’évolution chronique, volontiers découverte au stade de cirrhose[12]. La distribution de cette maladie est ubiquitaire, pouvant toucher de façon indiscriminée différentes régions et ethnies du globe, avec cependant des particularités cliniques et évolutives propres à chaque région. Ainsi, les formes ictériques seraient plus fréquentes en Alaska et les formes évoluées diagnostiquées au stade de cirrhose plus répandues chez les hispaniques et les afro- américains[7].
L’étiopathogénie de l’HAI n’est pas complètement élucidée. Plusieurs hypothèses ont été soulevées, incriminant des facteurs génétiques et environnementaux, notamment des agents infectieux tels que les virus de l’hépatite C (VHC), de l’hépatite E (VHE) et le cytomégalovirus (CMV), ou médicamenteux notamment les antibiotiques, les statines, les anti-TNF, l’acide tiénilique et la dihydralazine. Ces facteurs exogènes seraient responsables du déclenchement de la réaction auto-immune chez des patients génétiquement prédisposés[2,7,13,14]. Les antigènes (Ag) du soi partageraient des séquences antigéniques avec ces agents exogènes ; ainsi, après une première exposition aux Ag exogènes, le système immunitaire va réagir contre les Ag du soi, du fait de l’existence d’une homologie de séquence peptidique entre les deux types d’Ag, appelée mimétisme moléculaire, perpétuant ainsi les lésions inflammatoires et destructrices du parenchyme
hépatique[7]. À titre d’exemple, il existe un mimétisme moléculaire entre le cytochrome P450 2D6, le VHC et l’Herpès Simplex Virus (HSV), pouvant être responsable de la production des anti-LKM1[15].
Des cas familiaux d’HAI ont été rapportés. La prédisposition génétique est actuellement bien établie, notamment l’association avec les gènes du Système Majeur d’Histocompatibilité, situés sur le bras court du chromosome 6, en particulier ceux codant pour les Ag HLA DR3 et DR4[2,16]. Les molécules HLA jouent un rôle essentiel dans la présentation par les cellules présentatrices d’Ag, de peptides antigéniques aux lymphocytes T, à l’origine de l’initiation d’une réponse immunitaire adaptative. L’association de l’HAI à ces Ag HLA DR3/ DR4 est particulièrement retrouvée en Europe et en Amérique du Nord[2]. Elle est suffisamment robuste pour être intégrée dans les critères diagnostiques de l’AIHG modifiés en 1999[17]. D’autres variants génétiques, notamment les gènes CARD 10 et CTLA-4, confèreraient une susceptibilité à l’HAI de type 1[1,18,19]. Il existerait également une altération de l’activité des lymphocytes T régulateurs, plus souvent identifiée dans les formes pédiatriques, à l’origine de l’initiation et de la perpétuation de la destruction auto-immune des cellules hépatiques[20]. Les modèles animaux ont permis la réalisation d’avancées considérables dans le domaine de la pathogénie de l’HAI, en identifiant de nouvelles cibles antigéniques, ce qui permettra de mieux comprendre cette maladie et de développer de nouvelles armes thérapeutiques.
Manifestations cliniques et histoire naturelle de l’hépatite auto-immune :
L’HAI est caractérisée par une grande hétérogénéité clinique, allant de la forme asymptomatique à évolution indolente retrouvée dans 12-35 % des cas[7,21,22], à l’hépatite fulminante. Les formes aiguës sont retrouvées dans 25 % des cas[7,23]. Elles peuvent survenir sur un foie antérieurement sain, ou compliquer une hépatopathie chronique auto-immune.
Dans l’HAI aiguë, les auto-anticorps peuvent être absents, ce qui peut rendre le diagnostic difficile [23]. Les signes cliniques sont aspécifiques, à type d’asthénie, anorexie, nausées, vomissements, troubles du transit, douleur de l’hypochondre droit, ictère et parfois prurit dans les formes cholestatiques et arthralgies. Une aménorrhée est fréquemment retrouvée ; le rash cutané reste rare[7]. Le bilan biologique montre en général une cytolyse de gravité variable, pouvant dépasser les 50N et une élévation des GGT. Ces anomalies peuvent être associées ou non à une cholestase minime à modérée, à une hypergammaglobulinémie avec élévation sélective des immunoglobulines G (Ig G) et à une insuffisance hépatique[7,17]. Elle ne diffère aucunement des hépatites aiguës d’autres étiologies.
Le taux de gammaglobulines (G.glob) est normal dans un tiers des cas d’HAI aiguë, et le BAI négatif dans 9-17 % des cas, ce qui peut retarder le diagnostic[7,24]. Leur élévation en l’absence de cirrhose est un signe d’une grande valeur diagnostique dans l’HAI. C’est également un excellent marqueur de l’activité de la mala- die, que l’on peut utiliser pour le suivi sous traitement[2].
Les formes aiguës graves sont caractérisées par une baisse du facteur V<50 %, associée à une encéphalopathie hépatique dans les formes fulminantes. Il est à noter que l’activité biochimique n’est pas corrélée à la sévérité de l’atteinte histologique. Les transaminases peuvent baisser spontanément alors que l’activité histologique persiste ou s’aggrave[7].
Un tiers des patients présentent déjà une hépatopathie chronique au moment du diagnostic, ce qui alourdit le pronostic de cette maladie[22,25,26]. Les patients présentent souvent des signes aspécifiques tels qu’une asthénie, des arthralgies, parfois un ictère témoignant de la gravité de l’atteinte hépatique ou de l’association à une cholangite biliaire primitive (CBP) ou à une cholangite sclérosante primitive (CSP). L’examen clinique peut retrouver une hépatomégalie, une splénomégalie, des signes d’hypertension portale (HTP) et d’insuffisance hépatocellulaire au stade de cirrhose. Le bilan biologique retrouve, à des degrés variables, une cytolyse hépatique, une cholestase et une insuffisance hépatique, associées ou non à des signes d’hypersplénisme. Le taux de G.glob est élevé dans 85 % des cas[3]. Les examens d’imagerie médicale, notamment l’échographie-doppler hépatique, peuvent mettre en évidence une dysmorphie hépatique, associée à des signes radiologiques d’HTP. La cirrhose auto-immune peut être d’emblée révélée par une complication à type d’hémorragie digestive liée à l’HTP, de syndrome œdémato-ascitique ou d’encéphalopathie hépatique.
L’HAI évolue le plus souvent par poussées, entrecoupées de rémissions plus ou moins prolongées. En l’absence de traitement, elle peut évoluer vers la cirrhose, avec un risque de carcinome hépatocellulaire de 1,1 % par an, les facteurs de risque associés à la greffe néoplasique étant le sexe masculin et la cirrhose[27,28,29]. En l’absence de traitement, l’évolution de l’HAI peut être fatale, avec un taux de survie à 5 ans et 10 ans respectivement de 50 % et 10 %[30,31].
Quelle que soit la forme clinique, des manifestations extra-hépatiques auto-immunes (MEHAI) sont parfois retrouvées, en association avec l’HAI, pouvant parfois la révéler (Tableau 1). Ces MEHAI peuvent être présentes chez le patient ou les parents du premier degré. On estime à 20 % le taux de patients avec une ME- HAI concomitante ou consécutive à l’HAI[32]. Dans une étude ayant analysé 278 patients atteints d’HAI, 40% étaient porteurs d’une MAI. Les plus fréquentes étaient une thyroïdite auto-immune (10 %), un vitiligo, une polyarthrite rhumatoïde (2 %), un syndrome de Sjögren, une colite ulcéreuse (1,5 %) et une maladie cœliaque (1 %)[33].
Tableau 1 : Maladies auto-immunes associées à l’HAI
La poussée d’HAI peut survenir durant la grossesse, plus fréquemment après l’accouchement, du fait de l’état d’immunotolérance induit par la grossesse. Il peut s’agir d’une poussée inaugurale, mais le plus souvent, c’est une exacerbation dans le post-partum d’une HAI connue[34,35]. La morbidité fœtale peut être augmentée lorsque les femmes enceintes présentent une poussée durant la grossesse. Dans une étude du King’s College Hospital ayant inclus 81 grossesses chez 53 femmes at- teintes d’HAI, la morbidité fœtale était de 20 %, nécessitant une hospitalisation des nouveau-nés en unité de soins intensifs dans 11 % des cas. La mortalité infantile était plus élevée chez les patientes au stade de cirrhose. Dans cette étude, la morbidité maternelle était de 38%, à type de poussée d’HAI dans 33% des cas, avec décompensation d’une cirrhose dans 11 % des cas [36].
Des cas d’HAI ont été décrits suite à un traitement par interféron alpha pour hépatite virale C, survenant le plus souvent après éradication virale, particulièrement chez des patients ayant des Ac anti-LKM1 positifs avant l’initiation du traitement [37,38]. Un bilan d’auto-immunité doit donc être demandé avant d’entamer le traitement antiviral, et à chaque fois qu’une cytolyse non liée à une élévation de la charge virale C survient durant ou après le traitement. Dans ce cas, une biopsie hépatique peut être nécessaire, bien que les signes histologiques d’HAI soient parfois difficiles à distinguer des anomalies histologiques observées au cours de l’hépatite virale C.
