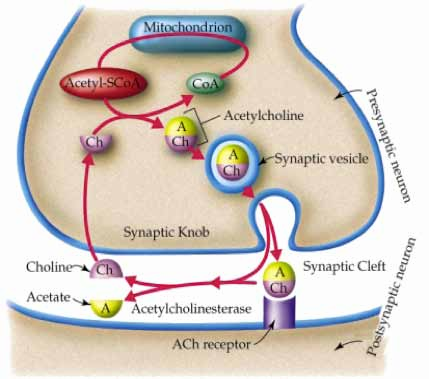
Pr Z. KACI,Pr N. BOUDJERRA, Service d’Hématologie, CHU Béni Messous, Alger
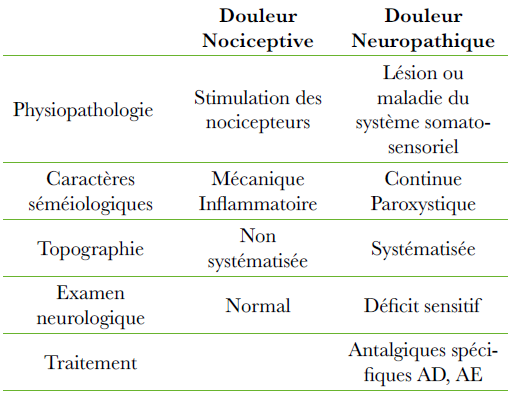
Abstract : The Myelodysplastic syndrome (MDS) is a clonal disease with transformation of hematopoietic stem cells and proliferation, differentiation and apoptosis abnormal.
SMD affects most of the time the old subjects. In 90 % of the cases, the symptoms are bound to an anaemia in the foreground, it explains the terminology of refractory anaemia.
The diagnosis is cytological evoked on the blood smear and confirmed by Smear of bone marrow. There are two classifications FAB and WHO.
The international MDS Workshop (IPSS) defines 4 groups of risks: low risk, Intermediary 1, Intermediary 2 and high risk.
The evolution towards acute leukeamia is noted in 30 % of the cases. The treatment is symptomatic and specific. The therapeutic decisions are based on the age, the general state and the comorbidity of the patient as well as on the scale of the risks (IPSS).
Résumé : Le SMD est une maladie clonale avec transformation des cellules souches hématopoïétiques et prolifération, différentiation et apoptose anormales. Le SMD affecte le plus souvent les sujets âgés. Dans 90% des cas, les symptômes sont liés à une anémie au premier plan, cela explique la terminologie d’anémie réfractaire.
Le diagnostic est cytologique évoqué sur le frottis sanguin et confirmé par le myélogramme. Il existe deux classifications : FAB et OMS.
L’International MDS Workshop (IPSS) définit 4 groupes de risque : Risque faible, Intermédiaire 1, Intermédiaire 2 et Risque élevé.
L’évolution peut se faire dans 30 % des cas vers une leucémie aiguë. Le traitement comporte des soins de support, une chimiothérapie et une allogreffe de moelle osseuse.
Les décisions thérapeutiques sont basées sur l’âge, l’état général et les comorbidités du patient ainsi que sur l’échelle des risques (IPSS).
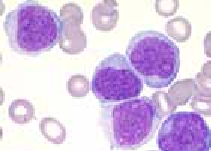
Les syndromes myélodysplasiques sont des anomalies qualitatives et quantitatives du fonctionnement médullaire, se traduisant par des degrés divers d’hématopoïèse inefficace avec probabilité variable d’évolution en leucémie aiguë. Il s’agit de désordres clonaux. Ils sont caractérisés par l’existence d’anémie, de neutropénie, de thrombopénie, isolées ou diversement associées, contrastant avec une moelle normo ou hyper cellulaire.
Épidémiologie :
Ces affections touchent principalement le sujet âgé avec une augmentation de fréquence après 65 ans et un âge moyen de 60 à 75 ans. Le sexe ratio est de 1. L’incidence est de 3–5 cas pour 100 000 habitants. Elle est de 20 cas/100 000 chez les patients de plus de 70 ans.
Syndromes myélodysplasiques primaires :
Les anomalies génétiques et épigénétiques semblent
Anomalies moléculaires
Type
Fréquence (%)
Anomalies chromosomiques Fréquentes : anomalies numériques ou structurelles : monosomie 7, 5q−, 7q−, 20q− ; trisomie 8, 14, ou 19 ; anomalies complexes, perte du chromosome X Non fréquentes : translocations « typiques de la LAM » : t(6;9), t(8;21), inv(16) ou t(9;22) ; translocations t(1;3), t(5;12).
40
10
Anomalies génétiques Activation des oncogènes (N-ras, moins fréquemment K-ras, H-ras). Mutations p53
10-15
5-10
Anomalies épigénétiques Hyperméthylation/ inactivation (par ex., p15) Surexpression de bc12
40-70
30
Syndromes myélodysplasiques secondaires :
Après une chimiothérapie (en particulier par agents alkylants).
Rayonnements ionisants (radiothérapie, exposition aux radiations).
Benzène et autres solvants organiques.
Insecticides
Signes cliniques :
Les signes sont ceux d’une insuffisance médullaire. Dans 90% des cas, les symptômes sont liés à une anémie au premier plan, cela explique la terminologie d’anémie réfractaire. Les manifestations infectieuses ou hémorragiques sont souvent plus tardives. L’examen clinique est pauvre.
Signes biologiques :
On retrouve des anomalies quantitatives et qualitatives des éléments sanguins et médullaires.
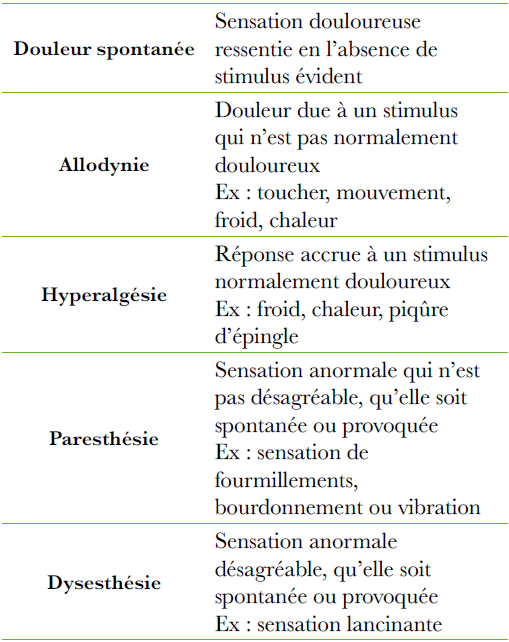
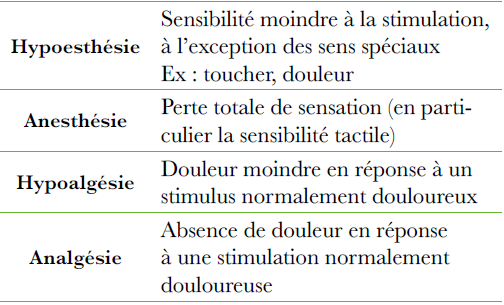
Sidéroblastose : Coloration de Perls : Type 1 (1à 5 granules) -Type 2 (5 à 10 granules) -Type 3 (en couronne)
Microscope électronique : fer augmenté dans les mitochondries.
Anémie arégénérative normochrome macrocytaire.
Aniso-poïkilocytose, ponctuation basophile
Lignée granulocytaire : Dysgranulopoïèse Anomalies de maturation de la lignée granuleuse (dégranulation des myélocytes et métamyélocytes, promyélocytes anormaux, excès de blastes et myéloblates).
Neutropénie
Polynucléaires dégranulés, un noyau segmenté (anomalie de Pelger acquise).
Lignée plaquettaire : Dysmégacaryocyto- poïèse
Petits mégacaryocytes basophiles à noyau non lobulé (micromégacaryocytes)
Grands mégacaryocytes multinucléés
Thrombopénie.
Plaquettes géantes ou dégranulées.
Anomalies fonctionnelles (agrégation)
La biopsie ostéomédullaire n’a pas d’utilité diagnostique.
Classification des SMD :
Classification FAB (French-American-British Cooperative Group, 1982) :
AR
Anémie réfractaire
ARSC
Anémie réfractaire avec sidéroblastes en couronne
AREB
Anémie réfractaire avec excès de blastes
AREB-T
AREB en transformation (en leucémie aiguë)
LMMC
Leucémie chronique myélomonocytaire
Classification selon l’OMS (2001) :
AR/ AR-SC
Anémie réfractaire (avec/sans sidéroblastes en couronne)
CRDM/ CRDM-SC
Cytopénie réfractaire avec dysplasie multi-lignée (avec/sans sidéroblastes en couronne)
AREB-1
Anémie réfractaire avec excès de blastes (5–9 %)
AREB-2
Anémie réfractaire avec excès de blastes (10–19 %)
5q–
Syndrome 5q−
Inclassée
Syndrome myélodysplasique, non classé.
– Maladies myélodysplasiques/ myéloprolifératives
Diagnostic :
Le diagnostic est cytologique évoqué sur le frottis sanguin et confirmé par le myélogramme.
Facteurs pronostiques :
Pourcentage de blastes médullaires :
Un taux supérieur à 10 % constitue un mauvais pronostic.
Pronostic des syndromes myélodysplasiques (selon la classification FAB)
Type
Risque de transformation (%)
Temps de survie (moyenne)(mois)
AR
10
37
ARSC
5
50
AREB
25
10
AREB T
50
5
LMMC
20
22
Score de risque d’après l’International MDS Workshop IPSS (système international de score pronostique) :
Facteurs pronostique
Score
Score
Score
Score
Score
0
0,5
1,0
1,5
2,0
Pourcentage de blastes
< 5%
5 – 10%
–
11-20%
21 –30%
Caryotype
Bon
Intermédiaire
Mauvais
Lignées cellulaires atteintes
0 – 1
2 – 3
Blastes donnés en pourcentage de la population des cellules de la moelle osseuse
Bon : caryotype normal, −Y, 5q−, 20q−.
Mauvais : caryotype complexe, anomalies du chromosome 7.
Intermédiaire : toutes autres anomalies.
Nombre de lignées cellulaires atteintes (granulo-/ erythro-/thrombopoïèse)
Score de risque d’après l’International MDS Workshop (IPSS) : groupes de risque
Groupe de risque
Score global
Risque de transformation maligne (années)
Médiane desurvie (mois)
Risque faible
0
>18
65
Intermé- diaire 1 (int1)
0,5 – 1,0
8
40
Inter- médiaire 2(int2)
1,5 – 2,0
3
14
Risque élevé
>2,5
0,5
5
Causes de décès les plus communes :
Infections, hémorragies.
Complications après transformation en leucémie aiguë myéloblastique.
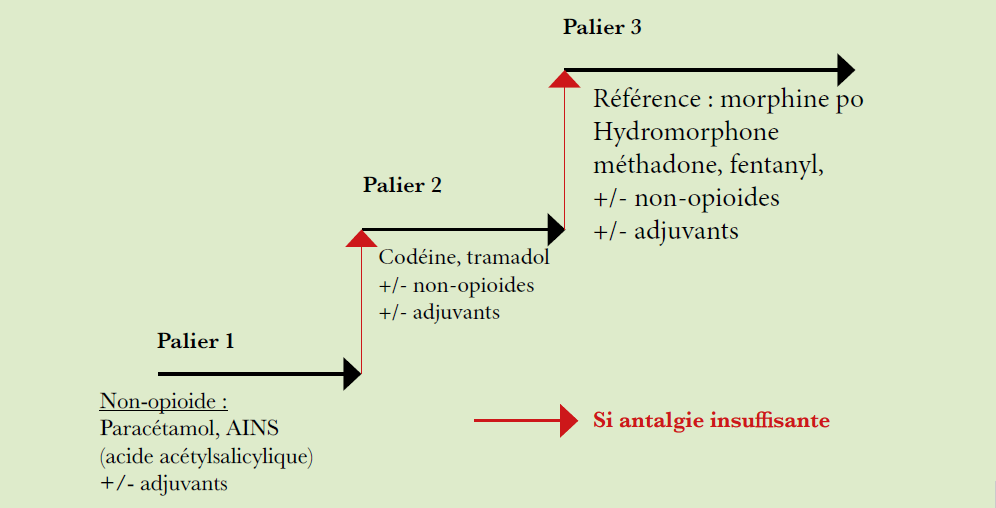
Principe de traitement :
Les décisions thérapeutiques sont basées sur l’âge, l’état général et les comorbidités du patient, ainsi que sur l’échelle des risques (IPSS).
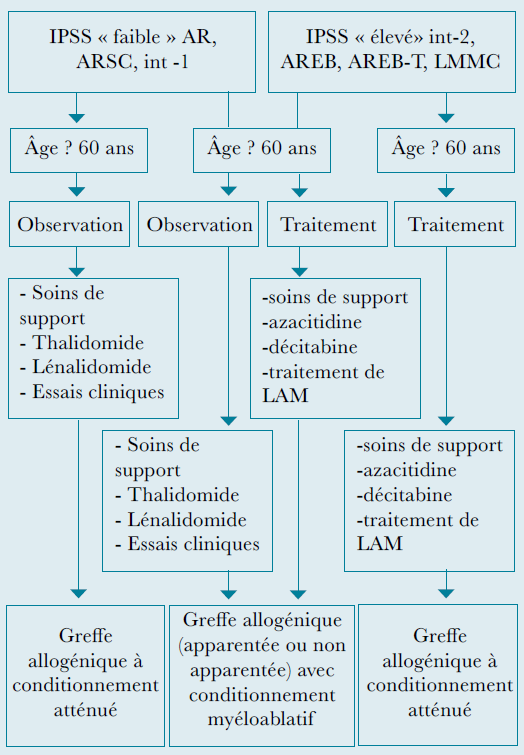
Un traitement à visée curative est possible chez les patients de moins de 60 ans, il s’agit d’une allogreffe de cellules souches hématopoïétiques.
Les patients âgés de 60-70 ans et plus sont généralement traités en intention palliative, par des traitements symptomatiques et des soins de support.
Transformation en leucémie aiguë : traitement par polychimiothérapie comme pour une leucémie de novo chez les jeunes patients, suivi d’une allogreffe de moelle.
Soins de support :
Transfusion de globules rouges, transfusion de plaquettes, chez les patients symptomatiques.
Traitement des complications infectieuses (antibiotiques, antifongiques).
Traitement de l’hémosidérose secondaire par des ferrioxamine mesylate, déferasirox ou déferiprone.
L’administration de facteurs de croissance (EPO, G-CSF) n’influence pas le temps de survie, leur utilisation palliative améliore la qualité de vie du patient.
Traitement des SMD à faible risque :
Inhibiteurs de l’ADN-methyltransferase (decitabine, 5-azacytidine).
Thalidomide, lenalidomide (traitement par lenalidomide, particulièrement dans le syndrome 5q–).
Traitement immunosuppresseur : cyclosporine, globuline antithymocytaire (ATG), globuline antilymphocytaire (SAL), particulièrement en cas de SMD hypoplasique.
Traitement expérimental : induction de la différentiation par acide rétinoïque ou inhibiteurs de l’histone deacétylase (acide tout trans-retinoïque, phenylbutarate, acide valproïque, SAHA/vorinostat, depsipeptide).
Traitement des SMD à risque élevé :
Inhibiteurs de l’ADN méthyltransferase (decitabine, 5-azacytidine). 50% de rémission avec l’administration de decitabine (réponse précoce sur la lignée plaquettaire), dans 30% des cas, réponse sur les 3 lignées et rémission cytogénétique.
Allogreffe avec conditionnement myéloablatif ou conditionnement d’intensité réduite (dépendant de la disponibilité de donneurs compatibles, de l’état général et de l’âge du patient).
Traitement expérimental : induction de différentiation par acide rétinoïque ou inhibiteurs de l’histone deacétylase (acide tout trans-retinoïque, phenylbutarate, acide valproïque, depsipeptide).
Diagramme thérapeutique des syndromes myélodysplasiques :
Conclusion :
On sépare les différents SMD en deux groupes de pronostic et de traitement différents : les SMD de faible risque et les SMD de haut risque. Une prise en charge précoce permet de mieux apprécier les risques d’évolution et d’adopter la meilleure attitude thérapeutique. L’objectif principal des traitements est de prolonger la survie en allongeant le délai entre le traitement reçu et la progression de la maladie. Les thérapeutiques actuelles ont permis d’atteindre cet objectif de manière statistiquement significative. De plus, quel que soit le traitement spécifique mis en place, les traitements symptomatiques et les transfusions gardent une place majeure.
Références :
Aul C, Giagounidis A, Germing U et al. A. Evaluating the pro- gnosis of patients with myelodysplastic syndromes. Ann Hematol 2002; 81:485–97
Bowen D, Culligan D, Jowitt S et al. Guidelines for the diagno- sis and therapy of adult myelodysplastic syndromes. Br J Hematol 2003; 120:187–200
Corey SJ, Minden MD, Barber DL et al. MDS: the complexity of stem-cell diseases. Nat Rev Cancer 2007; 7:118–29
Golshayan AR, Jin T, Maciejewski J et al. Efficacy of growth factors compared to other therapies for low risk MDS. Br J Hae- matol 2007,137:125–32
Howe RB, Porwit-MacDonald A, Wanat R et al. The WHO classification of MDS does make a difference. Blood 2004; 103:3265–70
Ardiman JW, et al. The 2008 revision of the World Health Orga- nization (WHO) classification of myeloid neoplasms and acute leu- kemia: Rationale and important changes. Blood. 2009; 114:937.
Malcovati L, Germing U, Kuendgen A et al. Time-dependent prognostic scoring system for predicting survival and leukemic evo- lution in MDS. J Clin Oncol 2007; 25:3503–10
Olney HJ, LeBeau MM. Evaluation of recurring cytogenetic ab- normalities in the treatment of MDS. Leukemia Res 2007;31:427– 34
Valent P, Horny HP, Bennet JM et al. Definitions and standards in the diagnosis and treatment of MDS: Consensus statements and report from a working conference.Leukemia Res 2012; 31:727–36
Norbert. G, Ulrich G. Advanced workshop on myelodysplastic syndrome. March2015
Dr Zohra OUCHENANE,Dr Sihem KEBAILI,Pr Noureddine Sidi MANSOUR. Service d’Hématologie, CHU Constantine
Abstract : Chronic myelogenous leukemia (CML) is a clonal hematopoiesis malignancy affecting hematopoietic stem cells, characterized by an acquired chromosomal abnormality translocation t(9,22) (q 34,q11) defining the Philadelphia chromosome ; the BCR-ABL fusion gene has a abnormal tyrosine kinase activity, thus the CML constitutes a unique physiopathological model, the progress made in the last years in the field of molecular biology, his treatment has been disrupted by the introduction of tyrosine kinase inhibitors specific BCR-ABL, such as imatinib, dasatinib, nilotinib, bosutinib and lately Ponatinib.
Résumé : La leucémie myéloïde chronique (LMC) est une hémopathie maligne clonale affectant les cellules souches hématopoïétiques, caractérisée par une anomalie chromosomique acquise : la translocation t(9,22) (q34,q11), définissant le chromosome Philadelphie ; le gène de fusion BCR–ABL présente alors une activité tyrosine kinase anormale, de ce fait la LMC constitue un modèle physiopathologique unique, et avec les progrès accomplis ces dernières années dans le domaine de la biologie moléculaire, son traitement a connu un bouleversement par l’introduction d’inhibiteurs de tyrosine kinase spécifiques de BCR-ABL, tels l’imatinib, le dasatinib, le nilotinib, le bosutinib et dernièrement le Ponatinib.
Abréviations : BCR=breakpoint cluster region ; ABL=Abelson ; ARNm=ARN messager ; STAT=signal transducer and activator of transcription ; PI3k=phosphatidyiinositol3-kinase ; JAK=januskinase.
Introduction – Définition :
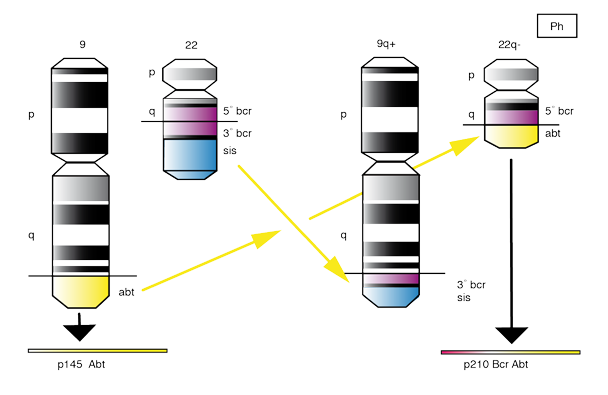
La leucémie myéloïde chronique (LMC) est un syndrome myéloprolifératif des cellules souches hématopoïétiques, appartenant au groupe des syndromes myéloprolifératifs. Cette maladie clonale est caractérisée par la présence d’un chromosome anormal, le chromosome Philadelphie (Ph). Il s’agit dans sa forme classique d’une translocation entre le chromosome 9 et le chromosome 22 t(9;22) (q34;q11), mettant en continuité les gènes BCR et ABL, ceci a pour conséquence la production d’un ARN de fusion et d’une protéine chimérique BCR-ABL tyrosine kinase anormale [1].
Historique : des dates sont importantes dans la LMC :
Décrite pour la première fois en 1945 par Bennett en Ecosse et Virchow en Allemagne.
Mise en évidence en 1960 par Peter Nowell et David Hungerford d’un chromosome 22 anormalement raccourcis portant le nom de la ville d’origine (Philadelphia)
En 1973, Rowley montre l’existence d’un échange de fragments entre les chromosomes 9 et 22 : il s’agit de la translocation respectivement en q34 et q11 : t(9,22) (q34, q11).
Durant les années 1980, la biologie moléculaire a montré que les points de cassure se situent au niveau des gènes ABL (oncogène Abelson) du chromosome 9 et BCR (Break point cluster région) du chromosome 22.
Dans les années 1990, George Daley et ces collaborateurs démontrent que le gène anormal BCR-ABL est responsable de la LMC et dont la protéine générée est à l’origine de son potentiel oncogénique [2].
Épidémiologie :
La LMC est peu fréquente. Elle représente approximativement 7 à 15% des leucémies de l’adulte selon les séries publiées. L’incidence varie en fonction des pays, en Algérie, l’incidence est plus faible mais croissante, elle passe de 0,19/100.000 habitants en 1994 à 0,44/100.000 habitants en 2009 et à 0,53/ 100.000 habitants en 2016. La prévalence en 2017 est de 1669 cas. La LMC est une
pathologie qui survient surtout entre 30 et 40 ans, mais peut se voir à tout âge, y compris chez l’enfant [3]. En Algérie, l’âge moyen au diagnostic est de 44 ans. Cette maladie touche préférentiellement les hommes avec un sex-ratio H/F de 1,12 [3].