Des cas d’HAI ont été rapportés dans les suites d’une transplantation hépatique (TH) pour hépatopathie d’une autre origine. Elle est appelée HAI de novo par la majorité des experts, hépatite à plasmocytes post-TH ou hépatite allo-immune post-TH par d’autres [39,40].
L’HAI peut survenir, dans 10-18 % des cas, dans un contexte de maladie héréditaire rare, la « Polyendocrinopathy Candidiasis Ectodermal Dystrophy » (APECED) qui est une maladie à transmission autosomale récessive liée à une mutation du gène régulateur de l’auto-immunité (AIRE) [41], associant une candidose cutanéo-muqueuse, une dystrophie ectodermique et une destruction auto- immune de différentes glandes de l’organisme [42,43]. L’HAI retrouvée dans ce contexte est caractérisée par la présence d’AC anti-LM (Liver Microsome) [43].
Diagnostic de l’HAI :
Devant des signes cliniques et biologiques évocateurs du diagnostic d’HAI, notamment un tableau d’hépatite aiguë ou chronique, avec une cytolyse marquée et une hyper G.globulinémie, un bilan d’auto-immunité doit être demandé. La positivité des aAC, portant sur les IgG et l’hépatite d’interface à l’histologie sont des piliers du diagnostic.
A. Auto-anticorps
Il s’agit essentiellement des Ac anti-nucléaires (AAN) présents dans 80 % des cas, des anti-muscles lisses (AML) présents dans 63 % des cas, de spécificité anti-actine et des AC anti-Liver Kidney Microsome 1 (anti-LKM1). L’association AAN et AML a une faible sensibilité (43%), mais une grande spécificité (99%) [44]. Les anti-LKM1 sont rarement positifs (3-38% des cas), mais ils sont hautement spécifiques [44]. Les AAN et les AML sont considérés positifs dans l’HAI, lorsqu’ils dépassent le seuil de dilution de 1/40 chez l’adulte et de 1/20 chez l’enfant. Le seuil de positivité des AC anti-LKM1 est de 1/10[2]. N’étant pas spécifiques de l’HAI, ces aAC peuvent être retrouvés dans l‘hépatite virale C, la CBP, la CSP, l’hépatite médicamenteuse immuno-allergique appelée DILI (Drug Induced Liver Injury) et la NASH (stéato-hépatite non alcoolique).
Lorsque ces aAC classiques sont négatifs, il faut procéder à la recherche d’autres aAC, moins spécifiques, notamment les AC anti-LiverCytosol1(anti-LC1), les Perinuclear Antineutrophil Cytoplasmic Antibodies (pANCA), souvent associés aux MICI et à la CSP et les anti-Soluble Liver Antigen/ Liver-Pancreas (anti-SLA/LP). Ces derniers, fréquemment associés au syndrome de Sjögren, sont caractérisés par une assez grande spécificité pour l’HAI (>90 %), mais une faible sensibilité, vu qu’ils sont détectables dans 20-50 % des cas seulement [45]. La recherche systématique de l’anti SLA-LP permettrait de réduire considérablement les cas d’hépatopathie cryptogénétique et d’HAI séronégatives. Ils sont prédictifs d’une forme sévère de la maladie et d’un pronostic péjoratif [45, 46, 47].
Les AC anti-actine ne sont pas toujours retrouvés chez les patients AML+. Les AC anti-α actine sont très spécifiques des HAI de type 1. Ils sont associés à une forme sévère de la maladie et à une faible réponse au traitement [48]. Les anti-LKM3 sont positifs dans 10 % des cas chez les patients HAI-2. Ils sont plus souvent associés à l’hépatite D [49]. Les anti-ASGPR (anti-Asia-loglycoprotein Receptor Antibody), seuls aAC spécifiques du foie, sont retrouvés dans 90 % des cas d’HAI. Ils sont associés à un fort taux de rechute [9].
Des AC anti-mitochondries (ACAM) de type M2 sont occasionnellement retrouvés au cours de l’HAI, dans 3% des cas environ, voire 34% selon les résultats d’une étude japonaise[50]. Leur présence, isolément, sans autre signe suggestif de CBP, n’implique pas forcément l’existence d’un chevauchement HAI-CBP ou le risque d’évolution vers un tel syndrome [51].
B. Classification des HAI
Les anti-LKM1 et les AAN-AML coexistent rarement chez le même patient (1-3 % des cas), ce qui a permis de classer les HAI en type 1 et type 2 [44].
L’HAI de type 1 représente 80-90 % des formes d’HAI, associée à l’HLA DR3, DR4 et DR13 [3]. Elle est caractérisée par la présence des AAN et/ou AML, parfois des pANCA.
Dans l’HAI de type 2, plus fréquente chez l’enfant et l’adulte jeune, associée à l’HLA DR3 et DR7, on retrouve l’AC anti-LKM1, anti-LC1, et rarement l’anti-LKM3[3]. Elle est souvent révélée par une hépatite aiguë, volontiers sévère, répondant mal au traitement, avec des rechutes fréquentes. Les pANCA sont retrouvés dans 50-90 % des HAI de type 2[9].
L’existence d’une HAI de type 3, caractérisée par la présence des AC anti-SLA/LP, est contestée, car considérée par certains experts comme une variante de l’HAI de type 1 [52].
C. Anatomo-pathologie
La ponction-biopsie hépatique (PBH) est un autre pilier du diagnostic, ayant pour objectifs de confirmer le diagnostic d’HAI et d’en évaluer la sévérité [17,53]. Du fait de la grande hétérogénéité et de l’absence de spécificité des anomalies histologiques, le fragment biopsique doit être confié à un pathologiste averti. L’étude anatomo-pathologique permet d’individualiser une hépatite d’interface, également appelée « peace-meal necrosis », caractérisée par un infiltrat inflammatoire mononuclée (avec ou sans plasmocytes portal et périportal), grignotant la lame bordante et s’étendant dans le lobule (Figures 2, 3). Ce peace-meal est d’autant plus important lorsque l’HAI est aiguë ou lorsqu’il s’agit d’une rechute. Il s’y associe une nécrose hépatocytaire, avec ou sans fibrose plus ou moins extensive, voire une cirrhose dans les formes évoluées. Les plasmocytes sont retrouvés dans 60-70% des cas[9]. Un aspect d’hépatocytes en « rosettes », caractérisé par une agrégation d’hépatocytes entourés de cellules inflammatoires est aussi fortement suggestif de l’HAI, de même que l’« emperipolèse » qui désigne le fait qu’une cellule géante phagocyte d’autres cellules, notamment des lymphocytes[9,54] (Figure 4). Aucune de ces lésions n’est spécifique de l’HAI, pouvant être retrouvées dans d’autres hépatopathies, notamment virales et médicamenteuses[3,6,7] ; néanmoins, l’HAI est dite typique en présence de l’association des 3 signes : l’hépatite d’interface, l’agencement des hépatocytes en rosettes et l’emperipolèse[55].
Figure 2 : (HEx20) Fibrose en ponts porto-portes avec une activité nécrotico-inflammatoire sévère
Figure 3 : (HEx20) : Infiltrat portal lympho-plasmocytaire et hépatite d’interfaceFigure 4 : (HEx20) : dégénérescence ballonisante des hépatocytes dont certains ont phagocyté un lymphocyte (emperipolèse) (Flèche) mieux visualisée à un plus fort grossissement (petite image en fenêtre)
Des signes cholestatiques peuvent également se voir à la PBH, notamment des altérations de l’épithélium biliaire ou une ductopénie, en dehors de tout syndrome de chevauchement, dans 10-15 % des cas[2]. D’autres anomalies peuvent être observées à l’étude histologique, notamment la présence de granulomes inflammatoires, d’une stéatose ou d’une stéato-hépatite.
Il existe des formes atypiques d’HAI, notamment les formes centrolobulaires, caractérisées par la prédominance des lésions inflammatoires dans la région centrolobulaire[56], qui posent le problème de diagnostic différentiel avec l’hépatite aiguë médicamenteuse immuno-allergique, et les formes à cellules géantes, fréquentes chez l’enfant[57].
En dehors du diagnostic, la biopsie hépatique trouve son intérêt dans l’évaluation histologique initiale de l’activité et de la fibrose hépatiques (gradées selon le score de Metavir, mais plus souvent selon le score de Knodell), et dans le suivi ultérieur du patient sous traitement immunosuppresseur. En effet, une biopsie réalisée 2 à 3 ans après l’obtention d’une rémission clinico-biologique complète est recommandée par la plupart des experts.
Elle intervient dans la décision de maintenir ou non le traitement. La persistance d’un infiltrat plasmocytaire ou d’une hépatite d’interface est prédictive de récidive à l’arrêt du traitement[53].
D. Scores diagnostiques
Du fait de la grande variabilité et l’absence de spécificité des signes cliniques, biologiques, immunologiques et histologiques, des scores diagnostiques ont été créés par l’IAHG en 1993[58], modifiés en 1999[17] puis simplifiés en 2008[55]. Ces scores ont pour but de standardiser la terminologie et les aspects diagnostiques et thérapeutiques de la maladie chez l’adulte (Tableaux 2, 3).