L’étiologie de la LMC semble inconnue. Il a été rapporté quelques observations de LMC familiale mais aucun facteur génétique n’a pu être identifié. Il existe des facteurs favorisants tels que les radiations ionisantes, l’exposition prolongée au benzène ou à certaines thérapies anticancéreuses.
Physiopathologie :
Réarrangement chromosomique de la LMC
Chromosome Philadelphie :
Le chromosome Philadelphie est présent dans la quasi-totalité des LMC et dans environ 25% des cas de leucémies aiguës lymphoblastiques de l’adulte. Il résulte d’une translocation réciproque équilibrée entre les bras longs des chromosomes 9 et 22.
La translocation t(9 ;22) (13)
Gènes impliqués dans la LMC :
Gène ABL et sa protéine :
Structure : Chez l’homme, ce gène est localisé au niveau du chromosome 9(q34), son extrémité 5’ étant orientée vers le centromère. Il comporte 11 exons dont 2 alternatifs (1a et 1b) séparés par un intron de 200 Kb où se situe la plupart des cassures observées dans la LMC.
La protéine ABL qui en dérive contenant l’exon 1a (majoritaire), a une localisation nucléaire prédominante et celle contenant l’exon 1b, a une localisation membranaire. C’est une protéine appartenant à la famille des tyrosines kinases (TK), de 1454 kDa et qui possède des domaines d’homologie SH (Srchomology). Ces domaines d’homologie sont le site catalytique responsable de la phosphorylation des résidus tyrosines.
Le gène BCR et sa protéine :
Structure : Le gène BCR se positionne sur le bras long du chromosome 22. Il s’étend sur 135 kb et comprend 23 exons, il est transcrit en ARNm qui code pour une protéine cytoplasmique de 160 kb d’expression ubiquitaire.
Le réarrangement BCR-ABL : la translocation t(9,22) entraine un réarrangement des gènes situés au voisinage des points de cassure et la constitution sur le chromosome 22 d’un gène de fusion hybride comportant la partie 5’ (N-terminale) du gène BCR et la partie 3′ (C-terminale) du gène ABL. La fusion se fait entre l’exon b2 ou b3 de BCR et l’exon a2 d’ABL. Ce gène BCR-ABL est transcrit en ARN messager de 8,5 kb, lequel est traduit en une protéine hybride BCR-ABL. Les points de cassure sur ABL sont très variables, et peuvent se situer en trois endroits :
M-BCR (major BCR) : La coupure se produit préférentiellement entre les exons b2 et b3 ou b3 et b4 et permet la formation des produits de fusion b2a2 et b3a2. Les ARNm produits codent pour une protéine de 210 kDa, observée au cours de la LMC.
m-BCR (minor BCR) : Le point de cassure se situe entre les exons e1 et e2. L’ARNm qui en résulte code pour une protéine chimérique de 190 kDa, retrouvée dans
les leucémies aigues lymphoblastiques Ph+, les syndromes myélodysplasiques et exceptionnellement la LMC.
u-BCR (micro break point) : Le point de cassure se situe entre les exons 10 et 20 du gène BCR (transcrit dérivé e19a2). L’ARNm code pour une protéine de 230 kDa, observée dans la leucémie chronique à polynucléaires neutrophiles (4).
Oncogenèse induite par BCR-ABL : la dérégulation de l’activité tyrosine kinase interfère avec les signaux cellulaires normaux impliqués dans le processus de prolifération, d’adhésion cellulaire, de différenciation et d’apoptose, en phosphorylant différents complexes protéiques. Les principales voies de signalisation affectées sont les voies RAS, JAK/STAT, PI3K [5]. Les conséquences induites par cette protéine chimériques sont [6] :
Inhibition de l’apoptose
Inhibition de l’adhésion des progéniteurs LMC au stroma
Dégradation des protéines de régulation par le protéasome
Modification de la régulation par les cytokines
Instabilité génique
Étude clinique LMC en phase chronique :
Circonstances de découverte : Le début est insidieux et les signes menant à la découverte de la maladie sont peu caractéristiques :
La découverte fortuite à l’occasion d’un examen systématique est retrouvée dans 40% des cas, la sensation de pesanteur ou de douleur au niveau de l’hypocondre gauche en rapport avec une splénomégalie est retrouvée dans 50% des cas, une altération de l’état général avec asthénie, amaigrissement, anorexie, fébricule et sueurs nocturnes, ou bien à l’occasion d’une complication inaugurale telle un priapisme, une crise de goutte, une lithiase urinaire.
Examenclinique:la splénomégalie (SPM)) est le signe majeur retrouvé dans 50 à 70% des cas, elle est ferme, indolore, mobile avec la respiration. Sa surface est lisse et le bord antérieur est crénelé. Sa longueur se mesure sur la ligne médio-claviculaire en cm de débord sous costal (débord splénique ou DS). Une hépatomégalie (HPM) modérée peut s’observer dans 10 à 40% des cas.
Biologie :
Bilan hématologique :
Hémogramme : l’étude de l’hémogramme et l’analyse du frottis sanguin (FS) suffisent pour évoquer la maladie.
Une hyperleucocytose franche comprise entre 20.10^9 leucocytes/l et 500.10^9 leucocytes/l ; faite d’une polynucléose neutrophile vraie (30% à 40%), d’une éosinophilie (5% à 10%), et d’une basophilie (3 à 10%).
Une myélémie > 20% (métamyélocytes, myélocytes, promyélocytes et rares myéloblastes <5%). Il n’y a pas d’hiatus de maturation.
Une anémie très modérée, normocytaire normochrome peu régénérative, peut se voir.
Une thrombocytose avec un taux plaquettaire entre 500 et 800.10^9 thrombocytes/l, parfois supérieur à 1000.10^9/l. La thrombopénie est rare en phase chronique.
Myélogramme :le frottis médullaire est très riche avec augmentation de la densité cellulaire prédominant sur la lignée granulocytaire (80 à 90%). Le médullogramme est inutile pour le diagnostic, mais il reste indispensable car il permet de définir le pourcentage des blastes et de déterminer ainsi la phase de la maladie et également la réalisation du caryotype.
Biopsie ostéo-médullaire (BOM) : on constate une moelle très riche avec raréfaction ou disparition des adipocytes, une hyperplasie mégacaryocytaire avec une fibrose modérée au début pouvant s’aggraver au cours de l’évolution.
Techniquesd’identificationdelatranslocation t(9,22) : Le diagnostic de la LMC repose sur la mise en évidence du chromosome Philadelphie (Ph) ou de son équivalent moléculaire le transcrit BCR-ABL.
Cytogénétique conventionnelle (caryotype) : c’est l’examen de référence pour mettre en évidence le chromosome Ph présent dans 95% des cas. Il est retrouvé dans les précurseurs granuleux, les mégacaryocytes, les érythroblastes, les monocytes ainsi que dans les lymphocytes T et B médullaires. Il permet également La recherche des variants du Ph, et les autres anomalies chromosomiques additionnelles (ACA) qui sont retrouvées dans 5 à 10% des cas au diagnostic [7,24].
FISH (Hybridation In Situ en Fluorescence) : recommandée en cas d’absence du chromosome Ph au caryotype conventionnel, ou de chromosome Ph masqué et en cas d’échec du caryotype médullaire (pas de mitoses).[8]
Dans la LMC, elle permet de mettre en évidence l’équivalent moléculaire du chromosome Philadelphie : le transcrit BCR- ABL à partir des cellules médullaires ou plus facilement sur un prélèvement sanguin, deux techniques sont utilisées : la RT- PCR (Reverse Transcriptase Polymérase Chain Réaction) classique qualitative et la RQ- PCR (quan- titative) ou RT- PCR en temps réel qui permet de quantifier la maladie résiduelle[9].
L’évolution naturelle de la LMC passe par 3 phases, la phase chronique, la phase accélérée et enfin la phase blastique. En absence de traitement, l’évolution est inéluctablement mortelle.
La phase chroniquesus-décrite, dure en moyenne 3 à 4 ans.
La phase accélérée correspond à la transition entre la phase chronique et la phase blastique. Sa durée est de 12 à 18 mois. Chez certains patients, cette phase passe inaperçue ou est inexistante [10].
La phase blastique c’est la dernière phase de la LMC dont l’espérance moyenne de vie est de 3 à 6 mois. La transformation aiguë s’effectue en transformation myéloïde chez 70% des patients et en transformation lymphoïde chez les autres [11]. On constate de nombreuses anomalies chromosomiques portant sur le nombre des chromosomes, décrites dans environ 80% des cas.
Pronostic :
Pour une meilleure évaluation du pronostic et afin de choisir le traitement le mieux adapté aux patients, il a été instauré un certain nombre de grilles d’évaluation qui permettent d’établir des scores pronostiques ou index pronostiques très fiables. Il s’agit du :
Score de Sokal : Quatre paramètres sont utilisés : L’âge, la taille de la rate en centimètres du rebord costal, le taux des plaquettes et le pourcentage des blastes circulants. Cet indice permet de séparer trois groupes (faible, intermédiaire, élevé) dont la médiane de survie est significativement différente [12].
Score d’EUTOS : Il s’agit d’un facteur prédictif de la RCyC à 18 mois. Il tient compte de deux paramètres, le débord splénique (DS) et le pourcentage des PN basophiles. Le calcul se fait selon la formule suivante : (DS x 4) + (% PNB x 7). Les patients sont classés en 2 groupes, haut risque (score >87) et bas risque (score= 87).
Score d’Hasford : Ce score est calculé à partir de données suivantes :
Age, taille de la rate en cm, pourcentage des blastes circulants, pourcentage d’éosinophiles et basophiles circulants ainsi que le taux des plaquettes [13].
Score de Gratwohl : Qui permet d’estimer la survie à 5 ans des patients qui sont proposés pour une allogreffe de moelle osseuse. Il dépend de l’âge, du stade de la LMC, de l’intervalle entre la greffe et le diagnostic, du sexe du receveur et du type de donneur. Sept cotes ont été ainsi établies de 0 à 6, sachant que 0 représente le pronostic le plus favorable [14].
Diagnostic différentiel :
Myélémies réactionnelles
Splénomégalie myéloïde (SM)
Thrombocytémie essentielle
Leucémie myélomonocytaire chronique
Leucémie à polynucléaires neutrophiles : Le chromosome Ph et le transcrit BCR-ABL sont absents.
Leucémie myéloïde chronique atypique (a-LMC): les critères diagnostiques selon l’OMS sont: Hyperleucocytose persistante, myélémie >10%, basophilie < 20% et monocytose < 1.109/L, blastose médullaire < 20%,signes de dysplasie affectant plusieurs lignées, absence de gènes de fusion BCR-ABL.
LAL à chromosome Ph : Elle constitue le diagnostic différentiel possible d’une LMC en phase de transformation aiguë [15].
Traitement :
Mesures d’urgence : On recommande une hospitalisation en cas d’hyperleucocytose symptomatique, pour faire une cytoréduction, mettre un hypo-uricémiant ou instaurer une hyperhydratation alcaline. Il peut exister des indications de cytaphérèse thérapeutique en cas de leucostase ou de priapisme.
Chimiothérapie conventionnelle : Hydroxy-urée: Le traitement cytoréducteur par l’Hydroxy-urée garde toute sa place en cas d’hyperleucocytose symptomatique et/ou de thrombocytose >100.109/L avant de
débuter un traitement spécifique de la maladie[16]. Interféron:Les IFN-α 2a et 2b recombinants, utilisés aujourd’hui dans le traitement de la LMC en phase chronique chez les patients qui ne pouvaient pas bénéficier d’une greffe mais les ITK sont désormais le traitement de première intention et l’IFN est quelquefois associé à l’imatinib en cas de résistance [17]. Greffe de moelle osseuse : Aujourd’hui, la greffe de cellules souches allogéniques est le seul traitement éradicateur de la LMC quel que soit le stade de la maladie. Grace au conditionnement et à l’effet greffon versus leucémie, elle permet l’élimination des cellules leucémiques et la reconstitution d’une hématopoïèse normale [18]. Cependant, malgré les progrès qui permettent de réduire la toxicité et la mortalité liées à la greffe (immunosuppresseurs, anti infectieux, conditionnements moins agressifs, nouvelles sources de CSH), elle s’accompagne toujours d’un taux de mortalité non négligeable qui limite son indication. Lesinhibiteursdetyrosinekinase(ITK):
Objectifs thérapeutiques et définition des critères de réponse aux traitements par les ITK :
Trois niveaux de réponses sont observés au cours du traitement de la LMC selon les recommandations de l’ELN (European Leukemia Net) [19] :
La réponse hématologique complète (RHC) : rate non palpable, taux de GB < 10.109/l, absence de myélémie, basophilie sanguine < 5% et taux de plaquettes < 450.000.109/l.
Laréponsecytogénétique(RCy):RCy complète : 0% de cellules médullaires Ph positives, RCy partielle : 1 à 35%, RCy majeure < 35%, RCy mineure : 36% à 65 %, RCy minime : 66% à 95 %, Absence de réponse > 95 %.
La réponse moléculaire : dite majeure (RMM) quand il y a réduction d’au moins 3 log du taux de BCR par rapport aux valeurs initiales, BCR-ABL/ ABL < 0,1% par RQ-PCR, la réponse est complète (RMC) si le transcrit BCR-ABL est indétectable.
Recommandations de l’ELN : Depuis le début du traitement par les antityrosines kinases, le groupe ELN n’a cessé de stratifier des recommandations quant à l’évaluation de la réponse sous ces différentes formes (hématologique, cytogénétique et moléculaire) et la recherche de signes annonciateurs de la progression de la maladie (tableau).
Recommandations de l’ELN 2013[20].
6 mois
RMM : BCR -ABL<1% et/ou Ph+ 0%
BCR-ABL : 1-10% et/ ou Ph+ : 1 -35%
BCR- ABL>10% et/ou Ph+> 35%
12 mois
BCR-ABL ≤ 0,1%
BCR-ABL > 0,1-1%
BCR-ABL>1%
Après et à tout moment
BCR-ABL ≤ 0,1%
ACA/Ph- (-7 ou 7q-)
Perte RHCPerte RCyCPerte RMM*MutationsACA/Ph+
Temps
Réponse Optimale
Alerte
Echec
Diagnos- tic
N/A
Sokal élevé ACA / Ph+
N/A
3 mois
RHC BCR-ABL ≤10% et/ou Ph+ ≤ 36%
BCR-ABL >10% et/ou Ph+ : 36-95%
Pas RHC et/ou Ph+>95%
Lesinhibiteursdetyrosinekinase(ITK):
Mésylate d’imatinib (GLIVEC) : son mécanisme d’action repose sur la neutralisation de l’activité tyrosine kinase de la protéine BCR-ABL par inhibition compétitive de l’ATP au niveau du site catalytique. Il en résulte une inhibition de l’autophosphorylation, une inhibition de la prolifération et l’induction de l’apoptose. La posologie varie selon la phase de la maladie, phase chronique 400 mg/j, phase d’accélération 600 mg/ j, 600 à 800 mg/j pendant la phase blastique [21].
Résistance primaire et secondaire à l’imatinib : Résistances secondaires : sont définies par des critères cliniques, cytogénétiques et moléculaires. Elles sont observées chez des patients ayant déjà obtenu une réponse à l’imatinib et leur fréquence augmente avec l’évolution de la maladie.
Résistances primaires : rares, correspondent à une maladie réfractaire d’emblée et aux critères d’échec définis dans le tableau des recommandations de l’ELN.
Mécanismes de résistance : Le mécanisme de résistance est bien souvent lié à une mutation biologique du gène BCR-ABL, dans le cas contraire, la cause est pharmacologique.
– Les mécanismes BCR-ABL dépendants sont :
La mutation du domaine BCR-ABL kinase : Elle représente 50 à 90% de la résistance secondaire. Plus de 50 mutations BCR-ABL ont été détectées chez des patients résistants. Elles intéressent toutes les régions de l’ABL kinase. Néanmoins 4 régions ont été mises en avant et qui sont : la boucle P, la thréonine 315, la méthionine 351 et la boucle A.
Amplification ou la surexpression de BCR- ABL : qui est observé dans 10% des cas.
– Les mécanismes BCR-ABL indépendants :
sont d’origine pharmacologique et sont :
Une augmentation du transport de l’imatinib qui dé- pend du gène MDR1, si celui-ci est muté ou amplifié, les protéines exprimées ABC1 et BCRP, responsables de l’efflux d’imatinib, vont voir leur activité augmenter et donc diminuer la concentration d’imatinib. Il y a également diminution de l’influx d’imatinib dans les cellules par inhibition de la pompe hOCT1,
Un taux d’imatinib plasmatique insuffisant par inobservance ou interaction médicamenteuse (mécanisme extracellulaire), ce qui souligne l’intérêt du dosage de l’imatinib,
Une activation d’autres voies de signalisation indépendantes de BCR-ABL (SRC kinases, Ras),
Une instabilité génomique avec une évolution clonale de la maladie constamment associée à l’apparition d’anomalies cytogénétiques additionnelles.
Inhibiteurs de 2ème génération :
Dasatinib (SPRYCEL) : c’est un inhibiteur puissant, 300 fois l’activité in vitro de l’imatinib, de BCR-ABL et la voie Src. Il est actif sur la plupart des mutations de la protéine BCR-ABL, sauf la T315I. Il est indiqué dans la LMC en phase chronique, accélérée ou blastique, en cas de résistance ou d’intolérance à un traitement antérieur incluant l’imatinib. La posologie recommandée est de 100 mg/ j en phase chronique, elle est de 140 mg/j dans les phases accélérée ou blastique.
Nilotinib (TASIGNA) : c’est un puissant inhibiteur de l’activité tyrosine kinase ayant une forte affinité pour le site ATP de la kinase. Il est 25 fois plus puissant in vitro que l’imatinib. Il est indiqué dans la LMC en phase chronique et accélérée, chez les patients résistants ou intolérants à l’imatinib [23]. Indiqué à la dose de 400 mg x 2/j à 12h d’intervalle.
Bosutinib : c’est un inhibiteur des Src kinases et de BCR-ABL. Il est 200 fois plus puissant que l’imatinib in vitro, mais inactif sur la T315I [22].
Inhibiteurs de 3ème génération : Ponatinib : La mutation T315I est retrouvée chez 4% à 19% des patients résistants à l’imatinib [23]. Le Ponatinib représente l’espoir de ces patients.
Conclusion :
Actuellement, le traitement de la LMC est de mieux en mieux codifié, les recommandations récentes de l’European Leukemia Net (ELN) permettent maintenant de mieux positionner la greffe de cellules souches et d’envisager la place des inhibiteurs de deuxième génération. La surveillance moléculaire et l’interprétation des résultats permettent d’ajuster avec précision les doses d’inhibiteurs.
Références bibliographiques :
Flandrin G. La nouvelle classification OMS des hémopathies malignes. Hématologie 2001;7: 136-41.
Nowell PC, Hungerford DA. A minute chromosome in human granulocytic leukemia. Science 1960 ; 132 : 1497.
Djouadi-Lahlou K. Etude épidémiologique nationale de la leu- cémie myéloïde chronique : travail coopératif et multicentrique sur une période de 16 ans. A propos de 1927 cas (1994-2009)
Abdennebi-Mansour N. Le traitement de la leucémie myéloïde chronique en première phase chronique par l’imatinib chez les pa- tients n’ayant pas de donneur HLA compatible. Thèse de doctorat en sciences médicales ; Alger ; Novembre 2010.
Herlet S. Les inhibiteurs de tyrosine kinase dans le traitement de la leucémie myéloïde chronique chez l’adulte, du Glivec aux traitements de deuxième génération, conséquence de la sortie de la réserve hospitalière pour le pharmacien d’officine. Thèse de docto- rat en sciences médicales ; Nancy; mars 2010.
Arkesteijn G-I-A, Martens A-C-M, Hagenbeek A. Bivariate flow ka- ryotyping in human Philadelphia-positive chronic myelocytic leukemia. Blood 1998;72: 282-286.
Fabarius A, et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML study IV. Blood 2011;26: 6760- 68.
Amare PS, Baisane C, et al. FISH: a highly efficient of mole- cular diagnosis and prediction for disease course in patients with myeloid leukemia. Cancer Genet Cytogenet2001;131: 125-134.
GabertJ, Beillard E, Vander Velden VH et al. Standardization and quality control studies of real time quantitative reverse trans- criptase polymerase chain reaction of fusion gene transcripts for residual disease in leukemia. A Europe Against Cancer Program. Leukemia;2003;17: 2318- 2357.
James W- Vardiman, Nang lee Harris and Richard D-Brun- ning. The world health organisation (WHO) classification of the myeloid neoplasms. Blood 2002;100: 2292- 2303.
Calabretta B, Perrotti D. The biology of CML-blast crisis. Blood 2004 ; 103 : 4010-4022.
Sokal JE, Cox EB, Baccarani M et al. Prognostic discremina- tion in good risk chronic granulocytic leukemia. Blood 1984;63: 789- 99.
Hasford J, Pfirman M, Helmann R, et al. A new pronostic score for survival of patients with CML treated with interferon alfa. J. Nat. Ins 1998;90: 850-8.
Gratwohl A, Herman J, Goldman JM et al. Risk assessment for patients with CML working party of the European Group for Bone Marrow Transplantation (EBMT). Lancet 1998 ; 352 : 1087-92.
Benakli M, Hamladji RM, Ahmed-Nacer R. leucémie myé- loïde chronique. Nouvelle revue médicale 2010 ;8 :46-56.
Hehlmann R, Berger V, Pfirrmann M et al. Randomised comparaison of interferon alpha and hydroy-urea, wiyh hydroxy urea monotherapy by the combination of interferon alpha and hydroxyl-urea. Leukemia 2003;17:1529-37.
Talpaz M, Kantarjian HM, Mc Credie KB, Trujillo JM, Kea- ting JM, Gutterman JU. Hematologic remission and cytogenetic improvement induced by recombinant human interferon alpha in CML. N. Eng. J. Med 1986;314: 1065-1069.
Goldman JM, Gale RP, Fefer A. Bone marrow transplantation for CML in chronic phase. Ann Intern Med; 1988 :806-814.
Baccarani M, Cortes J, Niederwieser D, Saglio G, Apperly J, Cervantes F, Deninger M, Gratwohl A, Guilhot F, Hochhaus A, Horowits M, Hugues T, Kantarjian H, Larson R, Rachid J, Simons- son B, Silveira RT, Gldman J, Helmann R. Chronic myeloid leukemia: An update of concepts and management recommendations of European Leukemia Net. Journ of clini oncolo 2009 ; 35 : 6041-51. : Baccarani ELN 2009
Baccarani M et al, Blood 2013 Aug 22 ;122(8) : 872-84.
Goldman JM, et al. Chronic myeloid leukemia- advances in biology and new approaches to treatment. N Eng J Med 2003;349: 1451-64.