Paramètres
Valeur
Score
Commentaire
Sexeféminin
Oui
+2
<1.5
+2
PAL/ASAT ou ALAT
1.5-3
0
>3
-2
>2N
+3
Ig G
1.5-2N 1-1.5N
+2 +1
<1N
0
>1/80
+3
Auto-anticorps
1/80
+2
Chez les enfants aAC, même à des
(AAN, AML, ou anti-LKM1)
1/40
+1
titres faibles, devraient être scorés +1
<1/40
0
Marqueurs de l’hépatite virale
Positifs Négatifs
-3 +3
Hépatites virales A, B et C
Antécédent de prise médicamen-
Présents
-4
Prise récente de médicaments à hépa-
teuse
Absents
+1
totoxicité avérée ou suspectée
Consommationmoyenned’alcool<25g/J>25 g/J
+2 -2
Histologie
Hépatite d’interface
+3
Infiltrat lympho-plasmo cytaire Hépatocytes en rosettesAucundecessignesAnomalies de l’épithélium
+1 +1 -5 -3
Altérations de l’épithélium biliaire spécifiques de la CBP/CSP Anomalies suggestives d’une étiologie
biliaire
Autres anomalies
-3
Autremaladieauto-immunechez le patient ou parent du 1er degré
+2
Séropositivité des autres aAC si ANA, AML, LKM1 négatifs
+2
pANCA, anti-LC1, anti SLA-LP, anti-ASGPR
HLA DR3 ou DR4
+1
Réponse au traitement ComplèteRechute
+2 +3
Score pré-thérapeutique
>15
HAI certaine
10-15
HAI probable
Score post-thérapeutique
>17
HAI certaine
12-17
HAI probable
Tableau 2 : Score modifié de l’IAHG 1999[2] ALAT : alanine aminotransférases ; ASAT : aspartate aminotransférases ; PAL : phosphatases alcalines ; aAC : auto-anticorps AAN : anticorps anti-nucléaires ; AML : anticorps anti-muscle lisse ; LKM : Liver Kidney Microsome ; Anti- SLA : AC anti-Soluble Liver Antigen; Anti-LC1 : anticorps anti-Liver Cytosol 1; ASGPR : récepteur de l’asialoglycoprotein pANCA : anticorps périnucléaires d’anti-neutrophile
Paramètres
Valeurs
Points
AAN ou AML
≥1/40 ≥1/80
1 2
Anti-LKM*
≥1/40
2
IgG
>N
1
>1.1N
2
Histologie hépatique
– Compatible avec le diagnostic d’HAI – Typique d’HAI
Tableau 3 : Score simplifié de l’HAI (IAHG 2008) [2] *maximum de points pour les auto-AC : 2 ; Score = 6 HAI probable, Score ≥7 HAI certaine
E. Diagnostic différentiel
Le problème du diagnostic de l’HAI se pose surtout lorsque les critères diagnostiques de l’HAI ne sont pas tous retrouvés et les scores insuffisants (indiquant une HAI probable). Dans ce cas, il est nécessaire d’éliminer, selon le contexte clinique et biologique, toutes les causes d’hépatopathie aiguë ou chronique, notamment :
Une hépatite entrant dans le cadre d’une maladie de système, comme le Lupus Erythémateux systémique (LES),
Une hépatite virale, par une sérologie virale complète (Ag Hbs, Ac anti-Hbc, Ac anti-HCV, IgM anti-HVA, IgM anti-HVE, ARN VHC ou ADN VHB),
Une hépatite toxique ou médicamenteuse, notamment l’hépatite aiguë immuno-allergique, associant nécrose centrolobulaire et positivité des AAN, entrant dans le cadre de la « Drug Induced Liver Injury » (DILI). Cette entité est difficile à distinguer d’une authentique HAI déclenchée par une prise médicamenteuse. La biopsie hépatique est la clé du diagnostic. Un travail récent a comparé les résultats de la PBH chez des patients ayant une DILI par rapport à ceux ayant une HAI idiopa- thique ; l’infiltrat portal de polynucléaires neutrophiles (PNN) et la cholestase intrahépatocytaire étaient prévalents dans la DILI (P < 0.02). La combinaison d’une inflammation portale, avec fibrose, PNN, plasmocytes et cholestase intrahépatocytaire serait, selon ces auteurs, prédictifs de DILI avec une précision diagnostique de 0,91 (AUROC 0,91)[63]. En pratique, il est difficile de distinguer ces 2 entités, et c’est le critère évolutif, à savoir la survenue d’une rechute après traitement immunosup- presseur, qui permet de trancher en faveur de l’HAI[3],
Une maladie de Wilson dans sa forme purement hépatique ; l’insuffisance hépatique et la cholestase peuvent être responsables d’une perturbation du bilan cuprique, et les auto-AC peuvent parfois être présents chez les patients atteints d’une maladie de Wilson,
Les autres hépatopathies dysimmunitaires, notamment une CBP ou une CSP, en cas de cholestase associée. Ces 2 affections peuvent être associées à l’HAI en cas de syndrome de chevauchement. Un dosage des ACAM et une bili-IRM doivent être réalisés pour rechercher un syndrome de chevauchement, particulièrement en
présence d’une anomalie de l’épithélium biliaire à l’histologie, de cholestase marquée ou de non-réponse aux corticoïdes[64]. Cependant, les ACAM peuvent être positifs au cours de l’HAI, et 10 à 15 % des patients avec cirrhose auto-immune peuvent présenter des rigidités des voies biliaires intrahépatiques, non liées à une CSP[65].
L’altération de l’épithélium biliaire, de même que la ductopénie retrouvée au cours de la CBP, peuvent aussi être observées dans 5 à 11 % des cas à la biopsie hépatique[64]. Ces formes cholestatiques d’HAI sont difficiles à distinguer de la CBP séronégative, de la CSP des petits canaux biliaires ou des syndromes de chevauchement,
Les atteintes hépatiques liées à la maladie cœliaque (MC) doivent aussi être éliminées, vu que les aAC peuvent se voir, à des titres faibles, au cours de la MC. En cas d’hépatite fulminante, il peut être difficile de faire le diagnostic d’HAI, surtout en l’absence d’une hyper G.globulinémie, d’auto-Ac ou de pathologie auto-immune associée[9]. Dans ce cas, il faut éliminer toutes les causes d’hépatite fulminante. Le recours au test thérapeutique aux corticoïdes est parfois nécessaire en pratique, après avoir écarté une cause virale.
Un algorithme a été proposé par Czaja pour le diagnostic de l’HAI, selon les aAC présents chez le patient et la biopsie hépatique[9] (Figure 1).
Figure 1 : Algorithme diagnostique devant une hépatite aigue ou chronique de cause inconnue [9] AAN : anticorps anti-nucléaires ; AML : anticorps anti- muscle lisse ; TG : transglutaminase ; AE : anti-endomysium ; LKM : Liver Kidney Microsome ; Anti-SLA : AC anti-Soluble Liver Antigen; Anti-LC1 : anticorps anti-Liver Cytosol 1; ASGPR : récepteur de l’asialoglycoprotein; pANCA : anticorps périnucléaires d’anti-neutrophile; CSS : coefficient de saturation de la sidérophiline; DILI : Drug Induced Liver Injury., NASH : Non Alcoholic Steato-Hepatitis
Traitement de l’HAI :
Le traitement de l’HAI repose essentiellement sur les corticoïdes (CTC) et les immunosuppresseurs. Selon les recommandations de l’EASL, le traitement est indiqué en cas d’HAI active et la décision de traiter doit être contrebalancée par le risque d’effets secondaires, particulièrement chez le sujet âgé[3]. Le bénéfice du traitement dans les formes minimes asymptomatiques n’est pas certain ; la survie à 10 ans dans ce cas-là, sans traitement, étant de 67-90 % [66,67]. Les patients présentant une HAI quiescente doivent donc bénéficier d’une surveillance rapprochée tous les 3-6 mois. La cirrhose, est une indication de traitement qu’elle soit active ou non[3].
Traitement conventionnel :
1. Patient naïf
L’objectif du traitement est l’obtention d’une réponse complète, définie par la normalisation des transaminases, des G.globulines, du taux d’IgG et des lésions histologiques[3,68]. En pratique, la réponse complète ne peut être obtenue chez tous les patients. Le traitement aura pour but, dans ce cas, de réduire au maximum les anomalies biochimiques avec un minimum d’effets secondaires[3].
Induction de la rémission :
Selon les recommandations des sociétés savantes (AASLD[53], BSG[69],EASL [3]), le traitement de référence repose sur la bithérapie associant les corticoïdes (CTC) (prednisone ou prednisolone) et l’azathioprine (AZT). L’AASLD recommandait, en 2010, de commencer par 30 mg/j de CTC associés à 50 mg/j d’AZT, avec réduction progressive des doses jusqu’à 4 semaines. Par la suite, les CTC seront maintenus à 10 mg/j et l’AZT à 50 mg/j jusqu’à la régression des anomalies cliniques, biologiques et histologiques[53] (Tableau 4).