Brummendorf TH, Cervantes F, Kim D. Bosutinib is safe and active in patients with chronic-phase chronic myeloid leukemia with resistance or intolerance to imatinib and other tyrosine kinase inhibitors. J Clin Oncol 2008;26: 372.
Cortes JE, et al. Phase I study of Ponatinib, best response to therapy CP-CML. Blood 2011; (11892): abstract 602.24 :
Pr Y. BERKOUK-REDJIMI, Dr H. AHMIDATOU,Pr N. BOUDJERRA. Service d’Hématologie – CHU Beni Messous.
Abstract : These last decades, the major advances in multiple myeloma comprehension and the development of clinical essays allowed considerable improvement in diagnosis, prognostic evaluation, therapeutic indications and survival of patients diagnosed with myeloma. In this article, we report the up-dates on this hematological malignancy. In terms of diagnosis, the International Myeloma Working Group (IMWG) updated the diagnostic criteria. In terms of prognosis, the score of International staging system (ISS) was revised and regarding treatment, new generations of molecules were introduced such as monoclonal antibodies, Imids and protesome inhibitors.
Résumé : Les avancées majeures de ces dernières décennies dans la compréhension du myélome multiple et la réalisation d’essais cliniques, ont permis d’améliorer considérablement la prise en charge et la survie des patients. En matière de diagnostic, l’International Myeloma Working Group (IMWG) a actualisé les critères diagnostiques. Le score pronostique, International staging system (ISS), a été révisé et en matière de traitement, de nouvelles générations de molécules ont été introduites comme les anticorps monoclonaux, les Imids et les inhibiteurs du protéasome. Nous rapportons dans cet article, ces différentes actualités sur cette hémopathie
Le myélome multiple (MM) est une prolifération clonale de plasmocytes au niveau médullaire, sécrétant ou non, une immunoglobuline monoclonale. Il représente 10 % des cancers hématologiques et 1 % de l’ensemble des cancers[1]. Ces dernières décennies, les avancées majeures dans la compréhension de cette maladie et la mise au point d’essais cliniques, ont permis d’améliorer considérablement le diagnostic, l’évaluation pronostique, les indications thérapeutiques et la survie des patients voire d’espérer une guérison pour certains. Dans cet article, nous avons regroupé les actualités sur le myélome, dans le domaine diagnostique, pronostique et thérapeutique.
Actualités dans la physiopathologie :
Il est désormais bien établi que plusieurs acteurs interviennent dans le MM : les plasmocytes et le microenvironnement cellulaire (cellules stromales mésenchymateuses, cytokines, lymphocytes T, ostéoblastes/ostéoclastes). Un élément crucial différenciant le myélome symptomatique du myélome asymptomatique est le nombre de lésions ostéolytiques, résultant d’un déséquilibre entre ostéoblastogenèse et ostéoclastogenèse. Le système RANK/RANK ligand joue en particulier un rôle central : son activation signe l’entrée dans la maladie (il est absent au cours des gammapathies monoclonales bénignes).
D’autres voies de signalisation dont le wingless (Wnt), dikkopf-1(DKK-1) ou secreted frizzled protein related-2 (sFRP-2) ont été récemment impliquées [2].
Actualité dans la biologie :
La présence d’un composant monoclonal sérique et/ou urinaire était un des critères majeurs du diagnostic du myélome multiple[3]. Actuellement, elle n’est plus nécessaire pour poser le diagnostic de myélome mais permet de prendre en compte les myélomes oligo ou pauci-secrétants[4].
Le ratio des chaines légères libres (Free light chain FLC) n’était pas parmi les anciens critères diagnostiques du myélome, il était un des critères d’évaluation thérapeutique. Il a été reporté que l’évolution des patients avec un myélome indolent et un ratio FLC ≥ 8, était associée à un risque de 40% de progression dans les 2 premières années suivant le diagnostic, ensuite, le seuil du ratio FLC associé à une probabilité de 80% de progression dans les 2 années suivant le diagnostic a été déterminé, il était de 100[5]. Un ratio FLC d’au moins 100 est un prédicateur de progression vers un myélome symptomatique et les patients asymptomatiques avec ce ratio doivent être considérés à haut risque et doivent être traités, c’est pour cela, que ce critère (ration FLC ≥ 100) a été inclus dans les nouveaux critères diagnostiques[5]. Un autre paramètre biologique actualisé, est la définition de l’insuffisance rénale, qui est un des critères CRAB, elle est précisée avec ajout d’une valeur seuil de clairance de la créatinine < 40 ml/mn[4].
Actualité dans l’Imagerie :
Les lésions osseuses ostéolytiques sont l’un des plus pro-éminents signes du myélome multiple. Elles sont présentes chez plus de 80% des patients au moment du diagnostic[6]. Les lésions observées sont très souvent une ostéoporose (signant l’ostéopénie), des lésions ostéolytiques (géodes ou lacunes) et des fractures[2]. La radiologie conventionnelle est la référence : crâne, rachis complet, bassin, thorax et grils costaux, humérus et fémurs[2].
Les nouvelles techniques d’imagerie morphologique (scanner corps entier faible dose) et d’imagerie fonctionnelle (l’imagerie par résonance magnétique IRM et la tomographie par émission de positons – TEP – scanner) ont été intégrées dans la nouvelle classification. Leurs sensibilités et spécificités sont nettement supérieures à celles des radiographies standards.
Au diagnostic, la tomodensitométrie (TDM) faible dose remplace les radiographies standards, car elle a l’avantage d’être rapide, de délivrer une faible dose d’irradiations et d’avoir un coût faible comparativement aux autres techniques. Le scanner faible dose est actuellement l’imagerie de référence pour l’identification de lésions lytiques depuis les recommandations européennes publiées en 2015[6] : en l’absence de lésions lytiques osseuses sur les radiographies, le scanner faible dose et/ou le TEP et/ou une IRM doivent être réalisés à la recherche de lésions focales. La mise en évidence d’au moins deux lésions ostéolytiques ≥ 5 mm au scanner faible dose ou au TEP est un critère suffisant pour définir une atteinte osseuse[4].
Nouveaux critères diagnostiques
Depuis 2003, le diagnostic d’un myélome multiple symptomatique se base sur l’existence de complications de la maladie (critères CRAB : anémie, insuffisance rénale, hypercalcémie, lésions osseuses). Il était donc nécessaire d’attendre l’apparition de ces complications pour envisager l’instauration du traitement par chimiothérapie[4]. Mais la faiblesse de ces critères et le fait que les patients classés asymptomatiques (myélome multiple asymptomatique ou indolent) et qui n’étaient pas traités, progressaient très rapidement vers un myélome symptomatique, a suscité les chercheurs à mener des études afin d’individualiser les facteurs de risque de progression.
En 2014, au vu des résultats obtenus, l’International Myeloma Working Group (IMWG) a décidé, d’intégrer 3 bio-marqueurs de myélome indolent (asymptomatique) à très haut risque dans la définition du myélome multiple : plasmocytose médullaire ≥ 60%, rapport K/L ou L/K ≥ 100 et au moins 2 lésions focales osseuses en IRM[4,6]. Après l’actualisation par l’IMWG en 2014, les nouveaux critères du myélome multiple sont[5] :
Plasmocytes médullaires monoclonaux ≥ 10 % ou plasmocytome osseux ou extramédullaire confirmé par biopsie.
Et au moins un des critères suivants :
Signes de lésions sur les organes cibles pouvant être attribués à la prolifération plasmocytaire sous-jacente (critères CRAB) :
Hypercalcémie : calcium sérique > 2,5 mmol/L (> 1 mg/dL) au-dessus de la limite supérieure normale ou > 2,75 mmol/L (>11 mg/dL)
Insuffisance rénale : clairance de la créatinine < 40 ml/min ou créatinine sérique > 177 µmol/L (> 2 mg/dL)
Anémie : hémoglobine (Hb)> 20 g/L sous la limite inférieure normale ou valeur Hb < 100 g/L
Lésions osseuses : au moins une lésion ostéolytique à la radio du squelette, CT ou TEP-CT
Et / ou la présence de l’un des marqueurs suivants :
Plasmocytes médullaires clonaux ≥ 60%
Ratio des chaines légères libres ≥ 100
Plus d’une lésion focale à l’IRM (au moins 5 mm)
La définition des myélomes indolents a elle aussi été modifiée, pour établir le diagnostic d’un myélome multiple indolent ou asymptomatique, il faut que les deux critères suivants soient satisfaits[5] :
Protéine monoclonale sérique (IgG ou IgA) ≥ 30 g/L ou protéine monoclonale urinaire ≥ 500 mg/ 24H et / ou plasmocytes médullaires clonaux compris entre 10 et 60% ;
Absence de critères caractéristiques du myélome décrits ci-dessus ou d’amylose.
Sont définis comme indolents à très haut risque de progression (80% à 2 ans), les patients avec au moins un des biomarqueurs de malignité. Patients qui doivent être considérés et traités comme des myélomes symptomatiques[4].
Actualité sur les facteurs pronostiques :
Dans le myélome multiple les facteurs pronostiques sont soit liés au patient (comme l’âge, le score de performans status ECOG et les comorbidités), soit liés à la maladie. Ces derniers sont des facteurs biologiques et cytogénétiques qui doivent être recherchés avant la mise en route du traitement du fait de leur caractère pronostique. Les facteurs biologiques (taux de β2-microglobuline et Albumine sérique) définis par l’International Staging System (ISS) (conférer le tableau 1) ont été révisés en 2015 par l’International Myeloma Working Group (IMWG[8]. A ces facteurs ont été ajoutés le taux de LDH et les anomalies cytogénétiques dits de haut risque comme la délétion 17p, t(44 ;14), t(14 ;16).
β2-microglobuline sérique < 3,5 mg/L et albumine sérique ≤ 35g/L ou 3,5 mg/L β2-microglobuline sérique < 5,5 mg/L quelque soit le taux d’albumine sérique
44
III
β2-microglobuline sérique ≥ 5,5 mg/L
29
Tableau 1 : stades de l’ISS
L’hybridation in situ en fluorescence (FISH) sur plasmocytes, permet de mettre en évidence les trois principales anomalies cytogénétiques qui sont associées à un pronostic péjoratif : la translocation (4;14), la délétion du bras court du chromosome 17 (17p) et la délétion 1p32[7,8]. L’hyperdiploïdie à type de trisomie 3 ou 5, est considérée comme un facteur de bon pronostic. Associée aux anomalies précédemment citées, elle peut atténuer leur caractère péjoratif. Par contre, la trisomie 21 peut assombrir le pronostic d’un patient sans la présence d’anomalie à caractère péjoratif (translocation ou délétion)[7].
Actualité sur la prise en charge thérapeutique :
Le myélome multiple reste à ce jour une hémopathie lymphoïde incurable. Toutefois, depuis les 40 dernières années, de nombreuses avancées ont été réalisées dans la prise en charge de cette pathologie et ce, notamment chez le sujet de moins de 65 ans.
La première avancée a été l’apport de la polychimiothérapie type VAD associant Vincristine Adriamycine et Dexamethasone, qui a permis d’obtenir de très bonnes réponses, voire des remissions complètes. Ce protocole, couplé à une intensification avec du Melphalan à fortes doses 200mg/m2, suivi d’une autogreffe de cellules souches périphériques, a permis d’augmenter la survie sans progression et la survie globale[9].
La seconde avancée a été l’introduction durant les années 90 d’immunomodulateurs comme les Imids type Thalidomide, Revlimid et Pomalidomide[9,10,11] et l’introduction d’inhibiteurs du protéasome comme le Bortézomib puis plus récemment le Carfilzomib et l’Ixazomib. Ces nouvelles molécules en traitement de première ligne, permettent d’obtenir des remissions complètes moléculaires avec des prolongations de la survie dépassant les 5 années et pouvant atteindre 10 années[11], grâce à des séquences d’induction (4 cures) suivies d’une consolidation (2 cures) avec entre les deux une intensification (Melphalan à fortes doses avec autogreffe de cellules souches périphériques).
Ces molécules ont trouvé également leur place en deuxième et troisième ligne chez le patient en rechute ou réfractaire[9,11].
La troisième avancée est en cours et cela depuis 2012, où une nouvelle classe thérapeutique s’est imposée dans le myélome multiple avec l’immunothérapie et les anticorps monoclonaux[9]. Le premier est un anticorps monoclonal de type IgG1 anti-SLAMF7 ou anti-CS1 exprimé par les plasmocytes malins, les lymphocytes NK et LT-CD8. Son efficacité en monothérapie a été décevante, mais il a montré tout son potentiel en association avec un Imid[9]. Quant à l’anticorps monoclonal anti-CD38, il semble être très efficace en mono chimiothérapie et est actuellement à l’étude en association avec un Imid ou un inhibiteur du protéasome chez des patients réfractaires ou en rechute. Ces dernières molécules, en plus de leur efficacité, ont l’avantage de présenter très peu d’effets secondaires[9].
Les multiples possibilités d’association thérapeutique entre Imid, inhibiteurs du protéasome et les anticorps monoclonaux ouvrent la perspective d’un traitement personnalisé avec de meilleurs taux de réponse voire une guérison chez le sujet de moins de 65 ans[9].
Les sujets âgés entre 65 et 75 ans ont également bénéficié des avancées thérapeutiques dans le myélome multiple et leur prise en charge est comparable à celle du sujet jeune sous réserves des comorbidités[12,13].
Les patients non éligibles à l’intensification ou âgés de plus de 75 ans, bénéficient également des nouvelles thérapeutiques. Il est cependant recommandé de diminuer les doses ou le rythme des injections. L’association actuellement recommandée est un Imid et Dexamethasone[12,13].
Conclusion :
La prise en charge actuelle d’un patient présentant un myélome multiple est calquée sur celle d’un patient avec une leucémie aigüe. Ce traitement comporte une phase d’induction, suivie d’une intensification puis d’une consolidation. L’association Imid + inhibiteurs du protéasome à de la Dexamethasone (trithérapie) est le traitement de référence en induction et la tendance est à la quadrithérapie avec les nouvelles classes de traitement (anticorps monoclonaux). Cette nouvelle approche thérapeutique permet d’obtenir de meilleurs taux de réponse et des survies sans progression pouvant dépasser les 10 années en l’absence de facteurs cytogénétiques de mauvais pronostic.
Ce qu’il faut retenir :
La présence d’un composant monoclonal sérique ou urinaire n’est plus nécessaire pour poser le diagnostic.
3 biomarqueurs de malignité s’ajoutent désormais aux critères CRAB : plasmocytes médullaires ≥ 60%, rapport κ/λ ou λ/κ ≥ 100 et au moins 2 lésions focales osseuses en IRM, permettant d’initier un traitement avant l’apparition des complications du myélome.
L’insuffisance rénale est précisée avec une valeur seuil de clairance de la créatinine < 40 ml/ mn.
En l’absence de lésion lytique osseuse sur les radiographies standards, le scanner faible dose et/ou le TEP, une IRM doivent être réalisés à la recherche d’une lésion focale.
Les anomalies cytogénétiques ont un poids pronostique et doivent être systématiquement recherchées au diagnostic.
L’ISS-révisé incorpore la cytogénétique et le taux de LDH
Intensification est actuellement incontournable dans le traitement du sujet de moins de 65 ans.
La consolidation post intensification avec autogreffe de moelle osseuse a un impact sur la survie.
Les nouvelles classes thérapeutiques arrivent en première ligne.
Le sujet âgé peut être traité comme le sujet jeune sous réserve des comorbidités.
Références :
J. Corre. Biologie du myélome multiple. Horizon Hémato, vol 6, N°1, 2016 : 18 – 9
C. Emille. Le myélome multiple : actualités biologiques. Option Bio, Septembre 2015, N° 531 : 20-1
S. Cerdá, B. Ballina, P. Escribano, L. Villalobos, J. Sánchez-Real, M. Fuertes et al. IMWG ’03 vs ’14 Diagnostic Criteria for Sympto- matic Multiple Myeloma: Will We Have to Solve a Problem? 15th International Myeloma Workshop, September 23-26, 2015: e122-3
O. Decaux. Nouveaux critères du myélome multiple et imagerie. Horizon Hémato, vol 6, N°1, 2016 : 20 – 1
S. Vincent Rajkumar et al. International myeloma working group update criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014, 15 : E538 – 48
E. Terpos, M. Kleber, M. Engelhardt, S. Zweegman, F. Gay , E. Kastritiset al. European Myeloma Network guidelines for the mana- gement of multiple myeloma-related complications. Haematologica. 2015 Oct; 100 (10) : 1254-66
Corre J. Biologie du myélome. Horizon Hémato. La revue des pra- tiques en Hématologie. Volume 6 numéro 01, Janvier/ Février/ Mars 2016 :18-19
Palumbo A et coll. Revised International Staging System for multiple myeloma: A report from International Myeloma Working Group. J.Clin Oncol,2015. 33(26) :2863-2869
Hebraud B. Myélome Multiple. Horizon Hémato. La revue des pratiques en Hématologie, supplément volume 06 numéro 01, février 2016 :47-48
Richardson PG et coll. Immunomodulatory drug,overcomes drug resistance and is well tolerated in patients with multiple myeloma. Blood 2002, Octobre 16; 100(09) :3063-3067
Roussel M. traitement des patients de novo eligibles a l’intensi- fication. Horizon Hémato. La revue des pratiques en Hématologie. Volume 6 numéro 01, Janvier/ Février/ Mars 2016 :11-13
Hulin C. Traitement des patients de novo non eligibles a une in- tensification/ autogreffe. Horizon Hémato. La revue des pratiques en Hématologie. Volume 6 numéro 01, Janvier/ Février/ Mars 2016 :15-17
Kint N, Delforge M. Concise review-treatment of multiple myélome in the very elderly: how do novel agents fit in ? Journal of geriatri- concology 7,2016 :384-389. Available online at : www.sciencedirect.com
Pr M.T ABAD Pr S. TAOUSSI EHS ELCC – Service d’Hématologie – Blida
Abstract : Hodgkin’s lymphoma is a relatively frequent hematologic malignancy in Algeria. It is an affection of the young adult, which affects the same proportions as men and women. The usual mode of presentation of the disease is dominated by the appearance of cervical lymph nodes axillary lymph nodes, and more rarely inguinal lymph nodes with or without general signs, which should lead to a lymph node biopsy and an anatomopathological examination with immunolabeling (CD30, CD15) for diagnosis. Radiological and biological clinical assessment should lead to staging, a necessary basis for indicating whether the disease is localized or generalized, a finding which will depend on the type of treatment. Under specific treatment, chemotherapy +/- radiotherapy, its prognosis is excellent since more than 70% of patients are cured. This means that between 20% and 30% of patients have either an extended illness or a poor prognosis factor and will not respond optimally to the treatment and will either resist (refractory) or relapse before one year. Then a very heavy treatment that can go as far as the autograft of hematopoietic stem cells.
Résumé : Le lymphome de Hodgkin est une hémopathie maligne relativement fréquente en Algérie. C’est une affection de l’adulte jeune, qui touche dans les mêmes proportions, autant les hommes que les femmes. Le mode de présentation habituel de la maladie est dominé par l’apparition d’adénopathies cervicales, axillaires, plus rarement inguinales, accompagnées ou non de signes généraux, situation qui doit conduire à une biopsie ganglionnaire et un examen anatomopathologique avec immunomarquage (CD30, CD15) pour assurer le diagnostic. Le bilan d’extension clinique, radiologique et biologique doit conduire à un staging, base indispensable pour indiquer si la maladie est localisée ou généralisée, constat dont dépendra le type de traitement. Sous traitement spécifique, chimiothérapie +/-radiothérapie, son pronostic est excellent, puisque plus de 70% de patients sont guéris. Cependant, près de 20% à 30% de patients ayant soit une maladie étendue soit des facteurs de mauvais pronostic ; ne vont pas répondre de façon optimale au traitement et vont soit résister (réfractaires) soit rechuter précocement (avant un an) et nécessiter alors un traitement très lourd pouvant aller jusqu’à l’autogreffe de cellules souches hématopoïétiques.
Mots-clés : Lymphome de Hodgkin, épidémiologie, extension, facteurs pronostiques.
Définition :
Depuis 2008, dans la Classification OMS des hémopathies lymphoïdes, le terme de « Lymphome de Hodgkin » remplace celui de Maladie de Hodgkin. Le lymphome de Hodgkin est une hémopathie maligne bien codifiée : elle est définie comme une tumeur maligne du tissu lymphoïde avec prolifération de cellules anormales, les cellules de Sternberg sur un fond de granulome inflammatoire et un envahissement capsulaire. C’est une affection dont le pronostic est excellent avec des taux importants de guérison allant de 70% dans les formes étendues de la maladie à 95% dans les formes strictement localisées.
Généralités :
Étiologie
Après plus de 180 ans depuis sa première description par Sir Thomas Hodgkin en 1832, la cause exacte de cette affection n’est pas connue même si des facteurs viraux sont mis en cause (Virus Epstein Bar : EBV).
Incidence
Les taux d’incidence standards en Europe sont estimés à 2-5 cas pour 100.000 habitants chez l’homme et à 1-2 cas chez la femme(1). Dans les pays en voie de développement le taux d’incidence est mal connu. Il serait faible par comparaison aux pays européens. Une première étude a été faite en 2006 en Algérie ; elle a porté sur une période de 10 ans (1996- 2005) et a montré une incidence de 0 ,7 cas pour 105 habitants et par an. En 2012, elle est passée à 1,8 cas pour 105 habitants(2). La fratrie de patients atteints du LH a un risque supérieur par rapport à la population générale d’être atteinte de cette maladie.
Répartition selon le sexe
En Algérie, le LH atteint de la même manière l’homme et la femme (sex-ratio=0,99), alors qu’en Europe la pré-dominance masculine est notée dans toutes les études. Répartitionselonl’âge
L’âge moyen au diagnostic est 31,7 ans (16-99) (hommes = 33,2 ans (16-99) ; femmes = 30,2 ans (16-88)). Il existe un seul pic de fréquence entre 16 et 34 ans. En Europe, un deuxième pic est observé vers 70 ans(1).
Délai diagnostique
Le retard pour le diagnostic est important puisque le délai diagnostique moyen est de 7 mois(1-48). Manifestations cliniques Circonstances de découverte
Le premier symptôme est représenté par :
Les adénopathies superficielles : 78,5%
Une toux rebelle au traitement symptomatique : 8,6%
Des signes généraux : 5,7% : fièvre au long court, amaigrissement, sueurs nocturnes
Localisation initiale des adénopathies:
Cervicale
(71%)
Médiastinale
(13,4%)
Axillaire
(6,8%)
Inguinale
(5,2%)
Abdominale
(3,1%)
Splénomégalie
(0,3 %)
Diagnostic de LH :
Il est facile dans la majorité des cas. Il est soupçonné sur une étude cytologique du produit de ponction ganglionnaire (GP) ; il est confirmé obligatoirement par une étude anatomopathologique sur une biopsie ganglionnaire superficielle ou profonde : étude morphologique et immunomarquage par CD 30, CD15.
La cellule de Sternberg est une grande cellule de 30- 40µ de diamètre, avec un noyau mono ou plurilobé, peu dense, parfois en miroir, possédant un à plusieurs nucléoles proéminents. Ces cellules présentent habituellement les marqueurs CD30 et CD15 et parfois le CD20. Le type histologique scléro-nodulaire (type 2) est le plus fréquent avec 75% des cas, suivi du type à cellularité mixte (type 3) dans 19,4% des cas.
Bilan d’extension de la maladie :
Il est indispensable pour évaluer l’étendue de la maladie, de définir le pronostic et par conséquent le traitement adapté surtout par rapport à l’indication de la radiothérapie.
Il est basé sur de nombreuses explorations cliniques, radiologiques, et biologiques. L’examen clinique complet doit répertorier toutes les atteintes ganglionnaires périphériques, mesurer la taille de la rate et du foie, identifier les signes généraux (fièvre au long court >38° C, sueurs nocturnes profuses mouillant le linge, amaigrissement récent de plus de 10% du poids du corps, prurit rebelle au traitement en absence d’affection dermatologique prurigineuse). Les examens radiologiques comprennent une radiographie thoracique de face, tomodensitométrie (TDM) thoraco-abdomino-pelvienne avec au mieux une tomographie par émission de positrons (TEP) au 18 fluorodésoxyglucose couplée à la TDM qui permet de mieux classer 10 à 30% des patients classés par la TDM seule en augmentant le stade de la maladie au diagnostic[7]. Le bilan biologique doit comporter un hémogramme, un frottis sanguin, une VS, un taux d’albumine, une électrophorèse des protides, un bilan hépatique avec un taux de phosphatases alcalines, les sérologies VIH, VHB, VHC, un bilan rénal, un bilan de l’hémostase. La biopsie ostéo-médullaire (PBO) est nécessaire pour préciser un envahissement médullaire.
Évaluation pronostique :
Le traitement du lymphome de Hodgkin doit être adapté non seulement au stade anatomoclinique mais aussi aux facteurs pronostiques.
La classification clinique de Ann Arbor modifiée par Costwolds en 4 stades (localisés I et II, étendus III et IV) est toujours en vigueur(3). Elle est la base du traitement passé et actuel du lymphome de Hodgkin.
Les facteurs pronostiques sont évalués par plusieurs classifications , celle de l’EORTC(4) distingue pour les stades I et II sus diaphragmatiques 2 groupes : favorable et défavorable, la deuxième classification basée sur le plus grand nombre de patients et qui concerne tous les stades est la classification allemande du GHSG (German Hodgkin Lymphoma Study Group), qui stratifie l’ensemble des 3 stades cliniques selon 3 groupes pronostiques : précoce, intermédiaire et avancé(5), cette classification en fait individualise le stade II B haut risque comportant soit une masse médiastinale volumineuse (≥10cm ou rapport médiastino-thoracique : IMT ≥0,35) et/ou une atteinte extra ganglionnaire et l’inclue dans les stades avancés au même titre que les stades étendus III et IV, ce qui influe sur l’orientation thérapeutique. Par contre l’utilisation du SPI (Score Pronostique International) dans les stades étendus III et IV(6) pour déterminer des groupes de risque et une stratégie thérapeutique adaptée au risque, ne fait pas consensus.
Traitement du lymphome de Hodgkin
Lymphome de Hodgkin au stade localisé favorable : il est recommandé une chimiothérapie par trois cycles d’ABVD, suivie d’une radiothérapie de type IFRT (aires ganglionnaires initialement atteintes) à 30 Gy en cas de rémission complète(7).
Lymphome de Hodgkin, stade localisé défavorable (selon classification GHSG) : il est recommandé d’utiliser 4 cycles d’ABVD associés à une radiothérapie IFRT à 30 Gy ou un schéma de 2 cycles de BEACOPP renforcé suivis de deux cycles d’ABVD en association à une radiothérapie IFRT à 30 Gy(8).
Dans les stades avancés ou disséminés regroupant (GHSG) tous les stades II B haut risque et les stades III et IV), le schéma classique est l’ABVD 6 à 8 cycles(9), l’autre schéma BEACOPP escaladé basé sur le concept de dose intensité développé par le GHSG comportant 8 cycles, a été actuellement réduit à 6 cycles permettant d’obtenir une survie globale (OS) comparable avec une diminution de la toxicité(10). La place de la radiothérapie dans ses formes avancées, durant un certain temps non recommandée, est actuellement utilisée à la dose de 30 Gy lorsque persistent des masses résiduelles de plus de 1,5 cm à la TDM, car elle s’est révélée efficace pour prévenir le risque de rechute(11).
La TEP occupe une place importante dans le suivi de la réponse immédiate (après 2 cures) au traitement dans l’ensemble des études cliniques récentes. Elle apporte selon le résultat (négatif ou positif) une réponse indispensable à la poursuite du traitement en termes de désescalade ou d’escalade thérapeutique(12).
Références :
A. Mohnereau , X. Troussard, M. Maynadie. Estimation nationale de l’incidence des cancers en France entre 1988 et 2012. Revue de l’Institut National du Cancer, Septembre 2013.
Abad M.T et al. Epidémiologie de la maladie de Hodgkin en Algérie. Première journée d’hématologie de Blida, 2006.
Lister. TA, Crowther. D, Suteliffe SB et al. Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Costwolds meeting J Clin Oncol 1989;7: 1630-6.
Tubiana. M, Henry Amar. M, Carde. P et al. Toward comprehensive management tailored to prognostic factors of parents with clinical stades I and II in Hodgkin’s disease. The EORTC Lymphoma Group Controlled Clinical Trials: 1964-1987 Blood 1998;73: 47-56.
Sieber. M, Engert. A, Diehl. V. Treatment of Hodgkin’s disease results and current concepts of the German Hodgkin’s Lymphoma Study Group Ann Oncol 2.000; 11 (SUPPL.1) 81-5.
Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med 1998; 339 :1506-14.
Fermé. C, Eghbali. H, Meerwaldt. J H et al. Chemotherapy plus involved-field radiation in early – stage Hodgkin’s disease N. Engl. J. Med 2007;357: 1916-27.
Von Treschow. B, Plutschow. A, Fuchs. M et al. Dose intensification in early infavorable Hodgkin’s lymphoma final analysis of the German Hodgkin’s Study Group HD14 trial J. Clin Oncol 2012;30: 907.
Gordon. LI, Hong. F, Fisher. RI et al. Rondomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced. stage Hodgkin lymphoma: an intergroup study coordinated by the Eastern Cooperative Oncology Group. J. Clin Oncol 2013;31: 684-91.
Engert. A, Haverkamp. H, Kobe. C et al. Reduced intensity Chemotherapy and PET- guided radiotherapy in patients with advanced stage Hodgkin’s lymphoma (HD 15 trial): a randomized, open -label, phase 3 non-inferiority trial Lancet 2012;379: 1791-9.
Borchman. P, Haverkamp. H, Diehl. V et al. Eight cycles of esca- lated –dose BEACOPP compared with four cycles of escaladed-dose BEACOPP followed by four cycles of base-line dose BEACOPP with or without radiotherapy in patients with advanced-stage hodgkin’s lymphome final analysis of the HD12 trial of the German. Hodgkin Study Group J. Clin Oncol 2011:29: 4234-42.
Raemaekers. JM, André. MP, Federico. M et al: Omitting radio- therapy in early position emission tomopaphy-negative stade I/II Hodgkin lymphoma is associated with an increased risk of early relapse. Clinical results of the preplanned interim analysis of the randomized EORTC/LYSA/FIL HD trial J.Clin Oncol 2014:32: 1188-94.
Pr Nadia BOUDJERRA Service d’Hématologie CHU Beni Messous, Alger
Abstract : Non-Hodgkin’s lymphomas (NHL) are cancers that originate from B or T lymphocytes cells, rarely from Natural Killer (NK) cells. They encompass an extremely heterogeneous group of diseases, the heterogeneity is based on the multiplicity of histologic subtypes, clinical presentations (nodal and/or extra nodal). Lymphoma incidence is constantly increasing; we see a better understanding of their development and therapeutic progress, a significant number of cases especially in the aggressive lymphomas, may be cured.
Résumé : Les Lymphomes non Hodgkiniens (LNH), sont des hémopathies malignes développés à partir de cellules lymphoïdes B ou T rarement NK entrainant une hypertrophie des organes lymphoïdes et non lymphoïdes, la localisation ganglionnaire est la plus fréquente.
Les LNH se traduisent par une diversité clinique importante, la complexité des classifications histopathologiques modernes et leur évolutivité variable.
L’intérêt porté aux lymphomes est lié à l’augmentation de leur incidence, à une meilleure connaissance de leur développement et aux progrès thérapeutiques permettant actuellement une guérison dans un nombre important de cas notamment dans les lymphomes agressifs.
Mots-clés : Lymphome non Hodgkinien, diagnostic, classification histologique, facteurs pronostiques.
Introduction :
Les Lymphomes non Hodgkiniens (LNH), sont des hémopathies malignes, caractérisés par une prolifération monoclonale des cellules lymphoïdes de type B ou T à différents stades de différenciation et d’activation. Cette multiplication des cellules est responsable d’une prolifération tumorale pouvant siéger au niveau des organes lymphoïdes (ganglions, rate, amygdales) ou d’organes non lymphoïdes (tube digestif, thyroïde, peau, cerveau …).
Les LNH se distinguent des lymphomes hodgkiniens par l’absence de cellules des Reed Sternberg (RS) à l’étude anatomo-pathologique ; ils se traduisent par une diversité clinique, morphologique et biologique reflétant la complexité des classifications histopathologiques modernes.
L’intérêt particulier porté aux lymphomes est dû à l’augmentation de leur incidence, à une meilleure connaissance de leur développement et aux progrès thérapeutiques permettant actuellement une guérison dans un nombre important de cas (1).
Nous distinguons 2 grandes catégories de lymphomes selon leur caractère évolutif, les lymphomes indolents qui sont d’évolution lente et les lymphomes agressifs dont l’évolution est rapide et fatale en l’absence de traitement.
Notions épidémiologiques :
Selon les registres nationaux des cancers, nous observons depuis les années 90, une augmentation de l’incidence des lymphomes, dans les pays développés où des registres ont été mis en place, on note 12 à 15 nouveaux cas pour 100.000 habitants avec une augmentation de 5 à 10 % par an (2) ; actuellement les LNH occupent la première place au sein des hémopathies malignes et se situent au 8ème rang des cancers au sein de l’Union Européenne (3). Les LNH se voient à tout âge ; selon la littérature, l’âge médian se situe à 60 ans avec un pic de fréquence compris entre 65 et 85 ans (1), en France, l’âge médian au diagnostic était situé en 2000 à 64 ans chez l’homme et 75 ans chez la femme (4). L’homme est plus fréquemment atteint que la femme avec un sex ratio M/F de 1,5 (5).
En Algérie, les LNH occupent la première place au sein des hémopathies malignes (6), l’incidence des LNH a considérablement augmenté ; selon une étude faite sur les lymphomes ganglionnaires diagnostiqués au niveau de tous les services d’hématologie, l’incidence est passée de 0,81 en 2003 à 2,24 en 2012 (7), l’âge médian au diagnostic est de 54 ans (16-95) avec un pic de fréquence entre 50 et 70 ans, et un sex ratio M/F de 1,2 (7).
Dans notre étude, les lymphomes agressifs représentent la majorité des LNH (67,5%), parmi eux 61 % sont des lymphomes diffus à grandes cellules de type B (LDGCB), ces taux sont beaucoup plus élevés en comparaison avec les séries européennes et celles de l’Amérique du Nord mais rejoignent les chiffres retrouvés en Asie et au Moyen Orient (8), les lymphomes T sont plus rares, ils représentent 8,6% (8).
L’étiologie des lymphomes est-elle connue ?
L’étiologie des LNH n’est pas connue, mais certains facteurs sont incriminés expliquant l’augmentation constante des lymphomes (1), on citera :
La diminution de la réponse immunitaire qui expliquerait l’augmentation de l’incidence chez les personnes âgées,
L’association fréquente à des infections virales comme le virus d’Epstein Barr, le virus de l’hépatite C, le virus HTLV1 et le VIH chez le sujet jeune,
Les facteurs environnementaux tels que : pesticides, herbicides, produits chimiques, teintures de cheveux, essence de bois,
La profession la plus incriminée, en Europe et en Algérie, reste celle d’agriculteurs (1, 10).
Comment fait-on le diagnostic du lymphome et de son type histologique ?
Le diagnostic d’un lymphome est anatomo-pathologique, l’examen histologique se fait après prélèvement d’un ganglion entier ou d’une biopsie d’une masse extra ganglionnaire, dans le cas de localisations profondes des prélèvements biopsiques échoguidés ou scanno-guidés à l’aide d’aiguille de large calibre, permettent de retirer des fragments pour une lecture histologique. Le prélèvement doit être acheminé rapidement au niveau d’un laboratoire anatomo-pathologique, il est inclus au préalable dans du formol à 10 %. Une étude morphologique et immuno-histochimique seront effectuées.
La variété du lymphome a été établie selon la classification OMS 2008, cette dernière s’applique maintenant à l’ensemble des proliférations malignes de cellules hématopoïétiques.
La classification OMS prend en compte :
L’architecture folliculaire ou diffuse
Le phénotype B ou T/ NK
La présentation clinique
Les lymphomes de type B représentent selon la littérature 85 % des LNH (1), en Algérie les lymphomes B représentent, sur une étude faite dans la région centre : 87,3 % (8), et sur une étude nationale : 91,4% (7). Parmi les LNH de type B :
Les lymphomes agressifs représentent un peu plus d’un tiers en Europe et aux USA et plus de la moitié en Algérie, les LNH agressifs sont représentés majoritairement par des LDGCB, un faible pourcentage est représenté par le lymphome du Manteau, le lymphome primitif du médiastin (LPM) et le lymphome de Burkitt (7).
Les lymphomes indolents sont représentés en grande partie par le lymphome folliculaire qui est cependant plus fréquent en Europe et aux USA (8), les autres lymphomes indolents sont représentés par : le lymphome lymphocytique, le lymphome de la zone marginale (LZM) splénique et ganglionnaire, le lymphome extra ganglionnaire de la zone marginale du tissu lymphoïde associé aux muqueuses (Malt).
Parmi les LNH de type T, on distingue : le lymphome T angio-immunoblastique (LAI), le lymphome T périphérique sans autre précision (NOS), le lymphome à grandes cellules anaplasiques de phénotype T ou nul ALK + ou Alk et le lymphome extra ganglionnaire NK/ T de type nasal (7).
Le diagnostic anatomo-pathologique du lymphome est un diagnostic difficile d’où l’intérêt d’un groupe de relecture de lames qui est mis en place en Algérie depuis 2006 par le Groupe d’Étude Algérien des Lymphomes (GEAL).
Quel bilan doit-on effectuer en présence d’un lymphome ?
L’examen clinique permet :
d’apprécier l’évolutivité de la maladie, de même qu’il nous renseigne sur le caractère agressif.
La recherche d’antécédents personnels permet de mettre en évidence la notion d’une affection auto-immune qui aurait précédé le tableau du lymphome ; une affection virale, ou une maladie associée qui peuvent entraver le traitement.
On recherche dans les antécédents des cas similaires dans la famille.
L’examen clinique permet d’apprécier la mesure du retentissement de la maladie sur l’activité, pour cela on utilise une échelle semi quantitative simple et reproductible décrite par le groupe coopérateur américain ECOG et reprise par l’OMS :
0 = activité normale
1 = présence de symptômes mais poursuite d’une activité ambulatoire
2 = incapacité de travailler, alitement dans la journée mais inférieur à 50
%3 = alitement plus de 50 % de la journée
4 = alitement permanent, nécessité d’une autre personne.
De mettre en évidence des signes généraux classiques : fièvre > 38°pendant au moins une semaine, amaigrissement : plus de 10 % du poids du corps pendant les 6 derniers mois, sueurs nocturnes mouillant le linge.
De rechercher les signes de diffusion de la maladie :
en examinant toutes les aires ganglionnaires superficielles, tous les ganglions palpés doivent être mesurés et notés sur un schéma, les caractères des ganglions seront recherchés (sensibilité, consistance, mobilité). Rechercher une splénomégalie et mesurer, si elle est palpable, son débord splénique, rechercher une hépatomégalie avec mesure de la flèche hépatique (FH) , une atteinte de la sphère ORL à la recherche d’une prolifération , une biopsie sera faite selon les cas, examen de la peau et du cuir chevelu, d’autres localisations plus rares seront recherchées (thyroïde, testicule, atteinte neurologique). L’imagerie permet de rechercher une atteinte profonde :
Radiographie thoracique de face et profil
Echographie abdomino-pelvienne, qui lorsqu’elle est positive, permet de suivre l’évolution sous traitement et de limiter les examens tomodensitométriques abusifs.
Tomodensitométrie qui est l’examen essentiel pour juger de l’extension ganglionnaire et viscérale de la maladie, elle est obligatoire dans le bilan initial, elle est réalisée, durant le traitement pour évaluer l’efficacité du protocole mis en place ; elle permet en fin de traitement de confirmer la bonne réponse et éventuellement la rémission complète chez les patients répondeurs.
TEP Scanner, la tomographie à émission de positons couplée au scanner est un examen d’une grande utilité dans certains types de lymphomes et ce, afin de déterminer l’extension initiale et surtout de juger de la réponse au traitement, cet examen est hautement recommandé dans les LDGCB.
L’IRM permet de compléter le scanner dans certains cas d’atteintes osseuses, pariétales et dans le lymphome cérébral.
Une échographie cardiaque est nécessaire, elle permet de calculer la fraction d’éjection ventriculaire (FEV), obligatoire pour l’utilisation des anthracyclines.
D’autres examens sont effectués afin de rechercher une atteinte lymphomateuse,
La biopsie ostéo médullaire (BOM) : indispensable dans tous les lymphomes, elle est réalisée sous anesthésie locale au niveau de l’épine iliaque antéro-supérieure ou postéro-supérieure, le prélèvement d’une carotte suffisante, en moyenne 2 cm, est nécessaire pour une bonne étude histologique. La lecture permet de mettre en évidence une infiltration lymphomateuse qui peut dans certains cas être différente de celle retrouvée au niveau du ganglion.
Une ponction lombaire est effectuée dans les lymphomes agressifs, elle permet de dépister une localisation méningée asymptomatique, un traitement local sera alors nécessaire.
Au terme de ces examens, le patient est classé dans l’un des stades de la classification anatomique qui dérive de la classification d’Ann Arbor utilisée dans le lymphome Hodgkinien.
Stade I : Atteinte ganglionnaire limitée à un seul territoire ou atteinte extra ganglionnaire isolée
Stade II : Atteinte ganglionnaire et extra ganglionnaire contiguë du même côté du diaphragme accessible à un même champ d’irradiation
Stade III : atteinte ganglionnaire ± atteinte splénique de part et d’autre du diaphragme
Stade IV : atteinte extra ganglionnaire et viscérale non contiguë ou 2 atteintes extra-ganglionnaires non contiguës
En Algérie, les stades étendus sont plus fréquents au diagnostic, 61 % pour les stades III et IV versus 39 % pour les stades localisés(7), ceci s’expliquerait par un retard au diagnostic.
Avant la mise en route d’un traitement, des examens biologiques doivent être effectués, d’une part pour établir un pronostic, ce qui nous permet d’adopter une conduite thérapeutique, et d’autre part pour moduler les doses de chimiothérapie.
Hémogramme à la recherche de cytopénies ou de cellules lymphomateuses circulantes ;
Un bilan hépatique qui permet d’évoquer une localisation ;
Un taux de LDH, élément essentiel pour le pronostic des lymphomes ;
Le dosage de la béta 2 microglobuline, élément intervenant aussi dans le pronostic ;
Sérologies virales (HIV, Hépatite C, Hépatite B, HTLV1…) dont les conséquences sur le traitement peuvent être importantes ;
Electrophorèse des protéines et dosage d’albumine qui permettent de refléter l’état nutritionnel du patient ;
Des examens à la recherche d’une comorbidité associée et des signes de retentissement fonctionnel sont effectués à savoir : bilan rénal, acide urique, ionogramme, glycémie ;
Bilan d’hémostase important, surtout si un dispositif intra veineux est envisagé ;
Chez les patients jeunes qui doivent recevoir une chimiothérapie agressive pouvant entrainer une stérilité, la congélation du sperme doit être proposée.
Au terme du bilan, comment doit-on choisir son traitement ?
La prise en charge thérapeutique dépendra de plusieurs facteurs ; outre le caractère évolutif (indolent ou agressif), le phénotype B ou T, ce dernier étant de mauvais pronostic, le type histologique (LDGCB, lymphome du Manteau, lymphome de Burkitt …), la présence ou non de comorbidités, il est important de classer le patient selon un index pronostique international (IPI) qui est différent selon le type de lymphome. Pour le LDGCB, le plus fréquent des lymphomes agressifs, l’IPI prend en compte 5 facteurs de risque : l’âge, le taux de LDH, l’indice ECOG, le stade clinique, le nombre de sites extra ganglionnaires.
Dans les lymphomes indolents, un index pronostic a aussi été établi notamment pour le plus fréquent d’entre eux, le lymphome folliculaire, cet index pronostique appelé FLIPI prend en compte 5 facteurs de risque : âge, stade clinique, taux d’hémoglobine, taux de LDH, nombre de sites ganglionnaires.
Selon le nombre de facteurs, les patients sont classés en groupes de risque : 4 pour l’IPI et 3 pour le FLIPI(1).
La polychimiothérapie de type CHOP (Cyclophosphamide : Endoxan® j1, Doxorubicine : Adriamycine® j1, Vincristine : Oncovin® j1, Prednisone : Cortancyl®) est celle qui est la plus utilisée, les travaux de Fisher ont conforté ces résultats. Les cures seront effectuées tous les 21 jours, 4 à 8 cycles seront nécessaires selon les cas. Les résultats ont été améliorés depuis l’association à la polychimiothérapie, d’une immunothérapie de type Rituximab, anticorps anti CD20.
Une surveillance clinique est nécessaire après chaque cure afin d’évaluer la réponse au traitement et de rechercher les signes de toxicité.
Une chimiothérapie plus agressive avec autogreffe de cellules souches périphériques (CSP) peut être utilisée chez le sujet jeune avec des facteurs de risque.
Dans les lymphomes indolents à faible masse tumorale une surveillance clinique est préconisée.
Conclusion :
De grandes avancées sont notées concernant la prise en charge des LNH dans le monde et en Algérie ; des études nationales sont régulièrement effectuées, ce qui nous a permis de constater une augmentation de l’incidence. Le diagnostic est plus fiable grâce à la mise en place de comité de relecture de lames, sous l’égide du groupe d’étude algérien des lymphomes GEAL. La prise en charge thérapeutique a été améliorée grâce à la mise en place de consensus thérapeutiques et l’arrivée des anticorps monoclonaux, type anti CD20 associé à la chimiothérapie.