Selon les recommandations européennes de 2015, l’AZT doit être prescrite à la dose de 1-2 mg/Kg/j, en commençant par 50 mg/j associée à 0.5 -1 mg/Kg/j de CTC. L’AZT doit être débutée à partir de la 3ème semaine, après amélioration des anomalies biologiques, pour éviter l’hépatotoxicité de l’AZT[3]. La réduction des CTC selon ce protocole est plus progressive, étendue sur 9 semaines au lieu de 4. Lorsque les transaminases reviennent à des valeurs normales, la dose peut être réduite à 7.5 mg/j. À partir du 3ème mois de traitement, si le bilan est toujours normal, la dose de CTC peut être réduite à 5 mg/j (Tableau 5).
D’autres protocoles d’induction de la rémission ont été proposés, notamment la majoration des doses de CTC à 1-2 mg/Kg/J en association avec l’AZT, ce qui a permis d’abréger le délai de rémission biologique chez des patients non cirrhotiques[70].
En cas de contre-indication ou d’intolérance à l’AZT (leucopénie < 2500/mm3, plaquettes <50.000/mm3, déficit en Thiopurine Methyltransferase (TPMT), néoplasie), un autre schéma thérapeutique est préconisé, basé sur les CTC à forte dose en monothérapie, avec réduction progressive de la dose, pour garder 20 mg au long cours en traitement d’entretien (Tableau 4).
Bithérapie
Monothérapie
Semaine
Corticoïdes (mg)
Azathioprine (mg)
Corticoïdes (mg)
1
30
50*
60
2
20
50*
40
3
15
50*
30
4
15
50*
20
Traitement d’entretien
10
50*
20
Tableau 4 : Protocoles thérapeutiques dans l’hépatite auto-immune chez l’adulte selon les recommandations de l’AASLD (2010)[Z8] *Dose d’azathioprine préférée par les experts Européens /1.2 mg/kg
Semaine
Prednisolone (mg/J)
Azathioprine (mg/J)
1
60
2
40
3
40
50
4
30
50
5
25
100
6
20
100
7-8
15
100
8-9
12.5
100
>10
10
100
Tableau 5 : Schéma thérapeutique de l’HAI chez l’adulte, selon l’EASL 2015[2]
Maintien de la rémission :
Lorsque la rémission est obtenue, elle sera maintenue par l’AZT à 2 mg/Kg/j ou les CTC à faible dose (dose minimale efficace pour maintenir des transaminases normales), selon le schéma utilisé. La monothérapie aux CTC est préconisée chez les sujets jeunes en âge de procréer et les patients exposés au risque néoplasique. Les patients doivent être surveillés à vie, tous les 3-6 mois,
pour dépister précocement les rechutes et les complications du traitement immunosuppresseur, notamment la cytopénie et le risque de néoplasie [3].
Durée du traitement :
Selon les recommandations européennes (EASL), le traitement doit être maintenu au moins 24 mois après normalisation du bilan biologique (transaminases et IgG). À ce moment-là, une biopsie hépatique devra être réalisée, particulièrement dans les formes histologiquement sévères au départ, avant de décider d’arrêter le traitement [2,3]. La régression des anomalies histologiques est appréciée au mieux par la biopsie hépatique, les examens non invasifs n’étant pas validés pour cette indication. En effet, le résultat du Fibroscan® peut être fortement impacté par l’inflammation hépatique. Il peut trouver sa place dans le cadre du suivi de l’HAI[3]. Le dernier sera interrompu progressivement, en l’absence d’activité histologique. Si celle-ci persiste (score d’Ishak
>3), le risque de rechute est quasi certain (≥80%) ; le traitement d’entretien doit être maintenu au long cours.
En l’absence de cirrhose et en cas de rémission complète, le traitement doit être arrêté. Cependant, certains auteurs optent pour le maintien du traitement au long cours, malgré la réponse complète, vu la grande fréquence des rechutes [71]. D’un autre côté, on estime à 20% la proportion de patients qui n’auraient jamais présenté de rechute à l’arrêt du traitement et que l’on aura exposés inutilement aux effets secondaires du traitement CTC et immunosuppresseur, en particulier le risque de néoplasie maligne [68].
– Résultats :
Le traitement immunosuppresseur actuellement disponible permet d’obtenir une rémission clinique et biochimique dans la grande majorité des cas (>80%) [71,69], bien que la rémission complète ne soit obtenue que dans 25 % des cas environ [2,3]. La survie à 5 ans des patients traités pour une HAI est actuellement ≥90 % [72].
Les résultats de la corticothérapie seule et de l’association CTC-AZT sont équivalents, en termes de réponses clinico-biologique et histologique [69]. Le bénéfice du traitement CTC dans l’HAI est connu depuis fort longtemps. Il a été démontré, par d’anciennes études contrôlées contre placebo, que les CTC pouvaient entrainer une réduction de la mortalité précoce de 56% à 14% [41]. L’association CTC-AZT n’a pas montré de supériorité en termes de survie, comparativement à la monothérapie CTC à forte dose [42], mais elle est préférée en raison de sa meilleure tolérance par rapport à la corticothérapie à forte dose en monothérapie.
La réponse clinico-biologique est en général rapide. Les facteurs agissant sur la rapidité de cette réponse au traitement ont été analysés chez 146 patients porteurs d’une HAI de type 1 ; cette rapidité était associée à un âge avancé >60 ans et à l’HLA DRB1 *04. En effet, dans cette étude, 94 % des patients âgés de plus de 60 ans avaient répondu en moins de 24 mois, vs 64 % des patients âgés de moins de 40 ans[71].
Une réponse histologique complète, avec restitution adintegrum est rare, surtout en cas de fibrose avancée, mais elle reste possible. Des cas de régression de la cirrhose ont même été rapportés[73]. Cette réponse histologique complète a été obtenue dans seulement 22 % des cas, après 56 mois de traitement chez 83 patients non cirrhotiques traités[74]. L’amélioration des lésions histologiques (disparition des signes d’activité avec ou sans amélioration de la fibrose) est par contre fréquente, elle survient dans 75 % des cas 3 à 8 mois après la réponse biologique (en moyenne 18 mois de traitement), et le traitement doit être maintenu jusque-là [75,76]. Dans une étude ayant analysé les résultats de toutes les séries publiées entre 1972 et 2013 concernant les HAI traitées, le taux d’amélioration de la fibrose hépatique était de 53-57 % [73]. La décision d’arrêter le traitement chez des patients cirrhotiques, qui n’ont pas présenté de réponse histologique, est laissée au bon jugement du praticien qui pourrait l’envisager en cas de stabilité clinique et biologique au-delà de 12 mois [69]. S’acharner à vouloir obtenir une réponse histologique peut exposer le patient au risque de toxicité médicamenteuse, notamment osseuse et endocrinienne.
Parmi les répondeurs, 28 à 87 % des patients vont présenter une rechute, définie par une élévation des ALAT>3N et/ou une élévation des IgG, après exclusion des autres causes d’élévation des transaminases[3,69]. La fréquence des rechutes souligne l’intérêt de suivre les malades régulièrement en consultation, et de façon rapprochée, même après l’obtention de la rémission. Ce taux est moindre, estimé à 28 %, en cas de normalisation des lésions histologiques lors du traitement de la première poussée[74]. Dans ce cas, 36 % des patients vont rester en rémission au-delà de 5 ans [77]. Les facteurs de risque de rechute sont la réponse lente au traitement d’induction de la rémission, la persistance de l’élévation des ALAT et/ou des G.Glob, la persistance de l’activité nécrotico-inflammatoire à la biopsie hépatique et la durée insuffisante du traitement [2]. La rechute survient, le plus souvent, dans les 12 mois suivant l’interruption du traitement, mais elle peut survenir bien au-delà de 20 ans de rémission [69]. En cas de rechute après l’arrêt du traitement, le même schéma thérapeutique peut être réinitialisé, avec augmentation de la dose d’AZT à 2 mg/Kg/j, avant l’arrêt des CTC. La réponse biochimique est obtenue dans plus de 90 % des cas après 4 mois de traitement et la réponse histologique, moins fréquente, est obtenue dans 59 % des cas[78]. L’évolution est en général favorable, à condition de débuter le traitement précocement. En cas de rechute, le traitement d’entretien au long cours est indiscutable.
Une intolérance au traitement corticoïde est observée dans 12 à 29 % des cas, particulièrement en présence d’une cirrhose[69]. Les effets secondaires les plus fréquemment notés sont l’obésité, l’ostéoporose, l’HTA et le diabète. Ce dernier a été rapporté chez 15-20 % des patients sous CTC au long cours. D’autres effets indésirables, tels que l’obésité cushingoïde, les troubles psychiatriques et la cataracte ont également été notés. L’os- téonécrose aseptique de la tête fémorale et la méningite sont plus rares. Des taux élevés de complications, atteignant 80 %, ont été notés chez les patients traités par CTC en monothérapie pendant plus de 2 ans, entraînant l’arrêt du traitement dans moins de 30 % des cas[79]. La bithérapie CTC-AZT, par contre, est associée à moins de complications induites par les CTC[69]. Il est recommandé de rechercher un diabète ou une HTA et de mesurer la densité osseuse avant de commencer un traitement CTC au long cours. Une supplémentation systématique par de la vitamine D et du calcium est également recommandée[3]. En cas d’intolérance ou de contre-indication aux CTC, il est recommandé d’associer le budésonide à l’AZT en l’absence de cirrhose[3,69].