Références bibliographiques :
Christian Gisselbrecht Les lymphomes non Hodgkiniens Hématologie collection FMC JL 2008
ParKin DM, Whelan S, FerlayJ and al. Cancer Incidence in five continentsVol VIII; IARC 2002
Ferlay J, Antier P, Boniol M and al. Estimates of the cancer incidence and mortality in Europe in 2006 – Ann Oncol 2007, 18: 581-92.
L, Esteve Y, Buvier AM and al. Incidence and mortality in France over the period 1978-2000 – Rev Epidemio santé publique 2003 ; 51 : 3-30 5- Monographie des lymphomes.
Revue du Praticien 2010 ; 60 : 29-79 6- Hamladji RM, Intérêt des premières études épidémiologiques effectuées sur une période de 10 ans – Revue Algérienne d’hématologie N° 00 Année 2009
Boudjerra N, Oukid S, Abad MT, and al. Etude descriptive de 2915 cas de lymphomes non hodgkiniens ganglionnaires de l’adulte, période 2007/2012 –
Revue Algérienne d’Hématologie N° 10-11 déc. 2015
Boudjerra N, Perry AM, Audouin J, and al. Classification of non- Hodgkin Lymphoma in Algeria according to the Health organization classification – Leukemia Lymphoma, April 2015, 56 (h): 965 970
Boudjerra N pour le Groupe d’Etude Algérien des Lymphomes GEAL : Épidémiologie des lymphomes. Fascicule de la Santé 2005 ; 3 : 16-20
Lebailly P, Niez E, Baldi I. Données épidémiologiques sur le lien entre cancer et pesticides. Oncologie 2007 ; 9 : 1-9
Dr A. KRIM, Pr M A. BEKADJA, Service d’Hématologie et de Thérapie Cellulaire, EHU 1er Novembre d’Oran.
Abstract : Acute myeloid leukemia is a malignant hemopa- thy, cytologically, cytogenetically and molecularly heterogeneous. Its annual incidence in Europe and in USA varies from 3 to 5 per 100.000 inhabitants ; while it is lower in Algeria. Its diagnosis is generally easy; essentially cytological. Cytogenetics and molecular biology presently constitute an essential assessment at the time of diagnosis in order to individualize the prognostic groups and adapt the therapeutic strategy.
Key-words : Leukemia, AML, Algeria, EHU 1st November
Résumé : Les leucémies aiguës myéloblastiques sont des hémopathies malignes hétérogènes du point de vue cytologique, cytogénétique et moléculaire. Leur incidence en Europe et aux USA, varie de 3 à 5 par 100.000 habitants par an ; alors qu’elle est plus faible en Algérie. Leur diagnostic est en général facile ; essentiellement cytologique. La cytogénétique et la biologie moléculaire constituent actuellement un bilan indispensable dès le diagnostic afin d’individualiser les groupes pronostiques et d’adapter la stratégie thérapeutique.
Mots-clés : Leucémie, LAM, Algérie, EHU 1er Novembre
Introduction :
Les leucémies aiguës myéloblastiques (LAM) sont des hémopathies malignes qui représentent le motif d’hospitalisation le plus fréquent dans les services d’hématologie. Elles constituent une urgence à la fois diagnostique et thérapeutique, car souvent elles engagent rapidement le pronostic vital. Leur prise en charge initiale est l’apanage du médecin généraliste qui joue un rôle essentiel dans le diagnostic et la prise en charge des complications, ensuite le relais est pris par les hématologues pour un traitement spécialisé.
Qu’est-ce qu’une leucémie aiguë ?
La leucémie aiguë est un groupe hétérogène d’hémopathies malignes, caractérisées par l’expansion clonale au niveau de la moelle osseuse de cellules immatures (blastes), appartenant soit à la lignée myéloïde, soit à la lignée lymphoïde. Ces cellules jeunes appelées « blastes » peuvent passer dans le sang ou envahir certains tissus (Ex. : ganglions, méninges, peau).
On distingue deux types de leucémies aiguës en fonction de la lignée atteinte : les leucémies aiguës myéloblastiques (LAM) et les leucémies aiguës lymphoblastiques (LAL).
Incidence des LAM et facteurs de risque ?
Les LAM représentent 80% des leucémies aigues de l’adulte(1). Elles sont rares chez l’enfant et surviennent en général avant l’âge de 2 ans ou après l’âge de 15 ans. En Algérie, les leucémies aiguës occupent le 3ème rang des hémopathies malignes, (18%) après les lymphomes non Hodgkiniens et lymphomes Hodgkiniens(2). Les LAM sont les plus fréquentes avec une incidence évaluée en 2010 à 0,91/100.000 habitants, l’âge médian étant de 44 ans, sans prédominance de sexe(3).
En Europe, l’incidence des LAM est plus élevée, de l’ordre de 3,9/100.000 habitants chez l’homme et de 3,35/100.000 habitants chez la femme en 2012 ; l’âge médian est de 70 ans avec une légère prédominance masculine(4,5).
Dans la majorité des cas, les LAM n’ont pas de cause connue et surviennent chez des sujets, jusque-là en bonne santé. Certains facteurs de risque sont néanmoins incriminés : l’exposition à des rayonnements ionisants ou à certains produits chimiques (en particuliers le Benzène et solvants dérivés, hydrocarbures aromatiques), les antécédents de chimiothérapie (notamment par les alkylants, les inhibiteurs de topo-isomérase II, les antimétabolites), certaines anomalies génétiques (dont la trisomie 21, la maladie de Fanconi) et des maladies hématologiques préexistantes (telles que notamment les syndromes myélodysplasiques et les néoplasies myéloprolifératives).
Quand penser à une LAM ?
La LAM est une pathologie d’installation aiguë dont l’expression clinique est la conséquence de la prolifération des blastes dans la moelle (signes d’insuffisance médullaire), puis de leur accumulation éventuelle dans un ou plusieurs organes (syndrome tumoral).
Il n’existe pas de signe spécifique. Le diagnostic est suspecté lors d’une complication clinique d’une ou de plusieurs cytopénies (altération de l’état général, syndrome anémique, syndrome hémorragique, état infectieux) et/ou devant un hémogramme anormal faisant suspecter une atteinte médullaire (cytopénies, blastose…).
Les signes révélateurs peuvent être :
Une symptomatologie liée aux cytopénies sanguines :
Plus au moins marquée, associant à des degrés différents :
– Syndrome anémique allant d’une pâleur modérée à une pâleur extrême avec des signes d’intolérance cardiovasculaire (dyspnée, tachycardie, obnubilation…)
Syndrome infectieux : en particulier une infection bactérienne trainante.
Et/ou un syndrome tumoral :
Les adénopathies sont rares (Figure 1)
La splénomégalie est retrouvée dans 15% à 20% des cas, et / ou hépatomégalie.
L’hypertrophie gingivale est fréquente dans certains types de LAM (notamment LAM4 et LAM5) (Figure 2)
Les localisations cutanées : nodules dermiques « leucémides » sont rares (Fig.3).
Les localisations neuro-méningées, y compris l’atteinte des nerfs crâniens (diplopie, anesthésie de la houppe du menton, strabisme, trouble de la déglutition…) sont également rares.
Les douleurs osseuses traduisent un envahissement médullaire massif.
Deux situations nécessitent le transfert d’urgence en milieu spécialisé :
Des signes de détresse respiratoire liés à la leucostase, conséquence des grandes hyperleucocytoses, possible au-delà de 100 g/l
Un syndrome hémorragique diffus, lié autant à la thrombopénie majeure qu’à une coagulopathie de consommation (CIVD et/ou fibrinolyse).
Le diagnostic des LAM est cytologique et repose sur l’étude morphologique du sang et de la moelle. Le bilan minimal doit comporter un hémogramme (numération formule sanguine « NFS » et frottis sanguin) et un myélogramme.
Hémogramme : il est anormal et permet très souvent de suggérer ou d’affirmer le diagnostic de leucémie aiguë, avec :
Une anémie normocytaire, normochrome, arégénérative dans 90% des cas, rarement isolée ;
Une thrombopénie dans 80-90% des cas, qui peut être isolée ;
Une neutropénie, associée ou non à une hyperleucocytose (30%) ;
Cette étude quantitative (NFS) est complétée par une étude qualitative « Frottis sanguin », ce qui permet de mettre en évidence la présence de blastes circulants en nombre variable (leur absence n’exclut pas le diagnostic de leucémie aiguë). (Figure 4, Figure 5)
Figure 4 : Frottis sanguin Figure 5 : Blastes
Myélogramme : réalisé en urgence dès l’admission en milieu hospitalier, il permet de confirmer le diagnostic et est également indispensable à l’identification de la leucémie aiguë.
Il met en évidence une blastose médullaire qui doit être supérieure ou égale à 20% sur l’ensemble des cellules nucléées. Il s’agit d’un geste peu invasif, qui consiste à prélever de la moelle osseuse (au niveau du sternum, os iliaque antérieur ou postérieur) par ponction et aspiration, pour pouvoir ensuite faire un frottis. (Figure6)
Figure 6 : Myélogramme
Autres examens biologiques :
L’étude cytochimique des frottis sanguins et médullaires est indispensable au diagnostic. Elle permet, grâce à des colorations spécifiques, de distinguer l’origine myéloïde ou lymphoïde des blastes.
L’étude immunophénotypique des blastes sanguins et/ ou médullaires est également indispensable au diagnostic. Cet examen repose sur l’utilisation d’anticorps monoclonaux dirigés contre les antigènes membranaires des blastes et cela à l’aide d’un cytomètre en flux multicouleurs. Ce qui nous permet d’avoir un diagnostic précis du type de leucémie aiguë et éventuellement de suivre la maladie après traitement (suivi de la maladie résiduelle).
L’étude cytogénétique des blastes : utile pour le diagnostic et surtout le pronostic des LAM .
Une fois le diagnostic établi, il faut classer les LAM en sous-groupes. Deux grandes classifications existent: la classification Franco-Américano-Britannique (FAB), qui permet de distinguer 9 types de LAM ; l’identification en particulier de la LAM3 est une urgence en raison du risque élevé de complications hémorragiques (Tableau 1). La classification OMS révisée chaque 4 ans (dernière révision en 2016), complète systématiquement la classification FAB et prend en compte les caractéristiques génétiques des blastes.
Nom
LAM 0
Indifférenciée
LAM 1
Myéloblastique sans différencition
LAM 2
Myéloblastique avec différenciation
LAM 3
Promyélocytaire
LAM 4
Myélo-monocytaire
LAM 4
Myélo-monocytaire avec éosinophiles Eo anormaux
LAM 5a
Monocytaires sans différenciation
LAM 5b
Monocytaires avec différenciation
LAM 6
Érythroblastique
LAM 7
Mégacaryocytaire
Tableau 1 : Classification FAB
Peut-on se contenter uniquement de l’étude cytologique et immunophénotypique des blastes dans les LAM ?
Notre connaissance des leucémies aiguës myéloblastiques s’est affinée au fur et à mesure des années et des avancées technologiques. Actuellement, les LAM sont un groupe hétérogène sur le plan cytologique, cytogénétique et moléculaire. Il s’agit d’hémopathies de pronostic variable d’où l’intérêt d’individualiser dès le diagnostic les groupes à haut risque qui nécessitent une intensification thérapeutique.
De ce fait, on ne peut pas se contenter uniquement de l’étude cytologique et immunophénotypique des blastes au diagnostic. La détermination des paramètres cytogénétiques et moléculaires est primordiale pour la décision thérapeutique.
En effet, l’influence majeure de la cytogénétique a été établie dans de nombreuses études de grandes cohortes(5,6,7). Ces études ont permis une stratification du risque en groupes pronostiques : favorable, intermédiaire et défavorable. Cependant, 40% à 50% des LAM ont un caryotype normal et sont classées de pronostic intermédiaire avec une disparité de réponse(8). Des progrès ont été réalisés dans la caractérisation moléculaire des LAM et de nombreuses classifications moléculaires se sont succédées pour affiner leur pronostic(9,10).
Afin d’homogénéiser les différentes études et d’intégrer les données moléculaires aux données cytogénétiques en matière de pronostic, un groupe d’expert sous l’égide de l’European Leukemia Net (ELN) a proposé une classification pronostique en 2010 (Tableau 2) et révisée en 2017 (Tableau 3). Cette classification distingue 3 grands groupes : favorable, intermédiaire et défavorable.
t(8 ;21)(q22 ;q22.1) ;RUNX1-RUNX1T1 Inv(16)(p13.1q22) or t(16 ;16)(p13.1;q22) ; CBFB-MYH11 Mutated NPM1 without FLT-ITD or with FLT3-ITDlow(c)
Intermediate
Mutated NPM1 and FLT3-ITDhigh(c) Wild type NPM1 without FLT3-ITD or with FLT3-ITDlow(c) ( w/o adverserisk genetic lesions) t(9;11)(p21.3;q23.3); MLLT3-KMT2Ad Cytogenetic abnormalities not classlfied as favorable or Adverse
Adverse
t(6;9)(p23;q34.1);DEK-NUP214 t(v;11q23.3);KMT2A rearranged t(9;22)(q34.1;q11.2) ; BCR-ABL1 inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2,MECOM(EVI1) -5 or del (5q) ;-7 ;-17/abn(17p) Complex karotype, emonosomal karytypef Wild type NPM1 and FLT3-ITDhigh(c) Mutated RUNX1g Mutated ASXL1g Mutated TP53h
Tableau 3 : Classification ELN 2017
Comment traiter les LAM ?
Les LAM sont des urgences thérapeutiques. Leur traitement comprend deux volets : un volet de traitement symptomatique et un autre de traitement de fond.
Traitement symptomatique : primordial et consiste à lutter contre les signes d’insuffisance médullaire.
Traitement de l’anémie : par des transfusions de culots globulaires iso-groupe iso-rhésus phénotypés, si le taux d’hémoglobine < 8 g/dl ou s’il existe des signes d’intolérance.
Traitement des hémorragies : par des transfusions de culots plaquettaires
À visée préventive :si le taux de plaquettes (PLQ) est inférieur à 10 G/L et en l’absence de facteur de risque ; ou le si taux de PLQ ≤ 20 G/L avec facteur de risque (situation de fièvre ≥38,5°C, infection, mucite grade ≥ 2, lésion hémorragique, chute brutale de la numération plaquettaire en 72 H) ; ou encore, si le taux de PLQ ≤ 50 G/L avec prise d’anticoagulant ou en présence d’une coagulopathie.
À visée curative :saignement extériorisé
Traitement des infections : une fièvre > 38,2°C avec neutropénie constitue une urgence thérapeutique.
Mesures préventives : isolement du patient dans une chambre individuelle stérile, alimentation protégée avec précautions d’hygiène.
Traitement curatif : La documentation de la fièvre (hémoculture, radiographie du poumon, ECBU, culture cathéter…) ne doit pas retarder l’introduction d’une antibiothérapie empirique à large spectre couvrant les bacilles Gram négatif et le streptocoque.
La réévaluation de la fièvre doit se faire après 48-72H en l’absence de signes de gravité (frissons, hypotension voire état de choc septique) Si la fièvre se prolonge, il existe un risque d’infection fongique.
• Traitement de fond :
1- Traitement du sujet jeune (16 ans – 60 ans) :
Le traitement des LAM (sauf la LAM3) comporte deux phases : une phase d’induction qui repose sur une polychimiothérapie conventionnelle, qui associe une anthracycline (Exemple : Daunorubicine ) à la cytarabine (pro- tocole « 3+7 ») (11) dont le taux de rémission complète varie de 60% à 80% (12) et une phase de consolidation envisagée seulement en cas de rémission, qui repose habituellement sur la chimiothérapie intensive suivie ou non d’une greffe allogénique ou autologue en fonction du risque de rechute.
Tous les patients reçoivent la même cure d’induction, puis en fonction du groupe pronostic, selon la classification de l’ELN 2010 qui subdivise les patients en quatre groupes (13) : le groupe favorable reçoit une consolidation par de l’aracytine à fortes doses, seule (14,15), pour le groupe défavorable : l’allogreffe de CSH reste le traitement de choix, quant aux groupes intermédiaires I et II, en raison de leur hétérogénéité, différentes approches thérapeutiques ont été proposées par plusieurs groupes d’étude (16), et il a été clairement établi la place de l’allogreffe surtout en cas de présence de la mutation FLT3-ITD (17).
2- Traitement du sujet âgé > 60 ans :
La chimiothérapie d’induction est similaire à celle utilisée chez l’adulte jeune, mais elle ne sera appliquée que si les risques de toxicité ou de résistance du clone leucémique n’apparaissent pas excessifs.
Certains patients (état général altéré, âge>80 ans, présentant des signes infectieux ou des comorbidités) sont jugés inaptes à recevoir ce type de chimiothérapie ; l’objectif est alors de leur assurer une qualité de vie, la meilleure possible et le patient se verra proposer un traitement de support couplé à une chimiothérapie palliative (hydroxyurée, l’Ara-C à faibles doses).
Expérience du service d’hématologie et thérapie cellulaire de l’EHU 1er novembre d’Oran :
Le service d’hématologie de l’EHU d’Oran recrute en moyenne 40 nouveaux cas de leucémie aiguë par an. Ces patients sont hospitalisés en chambre stérile, en unité d’allogreffe de moelle ou en unité d’hospitalisation conventionnelle. Le diagnostic des LAM est posé par les moyens biologiques suivants : hémogramme, myélogramme, cytochimie au Noir Soudan et immunophénotypage par cytométrie en flux. Le bilan pronostique comporte un bilan cytogénétique par caryotype conventionnel et la recherche par biologie moléculaire de la mutation FLT3-ITD (ce bilan est systématique depuis 2014).
Le traitement des sujets jeunes repose sur une induction standard « 3+7 » associant la Daunorubicine (DNR) à la cytarabine (ARAC). Les patients en rémission complète (RC) (définie par un taux de blastes dans la moelle < 5%, Polynucléaires neutrophiles > 1 G/L et PLQ > 100 G/L) reçoivent une consolidation (3 cycles d’ARAC Haute dose) et, pour ceux qui ont un donneur HLA compatible, une allogreffe de cellules souches hématopoïétiques (CSH) géno-identiques (donneur de la fratrie).
Les LAM de pronostic favorable reçoivent uniquement de la chimiothérapie exclusive.
Avec l’existence de deux centres de greffe de CSH en Algérie, celui du Centre Pierre et Marie Curie à Alger (depuis 1998) et notre centre à Oran (depuis Février 2013), il est désormais possible de proposer à nos patients souffrant de LAM une greffe de CSH. Selon l’expérience de notre service dans la prise en charge des sujets jeunes atteints de LAM, en comparant les deux périodes (2009-2012) et (2013-2015), c’est-à- dire avant et après l’ouverture du centre d’allogreffe de CSH à Oran, l’intensification de la dose de la daunorubicine et la proposition de l’allogreffe en consolidation à un plus grand nombre de patients, nous a permis d’améliorer de façon significative la survie globale (SG) de ces derniers (SG à 2 ans 29% vs 47%, p=0,02) (Figure 7).
Figure 7 : Courbe de survie globale des LAM Bras1 (2009-2012), Bras 2(2013-2015)
Conclusion :
Les LAM sont des pathologies hétérogènes dont le diagnostic est en général facile. Sans traitement, elles sont rapidement fatales, en raison du risque infectieux et
hémorragique. Leur traitement repose essentiellement, depuis plus de 40 ans, sur une polychimiothérapie type
« 3+7 ». Les chances de rémission complète varient en fonction de l’âge et des caractéristiques cytogénétiques et moléculaires de la maladie. L’allogreffe de CSH en consolidation pour les patients à risque intermédiaire et défavorable a permis de modifier le pronostic des LAM et d’avoir les meilleurs résultats en matière de survie. De nombreux essais cliniques sont en cours afin d’optimiser au mieux le traitement de la LAM en tenant compte des nouvelles données physiopathologiques.
Référence :
Yamamoto JF et al. Patterns of leukemia incidence in the united states by subtype and demographic characteristics, 1997-2002; Cancer Causes Control 2008; 19 :379-390.
R.M. Hamladji. Etat des lieux de la prise en charge des hémopathies ma- lignes en Algérie. Revue Algérienne d’Hématologie en 2013. N°8/9 septembre 2013-2014.
MA. Bekadja. Approche épidémiologique des leucémies aigues myéloïdes en Algérie. Revue Algérienne d’Hématologie, 2012, N°6-7, 6-10.
Visser. O et al. Incidence, survival and prevalence of myeloid malignancies in Europe.
Eur. J. Cancer 2012; 48(17):3257-66.
Grimwade. D, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromo- somal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010 Jul 22; 116(3):354–65.
Slovak.ML, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: A Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000 Dec 15; 96(13):4075–83.
Byrd JC, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leuke- mia Group B (CALGB 8461). Blood. 2002 Dec 15; 100(13):4325–36.
Mrózek K, et al. Cytogenetics in acute leukemia. Blood Rev. 2004 Jun; 18(2):115–36.
Renneville A, et al. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia. 2008 May; 22(5):915–31.
Dohner H Et al. Diagnosis and management of AML in Adults: 2017 ELN recom- mendations from international expert panel. Blood 2017; Volume 129, Number 4.
De Konchkovsky I et al. AML: a comprehensive review and 2016 update. Blood Cancer Journal (2016).
Sekeres MA, Elson P, Kalaycio ME, Advani AS, Copelan EA, Fader lS, et al. Time from diagnosis to treatment initiation predicts survival in younger, but not older, acute myeloid leukemia patients. Blood. 2009 Jan 1; 113(1):28–36.
Döhner Hetet al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European Leukemia Net. Blood. 2010 Jan 21; 115(3):453–74.
Bloomfield CD et al. Frequency of prolonged remission duration after high- dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res. 1998; 8(18):4173-9.
Pr Farid zIANI Orthopedic surgery department EHS Douéra Algiers Algeria
Abstract :
Objectives : This study aims to evaluate the cure rate of osteo-articular infections on material in orthopedic surgery treated with antibiotics within 6 months and 6 weeks in Algeria. It also identifies the factors associated to the treatment success.
Patients and methods : This is a national, multicenter, observational longitudinal study conducted in
10 orthopedic surgery departments. Eligible patients were ≥15 years old, underwent orthopedic surgery with implantation of osteosynthesis material, and developed osteo-articular infection within the year following the surgery. They were followed up for 6 months. Deep bacterial samples were considered for the diagnosis of osteo-articular infections.
Results : 101 patients were recruited: mean age 51.2 ± 21.5 years, 58.4% men. 88 (94.6%) of the 93 bacteriological samples performed were positive: 38 gram-positive (Staphylococcus aureus and coagulase-negative staphylococci) and 68 gram-negative (Enterobacteriaceae and Pseudomonas aeruginosa) bacteria were identified. The cure rate was 81.5% in 81 patients at 6 months and 58.8% at 6 weeks. Abscess (OR=8.24; 95%CI: 1.45 to 46.73) and at least one new surgery during the first 6 weeks following osteo-articular infection management (OR=1.60; 95%CI: 2.98 to 103.78) were significantly associated with treatment success and better cure.
Conclusions : Osteo-articular infections with gram-negative Enterobacteriaceae were frequent with a high 6-months cure rate. The identified factors for the treatment success echo the importance of continuously preventing and healing osteo-articular infections.
Objectifs : Cette étude évalue le taux de guérison sous antibiotiques des infections ostéo-articulaires en Algérie à 6 mois et 6 semaines. Elle identifie aussi les facteurs associés à la réussite du traitement et la guérison.
Patients et Méthodes : Cette étude nationale, multicentrique, observationnelle et longitudinale a été conduite dans 10 services de chirurgie orthopédique. Les patients éligibles étaient âgés de ≥15 ans, ayant subi une chirurgie orthopédique avec implantation de matériel d’ostéosynthèse et développé une infection ostéo-articulaire dans l’année suivant la chirurgie. Ils étaient suivis pendant 6 mois. Des prélèvements bactériologiques profonds ont permis l’établissement du diagnostic des infections ostéo-articulaires.
Résultats : 101 patients ont été inclus : âge moyen 51,2 ± 21,4 ans, 58,4% hommes.
88 (94,6%) des prélèvements bactériologiques étaient positifs: 38 bactéries Gram positives (Staphylococcus aureus et staphylococci coagulase-négatifs) et 68 Gram négatives (Enterobacteriaceae et Pseudomonas aeruginosa) étaient identifiées. Le taux de guérison était de 81,5% à 6 mois et 58,8% à 6 semaines. Les abcès (OR=8,24; IC 95% : 1,45-46,73) et au moins une nouvelle chirurgie durant les 6 semaines après le début de la prise en charge de l’infection ostéo-articulaire (OR=1,60; IC 95% : 2,98-103,78) étaient significativement associés au succès du traitement et à la guérison.
Conclusions : Les infections ostéo-articulaires aux entérobactéries Gram négatives étaient fréquentes avec un taux de guérison élevé à 6 mois. Les facteurs de réussite du traitement nous interpellent sur le rôle primordial de la prévention des infections ostéo-articulaires.
Mots-clés : Antibiothérapie, facteurs de risque, infection ostéo-articulaire, résistance bactérienne.
Introduction :
No surgery is completely «aseptic» despite all precautions taken. The surgical site infections are considered nosocomial when they occur within 30 days following surgery or if this requires the implementation of prosthesis or an implant, in the year following the surgery [1].
In elderly patients, the osteo-articular infection in orthopedic and trauma surgery constitutes often a severe complication (source of morbidity), functional sequels and sometimes a risk of mortality in acute disease (sepsis). Moreover, the management of an infected patient is heavy, and represents a major therapeutic cost due to repeated surgical interventions, repeated hospitalizations, long-term antibiotic therapy, sick leaves and severe consequences for the young population [2,3].
Osteo-articular infections on material are conventionally attributed to Staphylococcus aureus, which represents two-thirds of identified germs [4,5]. However, the predominance of gram-negative bacteria (GNB) has been reported by authors in Algeria [6]. Also, the infection rate depends on the type of surgery performed and its classification. Two classifications are currently used: namely the National Research Council (NRC) and Altemeier classifications. The former is the oldest classification with 5 types of interventions, depending on the
extent of contamination at the surgical site (hyper clean, clean, clean-contaminated, contaminated and dirty) [7]. As for Altemeier classification, it is used internationally for all the types of surgery with 4 classes of interven- tions (clean, clean-contaminated, contaminated and dirty), but seems less suitable for orthopedic surgery [7]. Currently, few multicenter studies in Algeria address this subject and no consensus or recommendations on prescribing curative antibiotics are available [6]. Therefore, the current study aim is to evaluate the rate of cure within the 6 months following antibiotherapy for osteo-articular infections on material in orthopedic surgery. The secondary objectives are to evaluate the rate of cure within the 6 weeks of treatment by antibiotic(s) and to identify risk factors for treatment failure (age [8], concomitant diseases such as neoplasia and diabetes [9], surgery history, concomitant treatment, the classifications of NRC and the American Society of Anesthesiology (ASA) of physical health, and clinical signs of infection at enrollment such as pain, fever and fistula [10]).
2. Materials and Methods
2.1. Study population and settings
This was a national, multicenter, observational and longitudinal study. It was conducted in 10 sites covering the main 3 regions of Algeria (center, east and west) from 13 January 2013 through 14 May 2014. Eligible patients were followed up at baseline (visit 1), then after 6 weeks (visit 2), and at 6 months (visit 3).
Were included patients aged over 15 years old, who underwent orthopedic surgery with implantation of osteosynthesis material regardless of the type, and who developed osteo-articular infection within the year following the surgery and prior to the inclusion in the study. Patients having fungal or mycobacterial infections, as well as acute and chronic blood-bore osteomyelitis were excluded.
2.2. Diagnosis of osteo-articular infections and data collection
Only deep bacterial samples were considered, and osteo-articular infections were diagnosed by the orthopedic department surgeon. Using the definition published by the Center for Disease Control (CDC) of Atlanta [7], the infections were defined according to the presence of at least one of the following conditions: clinical diagnosis by the surgeon, purulent discharge, isolated germ and obvious signs of infection at the surgery site [11].
While demographic data were collected during visit 1, the clinical, biological and bacteriological characteristics, as well as the medical and surgical management were recorded at the 3 study visits.
2.1. Statistical analysis
Cure rate within 6 months following the treatment of osteo-articular infection in orthopedic surgery was not available. Therefore, to have 10% precision to detect a cure rate of 50% at the 5% significance level, a minimum of 100 patients were required for the study.
Statistical analysis plan was finalized before database lock. Sample characteristics were summarized using mean, standard deviation, median, quartiles and extreme values for quantitative variables, and frequency distributions along with confidence intervals determined at 95% (95%CIs) for qualitative variables. Frequency distributions were compared between qualitative independent variables using the chisquared test. For continuous data, comparisons between 2 variables were made using Student t-test or Wilcoxon non-parametric test, whereas comparisons between more than 2 means were performed using one-way ANOVA or Kruskal-Wallis test for non-parametric data. The alpha risk was two-sided and set at 5%. The risk of treatment failure (or odds of cure) associated with each factor was quantified by the odds ratio (OR) along with their 95%CIs, the point being made at 6 months. In a second time, all significant factors or borderline significance (p<0.10) were introduced into a multivariate logistic model backward selection to assess the risk of each factor. Data were analyzed using SPSS software® version 18 for windows release (SPSS Inc. Released 2009. PASW Statistics for Windows, Version 18.0. Chicago: SPSS Inc.).
2.2. Ethical considerations
The study was approved by the ethics committees of the participating sites. It was performed in compliance with the guidelines for Good Epidemiology Practice. All
patients were included in the study after reading and
signing their informed consent. All data were collected and analyzed respecting the patients’ anonymity and confidentiality.
3. Results
3.1. Patients disposition and characteristics
At inclusion, 101 patients were recruited, of whom 97 patients were followed up until the 6-week visit, and 81 patients until the 6-month visit. 80.2% of the patients completed the 3 visits. Sixteen (15.84%) patients were lost to follow-up and 4 (3.96%) died.
The patients’ mean age at inclusion was 51.2 ± 21.5 years with a predominance of male (59 men/42 women, sex M/F ratio=1.40). Ten (9.9%) patients were older than 80 years old and 11 (10.9%) were obese.
Two (2%) patients had rheumatic arthritis, one (1%) patient had neoplasia, and 28 (27.7%) had diabetes. Also, 24 (23.8%) patient were previously operated on the same section. The NRC and the ASA classifications were reported for all participating patients. The patient distribution according to ASA showed that 35.6% (36 cases) of the patients were type 2 whereas 59.4% (60 cases) were ASA I. Five cases were ASA III (5.0%) and none was ASA IV (Table 1).
Risk Factor
N (%)
Age > 80 years
10 (9.9%)
Rheumatoid arthritis
2 (2.0%)
Corticosteroid therapy
2 (2.0%)
Neoplasia
1 (1.0%)
Immunosuppressive treatment
0 (0.0%)
Diabetes
28 (27.7%)
Immunodeficiency
1 (1.0%)
Obesity
11 (10.9%)
Surgery history on the same section
24 (23.8%)
Other infections
2 (2.0%)
Table 1 : Distribution of associated risk factors considered in the present study Abbreviations : ASA : American Society of Anesthesiologists; NCR : National Research Council.
The main reason of initial hospitalization was osteo-synthesis of fracture in 78 cases (77%) and the main type of surgery was bone plate in 28 cases (27.7%). Total hip replacement (9 patients, 8.9%) and knee replacement (3 patients, 3%) were observed in 11.9% of the patients. The time between surgery and inclusion was repor- ted for 100 patients, with an average of 5.0 ± 7.1 days (range 0–46 days).
Out of the 101 patients, 65 (64.4%) patients were cured at 6 months while 16 (15.9%) were not, data were missing for 20 (19.8%) patients.
2.1. Biological and bacteriological assessments
The biological assessment at baseline showed an increase in white blood cells in 55/88 patients (62.5%), an accelerated erythrocyte sedimentation rate in 59/65 patients (90.8%) and a C-reactive protein (CRP) increase in 60/70 patients (85.7%).
Overall, bacteriological samples were performed in 93 out of 101 patients: puncture bacteriological sampling was obtained in 27 (29%) patients versus 52 (55.9%) per surgical sample. Among the 93 bacteriological samples, 88 (94.6%) samples were positive. The results were negative in 3 (3.2%) cases and the information had not been specified in 2 (2.2%) cases. No significant link was detected between the type of sampling and identification of the bacteria type (p=0.783). The number of germs per patient varied from 0 to 3, where one microbial agent was detected in 72 (81.8%) patients, 2 bacteria in 14 (15.9%) patients and 3 in 2 cases (2.3%).
A total of 106 bacteria (38 gram-positive bacteria [GPB] and 68 GNB) were identified. The GNB were more common in the lower limb (68%) and the spine (62.5%) (Table 2). There was a significant predominance of GNB (64.2%) composed of 58.8% of Enterobacteriaceae (Escherichia coli, Proteus, Enterobacter, Klebsiella). Pseudomonas aeruginosa was identified for 28% of gram-negative germs. Staphylococcus aureus was the most common GPB (47.36%) followed by coagulase-negative staphylococci (Table 3).
Antibiograms were available for 83 pathogens. For Staphylococcus aureus, resistance to methicillin was 56%, resistance to rifampicin was 28.5%, 25% for fusidic acid and 20% for lincomycin. The resistance rates of coagulase-negative staphylococci were also 50% for methicillin and 25% for rifampicin. We noted a relevant resistance to fusidic acid (66.6%).The sensitivity was 100% for glycopeptide (vancomycin and teicoplanin) for both types of staphylococci. Regarding GNB, Pseudomonas aeruginosa resistance was 7.7% to quinolones, 50% to ceftazidime and 11.8% to imipenem. All pseudomonas strains were sensitive to colistin. The study of susceptibility and resistance for the most frequent enterobacteria (Proteus, Enterobacter and Klebsiella) showed a significant resistance to amoxicillin-clavulanic acid (87%) and to cefotaxim (66.6%). Resistance rate of enterobacteria to colistin was 36.8%. It was 28.9% for quinolones and 11.7% for imipenem. All enterobacteria tested were sensitive to ticarcillin and piperacillin.
Table 3 : Distribution of bacteria according to Gram classification
Location
Atleastone gram-positive bacterium
Atleastone gram-negative bacterium
At least one bacterium
Upper limb
5 (62.5%)
3 (37.5%)
8
Lower limb
28 (32.0%)
49 (68.0%)
72
Spine
3 (37.5%)
5 (62.5%)
8
Table 2 : Type of bacteria per infection’s location
Table 3 : Distribution of bacteria according to Gram classification
Abréviations : GNB (BGN): bactérie à gram négatif; GPB (BGP): bactérie à gram positif.
2.1. Curative antibiotherapy
The curative treatment was probabilistic in 29 patients and based on the antibiograms results in 72 patients. In the 3 remaining cases, initial treatment was probabilistic then modified according to antibiograms results. Overall, 67 (66.3%) patients received at least 2 antibiotics. The overall duration of antibiotic treatment, observed in 98 subjects varied from 6 to 90 days, with an average
of 30.8 ± 18.3 days. The number of antibiotics given to the patients varied from 1 to 4 antibiotics : one antibiotic was dispensed in 34 (33.7%) cases; 52 (51.4%) patients
received 2 antibiotics, 14 (13.8%) patients received 3 antibiotics and one (1%) patient was administered 4 antibiotics. Out of the 97 patients who performed the 6-week visit,
87 patients had a positive response to the treatment (presumption of cure or improvement of the general health status) 89.7% (95% CI: 80.6% to 93.6%). In parallel, 57 (58.8%) of the 97 patients were at a presumption of cure. The results of the primary endpoint show that the infection cure rate at 6 months based on the investigator’s judgment on visit 3 was 81.5% (95% CI: 72.0% to 89.0%) in 81 patients.
Risk factors for treatment failure The univariate analysis showed no significant relationship between the patient-related risk factors (such as age) and the treatment failure. As for factors related to the disease, the National Nosocomial Infections Surveillance (NNIS) index was significantly associated to the cure status (p=0.026) with index zero being predominant in cured patients. No significant relationship between the clinical signs of infections (fistula, pain and fever) and the cure status was observed, except for abscess (p=0.03) which showed better healing in the cured patients and inflammatory scar (p=0.057) which showed a lower cure rate in the cured patients. The association between the infection location and better healing was only significant for the lower limb (p=0.018). Moreover, the infection and surgery types were not significantly associated with the cure status. In parallel, no significant relationship at inclusion was established with biological assessment, except for CRP (p=0.018). Regarding factors related to treatment, no new surgery during the first 6 weeks (p=0.012) and no improvement at 6-weeks (p=0.058) following osteo-articular infection management were associated to treatment failure (Table 4).
As for the multivariate analysis, only abcess (OR=8.24 ;95%CI: 1.45 to 46.73) and a new surgery during the first 6 weeks following osteo-articular infection management (OR=17.60 ; 95%CI : 2.98 to 103.78) were retained as significant factors associated to treatment success.
Variable
Number (n)
Cured (n=65)
Not cured (n=16)
p-value* NNIS index
0
34
31 (47.7%)
3 (18.8%)
0.026*
1
23
18 (27.7%)
5 (31.3%)
2
1
0 (0.0%)
1 (6.3%)
Clinical signs of infection
Abscess
28
18 (27.7%)
9 (56.3%)
0.030*
Fistula
40
33 (50.7%)
7 (43.8%)
0.615
Pain
4
2 (3.07%)
2 (12.5%)
0.119
Inflammatory scar
52
45 (69.2%)
7 (43.8%)
0.057*
Fever
31
25 (38.5%)
6 (37.5%)
0.943
Location of infection
Upper limb
12
11 (16.9%)
1 (6.3%)
0.443
Lower limb
64
48 (73.8%)
16 (100.0%)
0.018*
Spine
7
7 (10.8%)
0 (0.0%)
0.335
Biological assessment a
Leukocytosis
69
57.9 ± 34.6
54.3 ± 25.8
0.859
ESR at 1 hour
53
17.0 ± 23.0
11.6 ± 5.9
0.811
CRP
50
30.5 ± 26.6
50.3 ± 30.8
0.018*
Factors related to treatment
Therapeutic approach
Probabilistic
23
18 (27.7%)
5 (31.3%)
0.777
Antibiogram
58
47 (72.3%)
11 (68.8%)
Cure status at 6 weeks following antibiotherapy for osteo-articular infections
No improvement
2
1 (1.5%)
1 (6.3%)
0.058*
Improvement
27
19 (29.2%)
8 (50.0%)
Presumed cured
48
43 (66.2%)
5 (31.3%)
New surgery performed before 6 weeks following antibiotherapy for osteo-articular infections
Yes
21
12 (18.5%)
9 (56.3%)
0.012*
No
58
51 (78.5%)
7 (43.8%)
New surgery performed between 6 weeks and 6 months following antibiotherapy for osteo-articular infections
Yes
19
13 (20.0%)
6 (37.5%)
0.108
No
60
51 (78.5%)
9 (56.3%)
Table 4 : Univariate analysis: risk factors for non-healing at 6 months
Niveau de significativité statistique de 10%. a : moyenne ± écart-type à l’inclusion.
Abréviations : CRP : protéine C réactive; ESR (VS) : vitesse de sédimentation ; NNIS : National Nosocomial Infections Surveillance.
2. Discussion
The bone and joint infection in orthopedic surgery is a serious source of morbidity, functional impairment and sometimes a risk of mortality by septicemia in acute phase [2]. Its management is heavy, and represents a major therapeutic cost for society due to repeated surgical interventions, long-term antibiotic therapy and absenteeism. Only few multicentric studies in Algeria addressed the osteo-articular infections [6].
The main objective of this study was to estimate the cure frequency during the 6 months following osteoarticular infection management in orthopedic surgery. The secondary objectives were to determine the response rate at 6 weeks of antibiotherapy. This study also allowed us to better know the epidemiology of these infected patients, their clinical and microbiological profile and to identify the risk factors for no healing.
Early infection seemed to be common in our study. The delay between the surgical intervention and the infection occurrence ranges from 2 to 90 days, with an average of 14.9 ± 14.1 days. No late infections of more than 30 days were reported, mainly because of a contamination of the surgical site during surgery or immediately in postoperative [7].
According to literature, risk of infection in orthopedic surgery can be attributed to factors such as age greater than 65 years, the existence of another infectious site in the patient, prolonged preoperative stay, obesity, corticosteroid therapy, a rheumatoid arthritis, diabetes, recent local radiotherapy, an eschar proximity and the occurrence of postoperative hematoma [3,5]. In our study, the patients were mostly male (sex ratio equal to 40), with a mean age of 51.2 ± 21.5 years and the risk factors were dominated by diabetes (27.7%), intervention history on the same segment (23.8%), less frequently obesity (10.9%) and being aged more than 80 years (9.9%). The postoperative complications were frequent (72% of cases), and dominated by postoperative hematoma and flow drain beyond 48 hours after inclusion. ASA score allows determining the preoperative patient health to assess the risk of postoperative infections and mortality. It also serves to determine the NNIS index [1,7]. The intervention was most often classified as type 1 or type 2 according to NRC [7]. The patients’ ASA score was I in 59.4% of cases, nonetheless 4 (3.8%) patients deceased. The causes of death were a digestive hemorrhagic stroke, a cardio-respiratory complication, a postoperative complication in elderly patients and renal failure with septic shock.
The microbiological data of the main studies done on materials infections showed that staphylococci were the most frequently isolated bacteria and infections were most often monomicrobial [3,7]. In our study, BGN were significantly predominant (64%), and composed of a high proportion of enterobacteria (Escherichia coli, Proteus, Enterobacter, Klebsiella) representing 58.8% of the strains. Pseudomonas was found for 28% of gram-negative germs. Staphylococcus aureus was the most frequent gram-positive bacterium (36%) followed by coagulase-negative Staphylococcus. These very alarming findings confirm the few bacteriological studies conducted in Algeria [6], and reflect the character of nosocomial infection because of a poor hygiene. Consequently, the fight against these infections must be developed and mastered [6].
As for antibiogram results, bacterial resistance was equally important for gram-positive and GNB. In parallel, antibiotics were prescribed because of their availability, whereas the poor knowledge of antibiotic practices in some cases needs some recommendations. Indeed, the prescription of antibiotics in the bone and joint infections complies with certain obligations[3] : documented infection; probabilistic antibiotherapy initiated after microbiological sampling; association of antibiotics ; high plasma levels to be reached by the antibiotics ; and use of molecules having a good bone diffusion, for a recommended duration of 6 weeks.
The rate of presumption of cure at 6 weeks was 58.8% whereas the rate of cure at 6 months was 81.5 %. These results cannot be compared to the cure results in literature because most of the studies were conducted in different settings and the healing criteria were difficult to be defined as infectious or functional [2,3]. Finally, we found 2 major risk factors significantly influencing the cure rate, namely the existence of an abscess and at least one surgical revision during the first 6 weeks.
2. Conclusions
This study provided data about the epidemiological and therapeutic aspects of osteo-articular infection on material in 10 orthopedic surgery departments in Algeria. The results showed a high proportion of infection with GNB (64%) where Enterobacteriaceae are the most frequent pathogens. The importance of multi-resistant bacteria is also highlighted for Staphylococcus aureus and coagulase-negative staphylococci and for Enterobacteriaceae. Ultimately, these observations shed the light on the importance of continuously preventing and healing osteo-articular infections.
References
Centers for Disease Control and Prevention (CDC): Draft guide- line for the prevention of surgical site infection Protocol Corrections, Clarification, and Additions. (Accessed on April 03, 2016)
Ziza jm, Zeller V, Desplaces N, Mamoudy P. Infections sur pro- thèses articulaires : conditions du diagnostic et traitement. Revue du Rhumatisme 2006;73:337–44.
Recommandations de pratique clinique-Infections ostéo-arti- culaires sur matériel (prothèse, implant, ostéosynthèse. Médecine et maladies infectieuses 2009;39:745–74.
Pääkkönen M, Kallio PE, Kallio Mj, Peltola H. Management of osteoarticular infections caused by Staphylococcus aureus is similar to that of other etiologies: analysis of 199 staphylococcal bone and joint infections. Pediatr Infect Dis j 2012;31(5):436–8.
Dumaine V. Proposition d’un protocole de suivi des infections avé- rées de site opératoire en chirurgie orthopédique et traumatologique Revue de chirurgie orthopédique 2007;93:30–6.
Atif ML, Bezzaouchaa A, Mesbaha S, Djellatoa S, Boubechoua N, Bellouni R. Évolution de la prévalence des infections nosocomiales dans un centre hospitalier universitaire en Algérie (2001 à 2005). Mé- decine et maladies infectieuses 2006;36:423–8.
Desplaces N. Infections nosocomiales en chirurgie orthopédique. En- cycl. Méd. Chir (Editions Scientifiques et Médicales Elsevier SAS, Paris, tous droits réservés), Appareil Locomoteur, 14-016-B-10, 2000, 11 p.
Prendki V, Zeller V, Passeron D, Desplaces N, Mamoudy P, Stir- nemann j, Marmor S, Ziza jM. Outcome of patients over 80 years of age on prolonged suppressive antibiotic therapy for at least 6 months for prosthetic joint infection. Int j Infect Dis. 2014;29:184–9. doi: 10.1016/j.ijid.2014.09.012.