Les effets secondaires de l’AZT (toxicité hépatique, pancréatique et médullaire) nécessitent une réduction de dose dans 5-10 % des cas, surtout chez les patients cirrhotiques. Le risque de myélosuppression est estimé à 6 % et celui de néoplasie maligne extra-hépatique à 2,7 cas par an, chez les patients recevant de l’AZT à 2 mg/ Kg/j[80,81]. Chez les patients intolérants à l’AZT, le métabolite de l’AZT, la 6mercaptopurine (6-MP) 1.5 mg/Kg/j, peut être prescrit, avec des résultats peu probants[82].
Le MMF est une bonne alternative à l’AZT dans ce cas, de même qu’une corticothérapie en monothérapie au long cours, en évaluant au préalable le risque d’effets secondaires [3].
1. Échec du traitement
La réponse partielle est définie par l’amélioration mais pas la disparition des anomalies cliniques, biologiques et histologiques. Sa fréquence est estimée à 14 %. Le cas le plus récurrent est la normalisation du bilan biologique, avec persistance de l’hépatite d’interface à la PBH de contrôle. Selon certains auteurs, on parle de réponse partielle si les transaminases baissent (<2N) sans se normaliser. L’alternative thérapeutique dans ce cas dépend du traitement initial [2,9] :
Chez les patients traités par Budésonide-AZT, il faudra remplacer le Budésonide par la prednisolone,
En cas d’association prednisolone-AZT, il faudra augmenter la dose d’AZT à 2 mg/Kg/j, en maintenant 5-10 mg/j de CTC,
Traitement non conventionnel.
Le but de ce traitement ne sera plus d’induire une rémission complète, mais de réduire l’activité inflammatoire et d’éviter la progression de la maladie, au prix d’une moindre toxicité des médicaments.
La non-réponse est définie par l’absence d’une réduction de plus de 25 % de – l’hypertransaminasémie à 2 semaines de traitement [2]. Elle est rare et retrouvée dans seulement 7 % des cas [83]. Des facteurs associés à une faible réponse au traitement ont été identifiés. Il s’agit essentiellement de l’âge jeune, la forme aiguë, l’hyperbilirubinémie sévère, le MELD score avancé et le HLA DRB1*03 [83,84]. Avant de rechercher des causes de non-réponse, il faut toujours commencer par évaluer la compliance au traitement des patients et éliminer toute autre cause de perturbation du bilan hépatique. Un syndrome de chevauchement est une cause fréquente de non-réponse au traitement. Une altération du métabolisme de l’AZT doit aussi être envisagée. Dans les centres spécialisés, il peut être utile de doser la concentration sanguine de prednisolone et/ou d’AZT et des métabolites actifs des TGN, avant d’envisager un changement de traitement [2]. Cependant, le bénéfice du dosage du taux de 6-Thioguanine pour déterminer la dose efficace d’AZT au cours de l’HAI est contesté [85].
En cas de non-réponse effective au traitement, une PBH doit être réalisée avant d’envisager une alternative thérapeutique. En l’absence de formes graves nécessitant le recours à la transplantation hépatique, différents schémas sont proposés :
Reprendre les CTC à 30 mg/j en association avec l’AZT à forte dose 150 mg/j, avec prolongement de la durée du traitement à 1 mois, puis diminution progressive de la dose par paliers de 10 mg de CTC et 50 mg d’AZT par mois. Garder un traitement d’entretien par CTC 10 mg/j et AZT 50 mg/j [69],
Reprendre les CTC à 60 mg/j associés à l’AZT 2 mg/Kg/j,
Si le patient est intolérant à l’AZT, commencer par
60 mg/j de CTC, avec diminution progressive de 10 mg chaque mois. Garder un traitement d’entretien par CTC 20 mg/j [69],
Traitement alternatif par les inhibiteurs de la calcineurine : ciclosporine et tacrolimus qui permettent de casser la poussée d’HAI dans plus de 80 % des cas (voir Traitements non conventionnels).
Tous ces traitements peuvent induire une réponse biochimique dans plus de 70 % des cas, mais la réponse histologique est faible, 14 à 20 %. Le traitement d’entretien est le plus souvent maintenu à vie, en dépit des effets secondaires. Malgré le traitement, le risque de progression des lésions vers une cirrhose reste très élevé, pouvant atteindre 82 % [83], et le taux de survie des patients est de 41 % à 5 ans [69]. La progression de la cirrhose est associée à la présence d’une nécrose lobulaire étendue avant traitement, à sa persistance durant le traitement, à une réponse lente aux corticoïdes et à la survenue de rechutes durant le traitement [69].
Traitements non conventionnels La ciclosporine, le tacrolimus, le mycophénolate mofétil et le budésonide, longtemps utilisés dans les formes réfractaires aux CTC, les cas d’intolérance ou de contre-indication des CTC, sont actuellement en cours d’évaluation en tant qu’alternative thérapeutique aux CTC et/ou AZT, chez des patients naïfs. Ces molécules ont été qualifiées de « Non-standard frontline therapies » pour cette indication. Elles sont actuellement utilisées en « off-label » dans l’HAI [69].
Budésonide Cette molécule est caractérisée par un effet de premier passage hépatique de 90 %. Elle ne doit pas être utilisée chez le patient cirrhotique, du fait de la réduction du métabolisme hépatique et de la présence de shunts péri-hépatiques, exposant le patient au risque d’effets secondaires inattendus [69]. De plus, des cas de thrombose porte ont été rapportés chez des porteurs d’une CBP stade IV et traités par le Budésonide associé à l’AUDC[86]. Ce traitement a été évalué dans l’induction de la rémission chez des patients porteurs d’une HAI, sans évidence de cirrhose. Il s’agit d’une étude multicentrique, prospective, contrôlée, randomisée et en double aveugle, ayant comparé 40 mg de prednisone + AZT 1-2 mg/Kg/j (Groupe 1) à l’association Budésonide 6-9 mg/j + AZT 1-2 mg/Kg/j (Groupe 2). Le but du traitement était d’obtenir une normalisation des transaminases à 6 mois. L’objectif primaire a été atteint dans 47 % des cas dans le Groupe 2 versus 18.4 % dans le Groupe 1 (p<0.001).
Une réponse biologique complète a été notée dans 60 % des cas dans le Groupe 2 versus 38.8 % dans le Groupe 1 (p=0.001). Les effets secondaires des stéroïdes étaient significativement moins fréquents dans le Groupe 2, 28 % versus 53.4 % dans le Groupe 1[87]. Dans cette étude, les résultats du Budésonide à long terme, particulièrement sur les lésions histologiques, ne sont pas rapportés.Le Budésonide, en association avec l’AZT 1-2 mg/Kg/j, est actuellement indiqué dans le traitement de la poussée modérée, avec fibrose et activité peu sévères, à raison de 9 mg/j en 3 prises, chez un patient susceptible de développer des effets secondaires des CTC sous traitement, en particulier chez l’enfant et l’adolescent. La dose sera réduite à 6 mg, puis 3 mg au long cours pour maintenir la rémission[2,9].
Ciclosporine Elle a été utilisée dans les formes réfractaires aux corticoïdes, sur un nombre réduit de patients, à la dose de 2-5 mg/Kg/j, avec des résultats probants en termes de réponse biochimique et de tolérance (>80 % de réponse biochimique) [88]. L’objectif de ciclosporinémie à atteindre est de 100-300 ng/ml. La ciclosporine a été évaluée chez 40 patients naïfs (en première intention), dans une étude randomisée contre prednisolone + AZT 2mg/Kg/j ; le taux de réponse biochimique complète était équivalent dans les 2 groupes de patients à 2 ans de traitement (47 % vs 50 %, p=1). Un seul cas d’effet secondaire grave avec décès du patient a été noté dans le bras CTC [89]. La ciclosporine semble efficace pour induire la rémission, mais l’effectif des études publiées est faible, et la réponse histologique n’a pas été évaluée. Son efficacité doit être contrebalancée avec son coût élevé et sa toxicité non négligeable.
Tacrolimus Il a été utilisé à la dose de 1-6 mg/j chez des patients réfractaires ou intolérants aux CTC avec des résultats similaires à ceux de la ciclosporine [90]. Le taux cible de Tacrolémie doit être à 3 ng/ml (1.7-10.7) [69]. Le tacrolimus a également été utilisé en première intention dans une petite série à faible effectif où la réponse biochimique a été obtenue dans 80 % des cas [91].
Mycophénolate Mofétil (MMF)Le MMF est un puissant inhibiteur sélectif de l’inosine monophosphate et de la prolifération lymphocytaire. Cette molécule a été utilisée en première intention, à la dose de 1.5-2 mg/Kg/j en association avec la prednisolone, durant 26 mois (3-92), chez 59 patients naïfs, pour maintenir la rémission. Le taux de réponse biochimique était de 88 % [92]. Néanmoins, le coût élevé de cette molécule en limite l’indication en première intention, vu la nécessité de traiter les HAI au long cours, parfois à vie. Le MMF a surtout été utilisé en deuxième intention, en cas d’intolérance ou de résultats insuffisants de l’AZT. Dans une étude rétrospective sur 21 patients traités par MMF pour une HAI réfractaire (N=12) ou intolérance au traitement (N=9), 20 ont présenté une réponse biochimique (95 %), sans réponse histologique. La dose de CTC a été réduite chez tous les patients traités par MMF (18.9 mg/j à 7.8 mg/j (P=0.01))[93].
Le MMF peut être utilisé en cas d’intolérance à l’AZT à la dose de 2 g/j[2]. Le risque de leucopénie sous traitement est important, et cette molécule ne doit pas être utilisée chez la femme enceinte, en raison du risque de malformations fœtales.
• Autres traitements
Le cyclophosphamide, le méthotrexate, l’infliximab, le rituximab ou le sirolimus ont été administrés dans quelques cas anecdotiques d’HAI réfractaires aux thérapeutiques usuelles[3]. Le rituximab, en association avec l’AZT, a été utilisé chez 6 patients qui ont été suivis pendant 72 semaines, après avoir reçu 2 injections de rituximab à 15 jours d’intervalle [bis] ; une nette réduction du taux de transaminases (90.0±23.3 U/L versus 31.3±4.2 U/L ; P=0.03) et une normalisation du taux d’IgG (16.4±2.0 g/L versus 11.5±1.1 g/L ; P=0.056) ont été notées chez ces patients, de même qu’une amélioration du score histologique à la biopsie hépatique. Le traitement a été bien toléré. Il n’y a pas, à l’heure actuelle, de recommandations concernant la place du rituximab et des autres traitements similaires dans l’HAI en pratique courante[2].
– Une vaccination contre l’hépatite A et B est recommandée par le consensus Européen[3].
Traitement des formes particulières :
• HAI aiguë grave/fulminante
Ces patients doivent être traités par de fortes doses de corticoïdes >1 mg/Kg/j, par voie intraveineuse. L’efficacité des CTC chez ces patients est controversée, vu que pour certains auteurs, elle ne modifie pas le pronostic. De plus, les critères de gravité de l’HAI fulminante, faisant indiquer la TH d’emblée, n’ont pas été bien définis. Le risque de sepsis chez ces patients n’est pas négligeable, justifiant l’utilisation prophylactique d’antibiotiques et d’antifongiques [94]. Selon les dernières recommandations de l’EASL, l’absence de réponse au 7ème jour de traitement CTC, particulièrement dans Les formes cholestatiques ou en cas d’aggravation du MELD score, doit faire envisager la TH en urgence [2]. La mortalité en cas de forme fulminante est de 19-45 %, avec un besoin de TH dans 10-80 % des cas.
• Déficit en TPMT
Ces patients doivent être traités par des CTC en monothérapie ou associés aux MMF [2,3].
• Femme enceinte
Le MMF doit être arrêté avant la conception, en raison du risque de tératogénicité. Par contre, l’azathioprine, les CTC à faible dose et la 6-MP peuvent être maintenus pendant la grossesse. Les patientes doivent être surveillées de façon rapprochée durant la grossesse et la première année du post-partum, particulièrement en cas de cirrhose, à cause du risque d’aggravation ou de poussée d’HAI durant cette période. En cas de poussée, une augmentation de la dose d’immunosuppresseurs peut être envisagée.
• Sujet âgé
Le choix des corticoïdes en monothérapie doit être évité chez le sujet âgé, étant donné le risque élevé de diabète et d’ostéoporose. Si aucune autre alternative n’est possible, il est préférable d’opter pour le Budésonide en association avec l’AZT. Une densitométrie osseuse devrait être réalisée avant traitement [3].
Transplantation hépatique (TH)
Elle reste indiquée en cas de cirrhose avancée et en cas d’HAI fulminante ne répondant pas au traitement corticoïde par voie intraveineuse. Contrairement aux autres hépatopathies, l’HAI représente moins de 5 % des indications de TH selon les registres de transplantation européen et américain, du fait de la bonne réponse aux immunosuppresseurs [2]. La survie des patients à 5 ans est supérieure à 80 % dans la plupart des séries[95]. L’HAI peut cependant récidiver en post-TH. Le traitement immunosuppresseur, après la TH, repose sur les CTC, l’AZT ou le MMF le plus souvent. Les CTC sont maintenus plus longtemps en comparaison avec les TH pour cirrhose virale [2].
Évolution – Pronostic :
La survie des patients traités par les immuno-suppresseurs est de 80-95 % à 10 ans [96,97]. La mortalité liée à la maladie était de 17 % dans une étude multicentrique tunisienne ayant inclus 83 cas d’HAI. Dans cette série, 57 % des patients étaient au stade de cirrhose, ce qui explique probablement ce taux élevé de mortalité. Un MELD score ≥12 est prédictif d’une mauvaise réponse au traitement et sa non amélioration sous traitement est prédictive d’un mauvais pronostic [83,84].
Le risque de carcinome hépatocellulaire (CHC) sur HAI est plus faible par rapport aux autres hépatopathies [98]. Il est estimé à 1,1 % par an et concerne exclusivement les malades qui ont développé une cirrhose [7]. Dans une étude japonaise, 180 patients avec HAI ont été suivis durant 80.2 mois en moyenne, et traités par immuno-suppresseurs. 3,3 % des patients ont développé un CHC. En analyse multivariée, les facteurs de risque de survenue du CHC étaient la cirrhose au diagnostic (OR 4.08) et l’hypertransaminasémie chronique (OR 3.66)[26].
Dans un autre travail publié en 2009, ayant colligé 243 patients atteints d’HAI, un CHC a été retrouvé dans 15 cas (6,2 %). Les facteurs de risque de survenue d’un CHC étaient la cirrhose (p=0.048) et la rupture de varices œsophagiennes (p=0.003). La durée séparant le diagnostic de cirrhose de celui de CHC était de 102.5 mois (12-195). La médiane de survie des patients porteurs de CHC dépisté était de 19 mois [6-36], nettement supérieure à celle des patients ayant un CHC symptomatique (2 mois [0-14]; p=0.042) [52]. Ces résultats soulignent l’intérêt d’un dépistage systématique semestriel du CHC (bien que non validé pour cette indication) chez les patients porteurs d’une cirrhose autoimmune[3].
Conclusion :
L’HAI est une maladie complexe, aux multiples facettes, qui doit être prise en charge dans des centres spécialisés. Actuellement, cette prise en charge repose encore sur l’association prednisolone–azathioprine, avec des taux de réponse satisfaisants. Le Budésonide, en association avec l’AZT, représente une bonne option thérapeutique chez des patients naïfs ou intolérants aux CTC, en l’absence de cirrhose. Ce traitement permet, à efficacité équivalente, de réduire considérablement les effets secondaires cortico-induits. Les anticalcineurines sont préconisés dans les formes réfractaires et le MMF en cas d’intolérance à l’AZT. L’HAI est de bon pronostic lorsque la prise en charge thérapeutique est adéquate.
Références :
Krawitt EL. Autoimmun hepatitis. N England J Med 2006; 354 :54-66
Manns MP, Lohse AW, Vergani D. Autoimmune hepatitis – Update 2015. J Hepatol. 2015 Apr;62(1 Suppl):S100-11.
EASL Clinical Practice Guidelines: Autoimmune hepatitis. J Hepatol. 2015 Oct;63(4):971-1004
Waldenström J. Leber, Blutportein und Nahrungseiweiss. Dtsch Ges Z Verdan Stoffwechselkr 1950;15:113-9.
Johnson PJ, McFarlane IG. Meeting report: International Autoim- mune Hepatitis Group. Hepatology 1993;18:998–1005.
Gatselis NK, Zachou K, Koukoulis GK, Dalekos GN. Autoimmune hepatitis, one disease with many faces: etiopathogenetic, clinico-labo- ratory and histological characteristics. World J Gastroenterol. 2015 Jan 7;21(1):60-83.
Hurlburt KJ, McMahon BJ, Deubner H, Hsu-Trawinski B,Williams JL, Kowdley KV. Prevalence of autoimmune liver disease in Alaska Natives. Am J Gastroenterol 2002; 97: 2402-2407.
Czaja AJ. Diagnosis and management of autoimmune hepatitis. Clin Liver Dis 2015 (19):57-79.
Ngu JH, Bechly K, Chapman BA, Burt MJ, Barklay ML, Gearry RB et al. Population-based epidemiology study of autoimmune hepatitis: a disease of older women? J Gastrenterol Hepatol 2010;25:1681-1686.
Floreani A, Liberal R, Vergani D, Mieli-Vergani G. Autoimmune hepatitis: Contrasts and comparisons in children and adults – a com- prehensive review. J Autoimmun. 2013 Oct;46:7-16
Czaja AJ, Carpenter HA. Distinctive clinical phenotype and treat- ment outcome of type 1 autoimmune hepatitis in the elderly. Hepato- logy 2006; 43:532-538.
Strassburg CP, Obermayer-Staub P, Manns MP. Autoimmunity in liver diseases. Clin Rev Allergy Immunol 2000; 18: 127-139
Manns MP, Kruger M. Genetics in liver diseases. Gastroentero- logy 1994; 106 : 1676-1697
Bogdanos DP, Lenzi M, Okamoto M, Rigopoulou EI, Muratori P, Ma Y, Muratori L, Tsantoulas D, Mieli-Vergani G, Bianchi FB, Ver- gani D. Multiple viral/self immunological cross-reactivity in liver kid- ney microsomal antibody positive hepatitis C virus infected patients is associated with the possession of HLA B51. Int J Immunopathol Pharmacol 2004; 17: 83-92.