Tice AD, Hoaglund PA, Shoultz DA. Risk factors and treatment out- comes in osteomyelitis. j Antimicrob Chemother. 2003;51(5):1261–8.
Baek S-H. Identification and preoperative optimization of risk factors to prevent periprosthetic joint infection. World journal of Or- thopedics. 2014;5(3):362–7. doi:10.5312/wjo.v5.i3.362.
Diagnostic de l’infection sur prothèse articulaire, Tirésias volume II. ISBN 2-9512472-2-2 Paris 2002,15–6.
Dr Mohamed Islam KEDIHA, Pr Meriem TAzIR, Service de Neurologie, CHU Mustapha Pacha, Alger.
Abstract : Alzheimer’s disease is a disease of memory and not related to aging. It is a progressive neurodegenerative affection manifested by a gradual unfolding of the cognitive functions and of the memory with impairment of the activities of daily living which conditions its severity. Psychiatric and behavioral manifestations can aggravate its evolution. It is the leading cause of dementia after age 65 and its incidence increases with life expectancy. Its diagnosis is based on clinical aspects, neuropsychological assessment, biological evaluation, imaging data and after eliminating all the causes of curable dementia. After the diagnosis has been announced, the management of this disease is based on several components, including: the drug treatment itself, which can be instituted as early as possible, the management of behavioral disorder events, cognitive and speech stimulation, and Guidance of families and in particular the main caregiver.
Key-words : Dementia, cognition, memory
Résumé :
La maladie d’Alzheimer est une maladie de la mémoire et non une maladie de la vieillesse. Il s’agit d’une affection neurodégénérative progressive se manifestant par une détérioration progressive des fonctions cognitives et de la mémoire avec retentissement sur les activités de la vie quotidienne qui en conditionne sa sévérité. Les manifestations psychiatriques et du comportement peuvent en aggraver son évolution. C’est la principale cause de démence après 65 ans et son incidence augmente avec l’espérance de vie. Son diagnostic repose sur les aspects cliniques, l’évaluation neuropsychologique, le bilan biologique, les données de l’imagerie et après avoir éliminé toutes les causes de démences curables. Après l’annonce du diagnostic, sa prise en charge repose sur plusieurs volets comprenant : le traitement médicamenteux proprement dit qui doit être instauré le plus précocement possible, la prise en charge d’éventuels troubles du comportement, la stimulation cognitive et orthophonique ainsi que la guidance des familles et en particulier l’aidant principal.
Mots-clés : Démence, cognition, mémoire
Introduction :
a maladie d’Alzheimer (MA), Alzheimer disease AD, est classiquement définie comme étant une affection dégénérative du cerveau associant des troubles prédominant sur la mémoire, des troubles cognitifs et/ou du comportement mais ayant un retentissement sur l’activité de la vie quotidienne des patients (1).
Un nouveau lexique de définition a été adopté par « le groupe international d’experts sur la recherche de nouveaux critères diagnostiques de la maladie d’Alzheimer » ; la pierre angulaire de ce lexique est de considérer MA uniquement comme une entité clinique et symptomatique englobant à la fois le stade prédémentiel et les différentes phases de la démence (2). L’évolution de la recherche a permis de mettre en évidence des biomarqueurs spécifiques de cette pathologie qui permettent d’asseoir un diagnostic plus précis. Ceci a donc modifié les critères de diagnostic, critères de l’IWG
« International Work Group for the diagnosis of AD » (3,4).
Le phénotype clinique est centré par une démence progressive qui comprend notamment une altération de la mémoire épisodique, associée à d’autres troubles cognitifs et à des lésions histopathologiques caractéristiques qui définissent la maladie : les plaques séniles et les dégénérescences neurofibrillaires.
Vu que les investigations neuropathologiques ne peuvent être pratiquées qu’en post-mortem, le diagnostic de la MA reste probabiliste, se basant sur des critères cliniques, neuropsychologiques, radiologiques et biologiques (biomarqueurs).
Historique :
La MA a été décrite et identifiée pour la première fois le 4 novembre 1906 par le psychiatre et neuropathologiste Allemand, Alois Alzheimer lors de la 37ème conférence des psychiatres du Sud-Ouest de l’Allemagne, tenue à Tubingen.
Il étudia le cas d’Augusta Deter, admise à l’hôpital de Francfort pour un tableau de démence associant : troubles de la mémoire, mutisme, désorientation et hallucinations.
La malade mourut en Avril 1906 et l’analyse de la pièce autopsique révéla la présence des deux lésions histologiques.
Dans la 8ème édition du manuel de psychiatrie en 2012,
on attribua la paternité de cette affection à Alzheimer. Durant les années 80, les constituants de ces lésions ont été identifiés (5) : la protéine Bêta-amyloïde (constituant majeur des plaques séniles), identifiée en 1984 par le biologiste américain G. Glenner ; et la protéine Tau (présente dans les dégénérescences neurofibrillaires) par le Belge jP. Brion en 1985.
Dans les années 90, plusieurs gènes, impliqués dans les formes familiales précoces, ont été identifiés : le gène APP (Amyloid Precursor Protein) sur le chromosome 21, le gène PS1 (Presenilline) sur le chromosome 1 et le gène PS2 sur le chromosome 14.
En 1993, l’allèle Apo E4 a été découvert. Il s’agit d’un facteur de susceptibilité génétique dans le développement de formes sporadiques de MA (6).
Depuis 20 ans, de nombreux progrès dans la recherche ont été réalisés avec notamment des tests diagnostiques plus spécifiques (biomarqueurs précoces) et l’identification de nouvelles cibles thérapeutiques grâce aux modèles expérimentaux.
Epidémiologie :
Les démences et la MA en particulier, sont devenues un problème majeur de santé publique en raison du vieillissement de la population et de l’augmentation de l’espérance de vie. Voir carte ci-dessous.
La prévalence des démences passe de 1.2% des 65-69 ans à plus de 28 % des plus de 90 ans et celle de la MA de 0.6 % à 22.2 % (7).
Cette maladie affecterait plus de 25 millions de personnes dans le monde (7).
L’espérance de vie en est passée, en Algérie, de 55 à 76.4 ans (source : ONS 2012) avec 2 millions et demi de personnes âgées de plus de 60 ans (7.4 % de la population) ; de ce fait, le nombre de patients atteints est en nette augmentation (plus de 100.000 personnes en seraient atteintes, données de la société Algérienne de neurologie en 2004).
De nombreux facteurs de risque sont incriminés et permettent de mieux comprendre la pathogénie de la maladie mais aussi de la prévenir afin d’en diminuer l’incidence.
La MA est une maladie multifactorielle, faisant intervenir des facteurs à la fois individuels (prédispositions génétiques) et environnementaux (16) :
L’âge : est le principal facteur de risque, l’incidence doublant par tranche d’âge de 5 ans après 65 ans (8) ;
Le sexe féminin : les femmes ont un risque plus élevé de développer une MA (9) ;
Les antécédents familiaux de démences : le risque est multiplié par 3 si un parent du premier degré est atteint, par 7 si deux ou plus sont atteints (10) ;
Le niveau d’éducation : association entre le niveau bas d’éducation et un risque accru de maladie d’Alzheimer (10) ;
Les activités de loisirs (jardinage, voyages, tricot ou bricolage) sont associées à un risque moindre de démence et de MA (11) ;
Autres facteurs : la dépression, le célibat, l’isolement social, les antécédents de traumatisme crâniens sont aussi incriminés ;
Les facteurs vasculaires (HTA, diabète, dyslipidémie…) sont associés à un risque accru de survenue de MA (12, 13) ;
Les facteurs génétiques : le risque relatif de développer une MA est plus important chez les sujets porteurs de l’allèle E4 de l’apolipoproteine E (14). Il existe également des formes monogéniques (moins de 1%) autosomiques dominantes, de transmission mendélienne (gènes PS1, PS2 et APP), responsables de formes à début précoce (avant 60 ans).
Histologie :
Deux lésions histopathologiques sont caractéristiques de la MA :
Les dégénérescences neurofibrillaires (DNF) : accumulation intra neuronale de fibrilles disposées en hélice,composées de protéine Tau anormalement phosphorylée. Les plaques séniles (PS) ou plaques amyloïdes : situées dans le domaine extra cellulaire avec comme principal constituant, le peptide amyloïde Beta.
Physiopathologie (1) :
Il est nettement établi, actuellement, que la production et l’accumulation du peptide bêta-amyloïde (A beta) sont essentielles à la pathogénèse de la maladie d’Alzheimer. Les mutations de l’Amyloïd Precursor Protein (APP) conduisent à une maladie d’Alzheimer de début précoce ; toutes les mutations actuellement connues associées à la maladie d’Alzheimer augmentent la production d’Ab ; Ab est neurotoxique in vitro et conduit à la mort cellulaire.
Les conséquences secondaires au dépôt d’A beta sont la formation de dégénérescences neurofibrillaires DNF, l’oxydation, l’excitotoxicité glutamatérique, l’inflammation et l’activation de la cascade de la mort cellulaire apoptotique ; c’est la cascade amyloïde.
L’extension progressive des DNF induit une perte neuronale et une atteinte des systèmes de neurotransmetteurs observée dans plusieurs noyaux sous corticaux : systèmes cholinergique et glutamatergique.
L’envahissement du tissu cérébral s’effectue selon la séquence suivante : région hippocampique (aires transentorhinale, entorhinale, hippocampique) qui est
la plus vulnérable du cerveau, puis extension vers tout le cortex temporal (pôle temporal, cortex temporal inférieur puis temporal moyen). À ce stade le processus dégénératif est encore infra-clinique et peut le demeurer plusieurs dizaines d’années. La phase clinique avec apparition des symptômes apparaît avec l’atteinte des régions corticales associatives (temporal supérieur, cortex préfrontal et cortex pariétal). En fin d’évolution, les lésions envahissent l’ensemble du cortex et de nombreux noyaux sous corticaux.
La « cascade Amyloïde » (Cummings 2004 – NEjM)
Manifestations cliniques (15,20) :
La MA est une maladie de la mémoire et non une maladie liée au vieillissement.
Les symptômes diffèrent d’une personne à une autre et ne sont pas de la même sévérité chez tous les malades ; cependant, les troubles de la mémoire sont ceux qui inquiètent fréquemment au début de la maladie. Il est cependant à préciser que la maladie ne commence pas forcément par des pertes de mémoire, mais également par d’autres troubles décrits ci-dessous.
Troubles de la mémoire :
Les défaillances de la mémoire comptent parmi les premiers signes de la MA et de la plupart des autres démences. Ces défaillances se manifestent d’abord par des petites perturbations de la vie quotidienne puis s’accentuent à tel point que des pans entiers du passé récent disparaissent. Enfin, peu à peu, la mémoire du passé plus lointain est atteinte. Ainsi, la mémoire se dégrade progressivement, touchant la mémoire des événements récents en premier. Le malade voit sa compréhension du monde morcelée par ces troubles de la mémoire. De plus, ils peuvent être bouleversants pour les proches quand la personne oublie le nom de son conjoint ou de son enfant, ou inquiétants quand elle oublie de fermer le gaz par exemple.
Le mode de début de ces troubles est progressif, dans les ¾ des cas, avec oublis de taches à effectuer, de rendez-vous ou de trajets, parfois une désorientation temporospatiale (à noter que la désorientation temporelle précède la désorientation spatiale).
L’interrogatoire permet aussi de faire sortir la notion de « plainte mnésique suspecte » cf. tableau
L’amnésie peut toucher la mémoire sémantique (connaissances générales du patient) qui reste longtemps préservée et la mémoire épisodique (ayant un rapport avec la propre histoire du patient).
Au total, la plainte mnésique peut revêtir 3 aspects :
La plainte bénigne, liée au vieillissement cognitif normal
La plainte avec baisse des performances cognitives et répercussion sur l’autonomie du patient
La plainte mnésique patente mais sans retentissement sur l’autonomie du patient, c’est le MCI ou Mild Cognitive Impairment, qui revêt tout son intérêt dans la prise en charge thérapeutique précoce de la MA.
Troubles du langage :
2ème élément sémiologique le plus retrouvé. Le patient va présenter un manque de mots avec une compréhension et une répétition qui restent longtemps normales, la lecture est préservée mais une dysorthographie peut se voir assez précocement.
Dans les formes extrêmes ou sévères, on observe une aphasie globale avec notamment une désintégration du langage oral et écrit, une expression jargonnée, une compréhension aléatoire et une écriture illisible.
Troubles praxiques :
Ils restent exceptionnels en mode d’entrée. Le patient aura des difficultés à réaliser des séquences gestuelles simples ou complexes comme plier une lettre ou une enveloppe, allumer une bougie avec une allumette… pouvant aller jusqu’à l’apraxie de l’habillage (difficultés à mettre un pantalon, à boutonner une chemise…)
Troubles gnosiques et visuo-spatiaux :
L’agnosie visuelle est fréquente avec, notamment, des difficultés à reconnaitre des images, puis des objets, puis des visages familiers (prosopagnosie), des difficultés à reconnaitre les parties de son corps (autotopoagnosie) et une anosognosie.
La désorientation topographique reste précoce pour le grand environnement, puis pour des espaces de plus en plus réduits.
Troubles des fonctions exécutives :
Apparition d’une difficulté voire impossibilité à interpréter correctement des proverbes, à résoudre des problèmes simples, un déficit du calcul mental, à élaborer une stratégie et à planifier les étapes d’une action … c’est le syndrome dysexécutif.
Troubles psycho-comportementaux :
Ils sont en rapport avec les lésions frontales et temporales de la maladie. Ils représentent la première cause de placement en institution.
Ils sont dits positifs quand le patient présente agitation, agressivité, délire et hallucinations ; et négatifs quand il y a apathie et dépression.
Ces troubles, après 70 ans, doivent faire rechercher un début de démence et faire pratiquer une évaluation des fonctions supérieures.
Troubles somatiques :
Il n’existe pas de signes neurologiques de focalisation dans la maladie d’Alzheimer sauf parfois en fin d’évolution ou l’on observerait des signes extra-pyramidaux, des myoclonies ou des crises d’épilepsie.
Évolution :
La MA évolue en 3 temps :
Une phase préclinique (dure 15 à 20 ans) : les lésions se constituent lentement et progressivement sans donner aucun signe clinique.
Une phase pré-démentielle (dure 2 à 4 ans) : les lésions débutant au niveau des centres hippocampiques avec apparition des premières difficultés (oublis des faits récents et modifications émotionnelles). L’autonomie du sujet reste largement respectée. C’est le stade MCI. Une phase démentielle : aggravation progressive des troubles initiaux, apparition d’autres déficits cognitifs (praxies, langage…) qui vont retentir sur l’autonomie du sujet et sa vie relationnelle (gérer ses comptes, sortir de chez soi, utiliser un téléphone, prendre correctement ses médicaments…)
Selon la sévérité de ce retentissement, on parlera de démence légère, modérée et sévère.
Bilan paraclinique :
Le diagnostic de la MA est un diagnostic d’élimination ; il faut donc avoir éliminé toutes les causes de démences curables pour pouvoir poser le diagnostic. De ce fait, ce bilan s’impose : dosage des folates, dosage de la vitamine B12, bilan thyroïdien avec dosage des anticorps anti TPO, sérologie syphilitique et de maladie de Lyme. Une IRM cérébrale, avec des coupes coronales, doit être pratiquée afin d’apprécier le volume des hippocampes mais également afin d’éliminer d’autres causes (processus intracrânien, hydrocéphalie chronique de l’adulte ou des séquelles d’accident vasculaire cérébral).
Le dosage des biomarqueurs(5) dans le liquide céphalorachidien, à savoir Tubulin Associated Unit Tau, la phospho-Tau et la protéine A beta 42 peuvent être réalisés en cas de doute diagnostique, en particulier chez les patients jeunes.
Diagnostic de la MA : (15)
Il est recommandé que ce diagnostic soit posé dès les premiers symptômes. Il nécessite une évaluation cognitive approfondie, réalisée dans le cadre d’une consultation mémoire spécialisée. Actuellement, presque tous les services de neurologie en Algérie en sont dotés.
Critèresdiagnostiques: il est recommandé d’utiliser les critères diagnostiques de la MA selon le DSM-IV-TR(15) ou le NINCDS-ADRDA(15).
Évaluation initiale : il faut effectuer un entretien avec le patient et son accompagnant qui serait capable de donner des informations fiables. Il faut alors relever les antécédents médicaux et familiaux du patient, les traitements antérieurs et actuels, le niveau d’éducation, l’activité professionnelle, l’histoire de la maladie et le retentissement des symptômes sur la vie quotidienne.
Évaluation cognitive globale : Une évaluation cognitive globale peut être effectuée à l’aide du Mini-Mental-State-Examination MMSE, dans sa version consensuelle (15) mais d’autres tests, selon le niveau d’éducation du patient, doivent être utilisés, parmi lesquels, on peut citer :
L’épreuve des 5 mots ;
Le test de fluence verbale ;
Le test de l’horloge ;
Le test des neuf images
Le test de mémoire associative ;
La batterie rapide d’évaluation frontale BREF ;
La DMS-48 … etc.
Évaluation fonctionnelle : Évaluer le retentissement des troubles sur les activités de la vie quotidienne à l’aide d’une échelle, IADL (Instrumental Activities of Daily Life), comportant 4 items : utilisation du téléphone, utilisation des transports, prise de médicaments et gestion des finances. La nécessité d’une aide à au moins un de ces items constitue un retentissement significatif de ces troubles sur l’activité quotidienne des patients.
Examen clinique : il doit apprécier l’état général (poids) et cardiovasculaire (TA, fréquence cardiaque, troubles du rythme), le degré de vigilance, les déficits sensoriels (auditifs et visuels) pouvant interférer avec la passation de test neuropsychologique.
L’examen neurologique reste longtemps normal dans la maladie d’Alzheimer. L’existence de signes neurologiques doit faire discuter d’autres diagnostics ou une comorbidité.
Étude génétique : le génotypage de l’Apo lipoprotéine E n’est pas recommandé comme test de dépistage de la MA. La recherche de la mutation de l’un des trois gènes actuellement en cause (APP, PSEN1, PSEN2) peut être réalisée chez des patients avec des antécédents familiaux de MA ou en cas de début précoce, et ce après consentement écrit.
Prise en charge de la maladie d’Alzheimer :
La prise en charge de ces patients passe par 5 étapes :
Annonce du diagnostic
Prise en charge thérapeutique médicamenteuse
Prise en charge thérapeutique non médicamenteuse
Prévenir et traiter les troubles du comportement
Guidance des familles
L’annonce du diagnostic : « Toute personne a le droit d’être informée sur son état de santé ». L’annonce peut être effectuée en plusieurs étapes selon la réceptivité de la personne. À un stade précoce de la maladie, avec des capacités cognitives suffisamment conservées pour que la compréhension soit possible, le diagnostic peut être communiqué à la famille ou à une personne de son choix.
À un stade tardif, l’annonce du diagnostic nécessite une écoute particulière du patient, un choix soigneux des mots et une prise en compte de ses capacités de communication.
2. Les médicaments de la MA(20) :
Un traitement spécifique doit être envisagé chez un patient atteint de la MA lorsque le diagnostic a été annoncé, en tenant compte du rapport bénéfice/risque. Il convient de discuter avec le malade et/ou l’accompagnant de l’attente raisonnable des effets du traitement et du risque d’effets secondaires. Ce traitement ne peut être instauré que par des neurologues, des gériatres ou des psychiatres.
2 classes thérapeutiques sont disponibles :
Les anticholinestérasiques : Donépézil, Rivastigmine et Galantamine. Ils agissent en bloquant l’acétylcholines-térase et augmentent donc les quantités d’acétylcholine dans la synapse. Voir image ci-dessous.
Indiqués uniquement dans les formes légères à modérées de la MA (MMSE entre 10 et 26).
Leur effet reste modeste sur la cognition mais favorable sur la prévention des troubles du comportement et le maintien de l’autonomie.
Donepezil existe en comprimés à 5 et 10 mg avec une posologie de 5 à 10 mg/jour en une prise.
Rivastigmine existe en gélules de 1.5, 3, 4.5 et 6 mg et en solution buvable. La posologie est de 6 à 12 mg en 2 prises.
Galantamine existe en comprimés à 4, 8 et 12 mg, en solution buvable et en forme à libération prolongée. Elle n’est pas encore commercialisée en Algérie.
Les anti glutamates : Mémantine. Agissent en bloquant les effets pathologiques de taux élevés de glutamates qui pourraient aboutir à un dysfonctionnement neuronal avec hyperphosphorylation de Tau. Elle est indiquée dans les formes modérément sévères à sévères de la maladie (MMSE entre 3 et 15). La posologie est de 20 mg/jour en deux prises et à atteindre progressivement en 3 semaines.
Contre-indications : Aucune contre-indication mais des précautions particulières en cas d’asthme et en cas de troubles de la conduction ou de la repolarisation cardiaque.
Effets secondaires : se voient dans 2 à 18 % des cas à type de nausées, vomissements et diarrhées (effets cholinomimétiques), bradycardie, syncopes, convulsions, insomnie et cauchemars.
Buts de la prescription : Rassurer le patient et sa famille « Ça se traite ». Stabiliser les troubles.
Améliorer et prévenir les troubles du comportement. Retarder l’usage des neuroleptiques.
Retarder l’institution.
Quand arrêter le traitement : En cas de mauvaise tolérance ou non compliance du patient. Si poursuite de la détérioration cognitive au même rythme et aucun bénéfice sur les capacités fonctionnelles. Si la démence devient très sévère.
Traitement par bithérapie : Dans les formes modérées à sévères de la MA, un traitement associant Donepezil et Memantine a été proposé mais aux effets contradictoires(22, 23) ; en effet, le seul bénéfice net de cette association étant celui sur les troubles du comportement mais pas sur la cognition.
1. Traitement des troubles psycho-comportementaux :
Ces thérapeutiques peuvent être envisagées après avoir
évalué leur bénéfice/risque. Il n’est pas recommandé d’utiliser des neuroleptiques classiques en raison du risque d’effets secondaires graves (atteinte extrapyramidale, AVC, pneumopathie d’inhalation…). Il est conseillé d’utiliser des antipsychotiques de dernière génération (Olanzapine, Risperidone) en cas de symptômes psychotiques sévères (hallucination, agitation, hétéro ou auto agressivité).
En cas d’insomnie, proposer du Zolpidem. En cas de syndrome dépressif associé, proposer des inhibiteurs de recapture de la sérotonine et des thymorégulateurs comme la carbamazépine en cas d’hostilité.
2. Traitement non médicamenteux :
Ils doivent être pratiqués par un personnel formé.
Prise en charge orthophonique : (18) Elle vise à maintenir et à adapter les fonctions de communication du patient (langage, parole et autres) et à aider la famille à adapter son comportement aux difficultés du malade. L’objectif étant de continuer à communiquer avec lui. Elle peut être prescrite à différents stades de la maladie.