Donaldson PT. Genetics in autoimmune hepatitis. Semin Liver Dis 2002; 22:353–64.
Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Can- cado EL et al. International Autoimmun Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 1999; 31:929-38.
De Boer YS, van Gerven NM, Zwiers A, Verwer BJ, van Hoek B, van Erpecum KJ, et al. Genome- wide association study identifies variants associated with autoimmune hepatitis type 1. Gastroentero- logy 2014; 147:e445.
Agarwal K, Czaja AJ, Jones DE, Donaldson PT. Cytotoxic T lym- phocyte antigen-4 (CTLA-4) gene polymorphisms and susceptibility to type 1 autoimmune hepatitis. Hepatology 2000; 31:49–53.
Liberal R, Longhi MS, Mieli-Vergani G, Vergani D. Pathogenesis of immune hepatitis. Best Practice & Research Clinical Gastroentero- logy 25 (2011) 653–664
Muratori P, Granito A, Quarneti C, Ferri S, Menichella R, Cassa- ni F, Pappas G, Bianchi FB, Lenzi M, Muratori L. Autoimmune hepa- titis in Italy: The Bologna experience. J Hepatol 2009; 50: 1210-1218
Grønbæk L, Vilstrup H, Jepsen P. Autoimmune hepatitis in Denmark: incidence, prevalence, prognosis, and causes of death. A nationwide registry-based cohort study. J Hepatol 2014; 60: 612-617
Takahashi H, Zeniya M. Acute presentation of autoimmune hepatitis: Does it exist? A published work review. Hepatol Res 2011; 41: 498-504
Ferrari R, Pappas G, Agostinelli D, Muratori P, Muratori L, Len- zi M, et al. Type 1 autoimmune hepatitis: patterns of clinical pre- sentation and differential diagnosis of the ‘acute’ type. QJM 2004;97:407–412.
Werner M, Prytz H, Ohlsson B, Almer S, Björnsson E, Bergquist A, Wallerstedt S, Sandberg-Gertzén H, Hultcrantz R, Sangfelt P, Wei- land O, Danielsson A. Epidemiology and the initial presentation of autoimmune hepatitis in Sweden: a nationwide study. Scand J Gas- troenterol 2008; 43: 1232-1240
Feld JJ, Dinh H, Arenovich T, Marcus VA, Wanless IR, Heathcote EJ. Autoimmune hepatitis: effect of symptoms and cirrhosis on natu- ral history and outcome. Hepatology 2005; 42: 53-62.
Yeoman AD, Al-Chalabi T, Karani JB, Quaglia A, Devlin J, Mie- li-Vergani G, Bomford A, O’Grady JG, Harrison PM, Heneghan MA. Evaluation of risk factors in the development of hepatocellular carcinoma in autoimmune hepatitis: Implications for follow-up and screening. Hepatology 2008; 48: 863-870.
Migita K, Watanabe Y, Jiuchi Y, Nakamura Y, Saito A, Yagura M, Ohta H, Shimada M, Mita E, Hijioka T, Yamashita H, Takezaki E, Muro T, Sakai H, Nakamuta M, Abiru S, Komori A, Ito M, Yat- suhashi H, Nakamura M, Ishibashi H. Hepatocellular carcinoma and survival in patients with autoimmune hepatitis (Japanese Natio- nal Hospital Organization-autoimmune hepatitis prospective study). Liver Int 2012; 32: 837-844
Montano-Loza AJ, Carpenter HA, Czaja AJ. Predictive factors for hepatocellular carcinoma in type 1 autoimmune hepatitis. Am J Gastroenterol 2008; 103: 1944-1951
Cook GC, Mulligan R, Sherlock S. Controlled prospective trial of corticosteroid therapy in active chronic hepatitis. Q J Med 1971; 40: 159-185.
Soloway RD, Summerskill WH, Baggenstoss AH, Geall MG, Gitnićk GL, Elveback IR, Schoenfield LJ. Clinical, biochemical, and histological remission of severe chronic active liver disease: a control- led study of treatments and early prognosis. Gastroenterology 1972; 63: 820-833
Gregorio GV, Portmann B, Reid F, Donaldson PT, Doherty DG, McCartney M, et al. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology 1997; 25:541–7.
Teufel A, Weinmann A, Kahaly GJ, Centner C, Piendl A, Worns M, et al. Concurrent autoimmune diseases in patients with autoim- mune hepatitis. J Clin Gastroenterol 2010; 44:208–213.
Samuel D, Riordan S, Strasser S, Kurtovic J, Singh-Grewel I, Koorey D. Severe autoimmune hepatitis first presenting in the early post partum period. Clin Gastroenterol Hepatol 2004; 2:622–4
Heneghan MA, Norris SM, O’Grady JG, Harrison PM, McFar- lane IG. Management and outcome of pregnancy in autoimmune hepatitis. Gut 2001; 48:97–102.
Westbrook RH, Yeoman AD, Kriese S, Heneghan MA. Outcomes of pregnancy in women with autoimmune hepatitis. J Autoimmun 2012;38:J239–44.
Todros L, Saracco G, Durazzo M, Abate ML, Touscoz G, Sca- glione L, Verme G, Rizzetto M. Efficacy and safety of interferon alfa therapy in chronic hepatitis C with autoantibodies to liver-kidney microsomes. Hepatology 1995; 22: 1374-1378
Dalekos GN, Wedemeyer H, Obermayer-Straub P, Kayser A, Barut A, Frank H, Manns MP. Epitope mapping of cytochrome P4502D6 autoantigen in patients with chronic hepatitis C during alpha-interferon treatment. J. Hepatol 1999; 30(3):366-75.
Mieli-Vergani G, Vergani D. De novo autoimmune hepatitis after liver transplantation. J Hepatol 2004; 40: 3-7
Finnish-German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD- type zinc-finger domains. Nat Genet 1997; 17: 399-403
Obermayer-Straub P, Strassburg CP, Manns MP. Autoimmune poly- glandular syndrome type 1. Clin Rev Allergy Immunol 2000; 18: 167-183
Obermayer-Straub P, Perheentupa J, Braun S, Kayser A, Barut A, Loges S, Harms A, Dalekos G, Strassburg CP, Manns MP. Hepatic autoantigens in patients with autoimmune polyendocrinopathy-can- didiasis-ectodermal dystrophy. Gastroenterology 2001; 121: 668-677
Czaja AJ. Performance parameters of the conventional serological markers for autoimmune hepatitis. Dig Dis Sci 2011;56:545–54
Baeres M1, Herkel J, Czaja AJ, Wies I, Kanzler S, Cancado EL, Porta G, Nishioka M, Simon T, Daehnrich C, Schlumberger W, Galle PR, Lohse AW. Establishment of standardised SLA/LP immunoas- says: specificity for autoimmune hepatitis, worldwide occurrence, and clinical characteristics. Gut. 2002; 51: 259-64
Ma Y, Bogdanos DP, Hussain MJ, Underhill J, Bansal S, Lon- ghi MS et al. Polyclonal T-cell response to cytochrome P450IID6 are associated with disease activity in autoimmune hepatitis type 2. Gas- troenterology 2006; 130: 868-882
Herkel J, Heidrich B, Nieraad N, Wies I, Rother M, Lohse AW. Fine specificity of autoantibodies to soluble liver antigen and liver/ pancreas. Hepatology 2002; 35: 403-408
Gueguen P, Dalekos G, Nousbaum JB, et al. Double reactivity against actin and alpha-actinin defines a severe form of autoimmune hepatitis type 1. J Clin Immunol 2006; 26: 495–505.
Strassburg CP, Obermayer-Straub P, Alex B, Durazzo M, Rizzetto M, Tukey RH et al. Autoantibodies against glucuronyl transferases differ between viral hepatitis and autoimmune hepatitis. Gastroente- rolgy 1996; 111:1576-1586.
Nezu S, Tanaka A, Yasui H, Imamura M, Nakajima H, Ishida H, Takahashi S. Presence of antimitochondrial autoantibodies in patients with autoimmune hepatitis. J Gastroenterol Hepatol 2006; 21: 1448-1454
Montano-Loza AJ, Carpenter HA, Czaja AJ. Frequency, behavior, and prognostic implications of antimitochondrial antibodies in type 1 autoimmune hepatitis. J Clin Gastroenterol 2008; 42: 1047-1053
Lohse AW, Mieli-Vergani G. Autoimmune Hepatitis. J. Hepatol 2011; 55: 171-182.
Manns MP, Czaja AJ, Gorham JD, et al. Diagnosis and manage- ment of autoimmune hepatitis. Hepatology 2010; 51: 2193-213
Dienes HP, Popper H, Manns M, Baumann W, Thoenes W, Meye- rzumbuschenfelde KH. Histologic features in autoimmune hepatitis. Z Gastroenterol 1989;27:325-330
Hennes EM, Zeniya M, Czaja AJ, Pares A, Dalekos GN, Krawitt EL, et al. Simplified criteria for the diagnosis of autoimmune hepati- tis. Hepatology 2008; 48: 169–176.
Singh R, Nair S, Farr G, Mason A, Perrillo R. Acute autoim- mune hepatitis presenting with centrizonal liver disease: case report and review of the literature. Am J Gastroenterol 2002; 97: 2670-2673
Ben-Ari Z, Broida E, Monselise Y, Kazatsker A, Baruch J, Pappo O, Skappa E, et al. Syncytial giant-cell hepatitis due to autoimmune hepatitis type II (LKM1+) presenting as sub-fulminant hepatitis. Am J Gastroenterol 2000; 95: 799-801
Johnson PJ, Mc Farlane IG, Meeting reports: International Au- toimmune Hepatitis Group 1993; 18: 998-1005.
Muratori P, Granito A, Pappas G, Muratori L. Validation of sim- plified diagnostic criteria for autoimmune hepatitis in Italian patients. Hepatology 2009; 49: 1782–1783
Qiu DK, Wang QX, Wang H, Xie Q, Zang GQ, Jiang H, et al. Validation of the simplified criteria for diagnosis of autoimmune hepatitis in Chinese patients. J Hepatol 2011; 54: 340–347.
Czaja AJ. Performance parameters of the diagnostic scoring sys- tems for autoimmune hepatitis. Hepatology 2008; 48: 1540–8.
Fujiwara K, Yasui S, Tawada A, Fukuda Y, Nakano M, Yoko- suka O. Diagnostic value and utility of the simplified International Autoimmune Hepatitis Group criteria in acute-onset autoimmune hepatitis. Liver Int 2011; 31: 1013–1020.
Suzuki A1, Brunt EM, Kleiner DE, Miquel R, Smyrk TC, An- drade RJ, Lucena MI, Castiella A, Lindor K, Björnsson E. The use of liver biopsy evaluation in discrimination of idiopathic autoimmune hepatitis versus drug-induced liver injury. Hepatology. 2011; 54: 931-9
Czaja AJ. Cholestatic phenotypes of autoimmune hepatitis. Clin Gastroenterol Hepatol 2014; 12:1430–8.
Lewin M, Vilgrain V, Ozenne V, et al. Prevalence of sclerosing cholangitis in adults with autoimmune hepatitis: a prospective magne- tic resonance imaging and histological study. Hepatology 2009; 50: 528–37.
Czaja AJ. Features and consequences of untreated type 1 autoim- mune hepatitis. Liver Int 2009; 29:816–823.
De Groote J, Fevery J, Lepoutre L. Long-term follow-up of chro- nic active hepatitis of moderate severity. Gut 1978; 19: 510–513
Czaja AJ. Current and prospective pharmacotherapy for autoim- mune hepatitis. Expert Opinion Pharmacother. 2014; 15: 1715-1728.
Gleeson D, Heneghan MA. British Society of Gastroenterology (BSG) guidelines for management of autoimmune hepatitis. Gut 2011; 60: 1611–1629.
Van Gerven NM, Verwer BJ, Witte BI, et al. Relapse is almost uni- versal after withdrawal of immunosuppressive medication in patients with autoimmune hepatitis in remission. J Hepatol 2013; 58: 141-7
Czaja AJ. Rapidity of treatment response and outcome in type 1 autoimmune hepatitis. J Hepatol 2009; 51: 161-7
Hoeroldt B, McFarlane E, Dube A, et al. Long-term outcomes of patients with autoimmune hepatitis managed at a non-transplant center. Gastroenterology 2011; 140: 1980-9
Czaja AJ. Review article: prevention and reversal of hepatic fi- brosis in autoimmune hepatitis. Aliment Pharmacol Ther 2014; 39: 385-406
Czaja AJ, Davis GL, Ludwig J, Taswell HF. Complete resolution of inflammatory activity following corticosteroid treatment of HBsAg negative chronic active hepatitis. Hepatology 1984; 4: 622-7
Czaja AJ, Carpenter HA. Decreased fibrosis during corticosteroid therapy of autoimmune hepatitis. J Hepatol 2004; 40: 646-52.
Murray-Lyon IM, Stern RB, Williams R. Controlled trial of pred- nisone and azathioprine in active chronic hepatitis. Lancet 1973; 1: 735–737.
Czaja AJ. Review article: permanent drug withdrawal is desirable and achievable for autoimmune hepatitis. Aliment Pharmacol Ther 2014; 39: 1043-58
Czaja AJ, Ammon HV, Summerskill WH. Clinical features and prognosis of severe chronic active liver disease (CALD) after corticos- teroid-induced remission. Gastroenterology 1980; 78: 518-23
Summerskill WH, Korman MG, Ammon HV, Baggenstoss AH. Prednisone for chronic active liver disease: dose titration, standard dose, and combination with azathioprine compared. Gut 1975; 16: 876-83
Johnson PJ, McFarlane IG, Williams R. Azathioprine for long- term maintenance of remission in autoimmune hepatitis. N Engl J Med 1995; 333: 958-63.
Ngu JH, Gearry RB, Frampton CM, Stedman CA. Mortality and the risk of malignancy in autoimmune liver diseases: a population-based study in Canterbury, New Zealand. Hepatology. 2012; 55: 522-9
Pratt DS, Flavin DP, Kaplan MM. The successful treatment of autoimmune hepatitis with 6-mercaptopurine after failure with aza- thioprine. Gastroenterology 1996; 110: 271-4
Montano-Loza AJ, Carpenter HA, Czaja AJ. Features associated with treatment failure in type 1 autoimmune hepatitis and predictive value of the model of end-stage liver disease. Hepatology 2007; 46: 1138-45.
Yeoman AD, Westbrook RH, Zen Y, Maninchedda P, Portmann BC, Devlin J, et al. Early predictors of corticosteroid treatment failure in icteric presentations of autoimmune hepatitis. Hepatology 2011; 53: 926–934.
Ferucci ED, Hurlburt KJ, Mayo MJ, et al. Azathioprine metabolite measurements are not useful in following treatment of autoimmune hepatitis in Alaska Native and other non-Caucasian people. Can J Gastroenterol 2011; 25: 21-7.
Hempfling W, Grunhage F, Dilger K, Reichel C, Beuers U, Sauer- bruch T. Pharmacokinetics and pharmacodynamic action of bude- sonide in early and late-stage primary biliary cirrhosis. Hepatology 2003; 38: 196–202
Manns MP, Woynarowski M, Kreisel W, Lurie Y, Rust C, Zuc- kerman E, et al. Budesonide induces remission more effectively than prednisone in a controlled trial of patients with autoimmune hepatitis. Gastroenterology 2010; 139: 1198–1206.
Fernandes NF, Redeker AG, Vierling JM, Villamil FG, Fong TL. Cyclosporine therapy in patients with steroid resistant autoimmune hepatitis. Am J Gastroenterol 1999; 94: 241–248.
Nasseri-Moghaddam S, Nikfam S, Karimiam S, et al. Cyclospo- rine-A versus prednisolone for induction of remission in auto-immune hepatitis: interim analysis report of a randomized controlled trial. Middle East J Dig Dis 2013; 5: 193-200
Aqel BA, Machicao V, Rosser B, Satyanarayana R, Harnois DM, Dickson RC. Efficacy of tacrolimus in the treatment of steroid refrac- tory autoimmune hepatitis. J Clin Gastroenterol 2004; 38: 805–809
Thiel DH, Wright H, Carroll P, et al. Tacrolimus: a potential new treatment for autoimmune chronic active hepatitis: results of an open- label preliminary trial. Am J Gastroenterol 1995; 90: 771-6
Zachou K, Gatselis N, Papadamou G, Rigopoulou EI, Dalekos GN. Mycophenolate for the treatment of autoimmune hepatitis: pros- pective assessment of its efficacy and safety for induction and main- tenance of remission in a large cohort of treatment-naive patients. J Hepatol 2011; 55: 636–646.
Sharzehi K, Huang MA, Schreibman IR, Brown KA. Mycophe- nolate mofetil for the treatment of autoimmune hepatitis in patients refractory or intolerant to conventional therapy. Can J Gastroenterol 2010; 24: 588–592.
Burak KW, Swain MG, Santodomino-Garzon T, et al. Rituxi- mab for the treatment of patients with autoimmune hepatitis who are refractory or intolerant to standard therapy. Can J Gastroenterol 2013;27:273-80
Ichai P, Duclos-Vallee JC, Guettier C, Hamida SB, Antonini T, Delvart V, et al. Usefulness of corticosteroids for the treatment of severe and fulminant forms of autoimmune hepatitis. Liver Transpl 2007; 13: 996–1003.
Carbone M, Neuberger JM. Autoimmune liver disease, autoim- munity and liver transplantation. J Hepatol. 2014; 60: 210-23.
Kogan J, Safadi R, Ashur Y, Shouval D, Ilan Y. Prognosis of symp- tomatic versus asymptomatic autoimmune hepatitis: a study of 68 patients. J Clin Gastroenterol 2002; 35: 75-81
Murray-Lyon IM, Stern RB, Williams R. Controlled trial of pred- nisone and azathioprine in active chronic hepatitis. Lancet 1973; 1: 735-737
Teufel A, Weinmann A, Centner C, Piendl A, Lohse AW, Galle PR, Kanzler S. Hepatocellular carcinoma in patients with autoim- mune hepatitis. World J Gastroenterol 2009; 15: 578-582