Stimulation cognitive (18) : Elle vise à limiter le handicap en stimulant les capacités restantes et elle est pratiquée par les orthophonistes. Elle doit être poursuivie si le patient est demandeur et arrêtée s’il se sent en situation de mise en échec. Les activités proposées sont des mises en situation ou des simulations de situations vécues (trajet dans le quartier, toilette, téléphone …), des exercices de
mémorisation, d’évocation de souvenirs, d’écriture et de langage.
Intervention portant sur le comportement (19) : La musicothérapie, l’aromathérapie, la stimulation multi sensorielle, la rééducation de l’orientation, la thérapie assistée d’animaux, le massage et la luminothérapie pourraient améliorer certains aspects du comportement.
Autres : Une bonne hygiène de vie avec un bon sommeil, un minimum d’anxiété et de stress sont bénéfiques. Réaménager le domicile en fonction des nouvelles capacités du patient (17).
Traitements préventifs à base de vitamine E et C dont l’association entrainerait une réduction de l’incidence de la maladie. L’activité physique de la cinquantaine (avec essoufflement et sueur 2 fois par semaine) réduit le risque de MA 20 ans plus tard (24).
3. Guidance des familles :
Les aidants doivent recevoir une information sur la maladie, sa prise en charge et sur l’existence d’associations de familles. Il leur sera proposé : une psycho-éducation individuelle ou en groupe ; un groupe de soutien avec d’autres aidants ; un support téléphonique ou par internet dans l’idéal ; une thérapie familiale ; Intérêt des structures d’accueil de jour ou d’hébergement temporaire pour les soulager (21).
Références bibliographiques :
1/ Cummings jL Alzheimer’s disease. The New England journal of Medicine. N. Eng j Med 2004; 351.56-67
2/ Dubois B, Feldman H, jacova C et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 2010; 9:1118-27. DOI: 10.1016/S1474-4422S(10) 70223-4
3/ Dubois B, Feldman H, jacova C et al Advancing research diagnos- tic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol 2014;13:614-19. DOI: 10.1016/S1474-4422(14) 70090-0
4/ Cummings jL, Dubois B, Molinero jL et al. International work group criteria for the diagnosis of Alzheimer disease. Med Clin North Am 2013 may; 97(3):363-8. DOI: 10-1016/j;mcna-2013.01.001
5/ Molinuevo jL, Blennow K, Dubois B, et al. The clinical use of cerebrospinal fluid biomarker testing for Alzheimer’s disease diagno- sis: a consensus paper for the Alzheimer’s biomarkers standardization initiative. Alzheimer and dementia 10(2014) 808-817. DOI: org/10- 1016/j.jalz.2014.03.003
6/ Rajah MN, Wallace LMK, Ankudowich E et al. Family history and Apo E4 risk for Alzheimer’s disease impact the neural corre- lates of episodic memory by early midlife. Neuroimage.clinical.2017 mar31;14:14.760-774. DOI: 10.1016/j.nicl.2017.03.016.
7/ Dartigues jF, Berr C, Helmer Cet al. Epidémiologie de la maladie d’Alzheimer. Médecine sciences 2002, 18 : 737-43
8/ Lobo A, Launer Lj, Fratiglioni L et al. Prevalence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurology 2000; 54(suppl 5): S4-9
9/ Fratiglioni L, Launer Lj, Andersen K et al. Incidence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurology 2000; 54(suppl 5): S10-5
Launer Lj, Andersen K et al. Rates and risk factors for dementia and Alzheimer’s disease: result from EURODEM pooled analyses. European studies of dementia. Neurology 1999; 52: 78-84
11/ Fabrigoule C, Letenneur L, Dartigues jL et al. Social and leisure activities and risk of dementia: a prospective longitudinal study. j.Am Genet soc 1995; 43: 485-90
12/ Hofman A, Ott A, Breteler MMB, et al. Atherosclerosis, apoli- poprotein E, prevalence of dementia and Alzheimer’s disease in the Rotterdam study. Lancet 1997. 349: 151-4
13/ Ott A, Stolk RP, Hofman A et al. Association of diabetes mellitus and dementia: the Rotterdam study. Diabetologica 1996 ; 39: 1392-7 14/ Breitner jCS, Folstein MP, Murphy EA, et al. Familial agragation in Alzheimer’s dementia. j. Psychiatr Res 1986; 20:31-43
15/ Groupe de travail de la HAS (recommandations professionnelles). Diagnostic et prise en charge de la maladie d’Alzheimer et des mala- dies apparentées. Revue neurologique 164(2008) 754-774. DOI : 10.1016/j ; NEUROL.2008.06.007
16/ Chan KY, Wang W, Wu jL et al. Epidemiology of Alzheimer’s disease and other forms of dementia in China 1990-2010: a systemic review and analysis. Lancet 2013 ; 381: 2016-23.
17/ Thèse de doctorat en médicine- Créteil. Marine Lousteau. L’hospitalisation à domicile est-elle une réponse adaptée à la prise en
charge des patients Alzheimer ? 2011
18/ Thèse de doctorat en sciences humaines et sociales. Emiline Lapre. Maladie d’Alzheimer et thérapies non médicamenteuses : éva- luation de la stimulation cognitive et de l’activité physique sur le fonc- tionnement exécutif. Soutenue le 10/12/2010 à Bordeaux
19/ Thèse de doctorat en psychologie- Montréal. Aline Moussand. L’utilisation de la musique comme support de nouveaux apprentis- sages dans le vieillissement normal et la maladie d’Alzheimer. Soute- nue en mai 2012
20/ Scheltens P, Blennow K, Breteler MMB, et al. Alzheimer’s di- sease. Lancet neurology 2016. DOI : 10.1016/S0140-6736
21/ Thèse de doctorat en médecine. Sarah Benhamadou. Maladie d’Alzheimer et démence apparentée : intérêt de la réhabilitation à domicile. Lille , le 3 avril 2013
22/ Wimo A. Long term effects of Alzheimer disease treatment. Lan- cet neurology 2015. DOI: 10.1016/S1474-4422.
23/ Howard R et al. Donepezil and Memantine for moderate-to- severe Alzheimer’s disease. The New England journal of Medicine. March 2012
24/ Rovio S et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurology 2005 nov; 4(11): 705-11. DOI: 10.1016/S1474-4422(05)70198-8
Pr Lyes CHERFI Anesthésie Réanimation, HCA, Ain Naadja, Alger
Abstract :
Anesthesia is a discipline that aims to ensure the patient’s comfort, during painful invasive procedures, especially surgical, but also diagnostic. To do it, several techniques exist roughly separated into general anesthesia and loco regional anesthesia. In reality, these two major groups of techniques represent several practices, the name of which is not always clear. Moreover, the combination of these two techniques is advocated in what is today called improved rehabilitation. Neither of these two groups of techniques is superior to the other in terms of efficiency and safety. The choice of technique depends on the type of surgery, the condition of the patient and the habits of the service. General anesthesia is the most common. It causes profound physiological changes and requires the partial or total substitution of many functions, in particular respiratory, neurological and cardiovascular, by artificial assistance. Security is therefore essential; including through extensive monitoring and clinical monitoring. Monitoring detects early and sensitive changes in major functions, helps the physician in the choice of drugs and their doses, detects the appropriate time for the use of antidotes and guides the protocol of their administrations.
Résumé : L’anesthésie est une discipline qui a pour but d’assurer le confort du patient, lors d’actes invasifs douloureux, en particulier chirurgicaux, mais aussi diagnostiques. Pour ce faire, plusieurs techniques existent, séparées grossièrement en anesthésie générale et en anesthésie locorégionale. En réalité, ces deux grands groupes de techniques représentent plusieurs pratiques, dont la dénomination n’est pas toujours claire. Par ailleurs, l’association de ces deux techniques est préconisée dans ce qui s’appelle aujourd’hui la réhabilitation améliorée. Aucun de ces deux groupes de techniques, n’est supérieur à l’autre, en termes d’efficacité et de sécurité. Le choix de la technique dépend du type de chirurgie, de l’état du patient et des habitudes du service. L’anesthésie générale est la plus pratiquée. Elle entraine des modifications physiologiques profondes et impose la substitution partielle ou totale de certaines fonctions, en particulier respiratoire, neurologique et cardiovasculaire, par une assistance artificielle. La sécurité s’impose donc ; notamment par le recours à un monitorage exhaustif et une surveillance clinique. Le monitorage permettra de détecter précocement et de manière sensible toute modification des grandes fonctions, d’aider au choix des drogues et de leurs posologies et enfin de déterminer le moment idoine pour recourir à l’usage des antidotes et le protocole précis de leur administration.
Mots-clés : Sécurité en anesthésie, techniques anesthésiques, antidotes, monitorage.
Introduction :
anesthésie est une discipline, dont le rôle principal est d’allier constamment la sécurité et le confort du patient. Ce n’est ni une spécialité à démarche diagnostique, ni thérapeutique. Elle permet le déroulement des actes invasifs thérapeutiques chirurgicaux ou non chirurgicaux (ponction-drainage, résection endoscopiques de polypes …) et des actes diagnostiques (colonoscopie, bronchoscopie, cholangio-pancréaticographie rétrograde par voie endoscopique ….).
L’anesthésie assure une insensibilité à la douleur et une amnésie et un relâchement musculaire (le plus souvent recherché). Afin d’atteindre ces objectifs, il existe plusieurs techniques. Elles sont séparées en deux grandes familles :
Anesthésie générale (AG), qui consiste à induire un sommeil profond, une amnésie et une analgésie intense et un relâchement musculaire.
Anesthésie locorégionale (ALR), qui consiste à bloquer la transmission de l’influx nerveux à un endroit donné du nerf, évitant ainsi temporairement l’arrivée du signal douloureux ou sa transduction en douleur consciente.
Dans les 2 cas, les produits utilisés ainsi que les techniques d’administration, induisent des modifications importantes des grandes fonctions et imposent souvent, comme dans le cas d’une anesthésie générale, le recours à une assistance respiratoire via une ventilation mécanique et parfois cardiovasculaire notamment par un support vasopresseur. Un réchauffement passif et une protection contre toute déperdition thermique, permettent de maintenir une température corporelle normale. Enfin, des apports hydroélectrolytiques permettent de se substituer au jeune imposé par la chirurgie et aux déperditions exagérées par l’évaporation et les pertes plasmatiques. Tout ceci ne peut être réalisé sans un contrôle étroit et permanent par l’équipe d’anesthésie-réanimation, aidée en cela par un monitorage exhaustif défini en fonction de l’état du patient et de l’importance de la chirurgie. L’anesthésie générale est donc un ensemble de techniques qui modifie le fonctionnement normal et habituel de l’homéostasie. Le rôle du médecin anesthésiste-réanimateur (MAR) est de recourir à des moyens de substitution partielle ou totale des fonctions altérées. Plus que l’ALR, l’AG induit le maximum de modifications. Nous allons limiter notre propos à cette dernière
Déroulement d’une AG :
Le nombre d’anesthésies générales réalisées en Algérie est difficile à apprécier. D’abord pour un problème de définition. En effet, beaucoup d’actes de sédation plus ou moins profondes ne sont pas comptabilisés, de même qu’il existe vraisemblablement une sous déclaration de l’activité anesthésique libérale. Enfin, la part de l’ALR varie d’une enquête à une autre(1). Nous estimons le nombre d’actes anesthésiques totaux à plus de 2 millions par an ; parmi lesquels l’AG couvre au moins 70% des actes.
Lors d’une AG, trois familles de drogues anesthésiques sont utilisées. Les morphiniques qui bloquent la sensibilité douloureuse et les narcotiques qui assurent un sommeil artificiel, déjà entamé par les morphiniques. Alors pourquoi induire le sommeil, si les morphiniques sont suffisamment puissants pour bloquer toute sensation douloureuse ? La question mérite d’être posée puisqu’une technique d’anesthésie basée uniquement sur les morphiniques, mais à très forte dose, a longtemps été utilisée, en particulier en chirurgie cardiaque. Il s’agit de l’anesthésie analgésiante. Cette technique, connait cependant beaucoup de contraintes, une très forte dose de morphiniques, implique des effets adverses puissants et un retard d’élimination de ces derniers. L’autre inconvénient est que le 2ème objectif de l’anesthésie qu’est l’amnésie est alors difficilement atteint. Rappelons que cet objectif représente la deuxième préoccupation des MAR(2). L’association d’une narcose pharmacologique est utile à plus d’un titre : l’amnésie, le confort du patient et la réduction des doses de morphiniques.
Les morphiniques sont par ailleurs impliqués dans la genèse des douleurs chroniques post-chirurgicales (DCPC)(3). La réduction des doses administrées permettra de réduire leurs effets adverses directs (bradycardie, légère hypotension, dépression respiratoire, nausées-vomissements ….), mais aussi l’incidence des DCPC. Par ailleurs, certaines équipes, notamment Belges ont proscrit les morphiniques en peropératoire. Ils s’aident en cela en associant une anesthésie générale à base de narcotiques avec ou sans curares et d’une ALR. Cette démarche semble extrémiste à l’exemple du ‘’tout morphinique’’ d’avant et ne peut être proposée pour une large pratique. Les curares ou myorelaxants représentent le troisième groupe de drogues utilisées lors d’une AG. Même s’ils ne sont pas toujours utilisés, ils restent indispensables dans la
majorité des cas. Leur usage facilite l’intubation endotrachéale, mais surtout le relâchement musculaire lors de la chirurgie thoraco-abdominale(4).
Enfin, le terme prise en charge anesthésique peri-opératoire est plus approprié, du fait de l’élargissement progressif du cahier des charges du MAR, qui doit prendre en charge le patient à anesthésier, plusieurs jours ou semaines avant, et aussi après l’acte opératoire. Ces trois étapes préopératoire, peropératoire et postopératoire, sont une suite logique, puisqu’il s’agit d’une continuité de soins, à commencer par la période préopératoire qui consiste à apprécier l’état du patient, sa capacité à subir l’agression chirurgicale, la définition de son statut, sa préparation éventuellement et enfin la définition de la stratégie de sa prise en charge. Tout ceci est défini lors de la consultation d’anesthésie, obligatoire et qui doit se tenir à distance de l’acte opératoire. Elle permettra, après un examen soigneux et l’analyse de certains examens paracliniques, d’apprécier le risque anesthésique et chirurgical en s’aidant de classifications simples mais largement validées, à l’exemple de la classification ASA(5), de réajuster certaines thérapeutiques (anti-thrombotiques, anti-diabétiques, anti-hypertenseurs …..), et de s’aider le cas échéant de certains avis et examens spécialisés (echocoeur, EFR …). C’est durant cette phase qu’est définie toute la stratégie anesthésique (protocoles anesthésique, analgésique, anti-infectieux, anti-thrombotique, règles du jeune ….).
La période peropératoire est le temps effectif de la réalisation de l’anesthésie. La pratique d’une anesthésie générale connait aussi trois temps, l’induction ; l’entretien et le réveil. L’induction anesthésique consiste à induire le sommeil par l’administration des drogues anesthésiques, mais aussi de mettre en route l’assistance respiratoire, le plus souvent par le recours à une ventilation mécanique via une prothèse endotrachéale, appelée communément l’intubation trachéale. De plus, cette période d’induction, comparée à juste titre au décollage d’un aéronef, est une période critique, susceptible de voir apparaitre un certains nombre de complications, tels que la réaction allergique imprévisible, l’impossibilité de réaliser l’intubation endotrachéale, le retentissement hémodynamique chez les sujets à risque etc. Pour cela la connaissance parfaite de l’état du patient s’impose, ainsi que la maitrise des techniques et de la pharmacologie des drogues anesthésiques. Enfin, la mise en route d’un monitorage adapté au patient et aux conséquences potentielles du type de chirurgie, est une étape primordiale. La phase d’entretien, la plus longue, consiste à entretenir un niveau d’endormissement et éventuellement de relâchement musculaire adéquats, nécessite la même surveillance, pour prévenir toute complication inhérente à l’anesthésie mais surtout à la chirurgie (hémorragie….). Le réveil, comparé à l’atterrissage, est aussi une étape cruciale, puisque ce dernier peut être retardé, par une élimination incomplète des drogues, et nécessiter des antidotes, guidés en cela par un monitorage spécifique (exp : monitorage de la profondeur du relâchement musculaire : NMT). Ce rapprochement avec l’aviation, a incité les professionnels de l’anesthésie (médecins, fabricants de matériel) à s’inspirer des avancées de l’aviation pour renforcer la sécurité des patients. Un des enseignements clés de cette comparaison est l’instauration de check-list quotidienne dans chaque salle opératoire, la rédaction de protocoles disponibles dans les lieux d’exercice (salles opératoires, salles de surveillances post-interventionnelles). Le réveil d’une anesthésie générale est donc une étape importante et délicate de l’anesthésie. De la qualité de ce réveil, découlera en partie le déroulement de la période postopératoire. Le réveil anesthésique peut être rapide, calme, sans incidents ou lent, incomplet et agité. La qualité du réveil dépend de l’état antérieur du patient, du déroulement de la chirurgie ainsi que de la qualité de l’anesthésie. Cette dernière a une grande influence sur les délais de recouvrements des différentes fonctions (respiratoire, conscience ….), et sur l’absence de complications (agitation, douleur, hypoxie, inhalation….). Le MAR peut aussi s’aider d’antidotes pour contrecarrer les effets résiduels de certaines drogues.
Enfin, la période postopératoire consiste en la poursuite des soins et de la surveillance du patient. Cette partie, que le MAR partage avec le chirurgien et le personnel paramédical est la plus longue. Elle consiste à remettre toutes les fonctions en l’état, à traiter les conséquences de l’acte anesthésique et chirurgical (douleur, nausées, vomissements, transit perturbé …), à assister certaines fonctions (déshydratation, dénutrition, troubles électrolytiques…), à surveiller et à prévenir la survenue de complications postopératoires précoces (troubles respiratoires, cardio-vasculaires…) et tardives (maladie thromboembolique, infections ….)
Quelles drogues anesthésiques utiliser ?
Nous ne considérerons que les produits disponibles en Algérie ou qui le seront dans un avenir proche.
Drogues utilisées pour l’endormissement (narcotiques) : sont regroupées sous le terme générique d’anesthésiant, appartiennent à plusieurs familles et sont parfois associées entre elles. Le choix de la ou des molécules dépend de plusieurs paramètres, liés au patient (tolérance cardio-vasculaire, allergie …) ou au type de chirurgie (durée, siège …). Nous distinguons :
Les narcotiques administrés par voie intraveineuse : appartiennent à plusieurs familles (Benzodiazépines, Barbituriques, Propofol, Kétamine …)
La voie IV est la plus utilisée ; adaptée au poids et à la tolérance du patient, la plupart des narcotiques entrainent un retentissement faible. Leur association (co-induction) est synergique. Elle permet de réduire les doses et le retentissement en particulier cardio- vasculaire.
– Les narcotiques administrés par voie inhalatoire (protoxyde d’azote (N20), halothane (Hal), isoflurane (Iso), sevoflurane (Sev), desflurane (Des)) L’anesthésie inhalatoire est l’ancêtre de toutes les anesthésies. D’abord archaïque à base de chloroforme et d’éther administrés directement sur une éponge posée sur la face du patient, avec énormément de complications (détresse respiratoire, inhalation, hypersécrétion…), elle a presque était abandonnée au profit de la voie intraveineuse, plus sécurisante et surtout moins hasardeuse. Depuis, de nouvelles molécules sont apparues, et leur administration nettement améliorée par des vaporisateurs de mieux en mieux précis. Ces deux dernières décennies, deux nouveaux halogénés ont étés développés, le sévoflurane et le desflurane. Leurs structures chimiques leur procurent une cinétique particulièrement intéressante. En effet, les halogénés sont caractérisés par leur solubilité dans le sang ; administrés par voie inhalatoire, leur diffusion vers le sang capillaire dépend du gradient alvéole-sang capillaire qui dépend de la concentration administrée et de la solubilité. Plus un halogéné est soluble, plus l’induction inhalatoire est lente, la dose est importante et l’élimination est retardée. À l’inverse, un halogéné peu soluble, maintient un gradient élevé, facilite la diffusion, limite l’espace de diffusion aux régions richement vascularisées, telle le cerveau, site d’action de ces médicaments. Ainsi, l’induction et le réveil anesthésiques sont rapides, la profondeur de l’anesthésie est facilement modulable lors de l’entretien, avec moins de retentissement cardiovasculaire. Le coût relativement élevé de ces drogues est facilement contrebalancé par les économies réalisées par le recours à un circuit avec bas débit de gaz frais, réduisant ainsi la quantité consommée par un rapport de 8/1 voire 10/1, sous condition d’un monitorage des gaz inhalés, disponible actuellement en série sur tous les nouveaux appareils d’anesthésie (6, 7, 8). Un autre grand avantage spécifique au sevoflurane est la possibilité de faire une induction anesthésique inhalatoire chez l’enfant et l’adulte. En effet, parmi les halogénés les moins solubles, seul ce dernier est toléré par le patient éveillé (gout doux sucré). Les autres ; l’isoflurane, mais surtout le desflurane ont un gout ocre et ne sont utilisables que sur patient déjà endormi pour entretenir l’anesthésie. Le sevoflurane est donc utilisable pour induire et entretenir l’anesthésie.
L’analgésie (morphiniques) : À part la durée d’action, peu de spécificités conditionnent le choix d’un morphinique. Ainsi nous distinguons les morphiniques à très courte durée d’action, le rémifentanil, de moins en moins utilisé depuis qu’il est mis en cause dans la genèse des DCPC. L’alfentanil est utilisé pour les chirurgies et actes invasifs de courte durée et enfin le fentanil et surtout le sufentanil sont les plus utilisés et répondent aux impératifs de la majorité des situations. Parmi les inconvénients de ces derniers, la durée d’action relativement longue et le risque d’accumulation, entrainant des retards de réveil et/ou une dépression respiratoire postopératoire précoce. Le monitorage de la profondeur de l’analgésie n’est pas encore de routine. Quelques systèmes existent déjà, mais ne sont pas encore validés et proposés par les sociétés savantes. Le risque d’accumulation est donc apprécié sur des éléments cliniques précis mais pas spécifiques. Le recours aux antidotes des morphiniques est rare en pratique et son utilisation ne doit pas nous affranchir d’une surveillance postopératoire clinique et instrumentale (saturation en oxygène).
Le relâchement (curares) : Les curares agissent au niveau de la jonction neuromusculaire, plus particulièrement au niveau de sa partie musculaire post-synaptique, appelée ‘’plaque motrice’’. À ce niveau existent des récepteurs spécifiques à l’acétylcholine (ACH), neuromédiateur de la transmission neuro-musculaire. L’ACH est produite dans les terminaisons nerveuses des moto-neurones et stockée dans des vésicules. Ces dernières sont mobilisées par l’arrivée d’un influx nerveux (IN) et déversent leur contenu dans l’espace ou fente synaptique. L’ACH se fixe alors sur des récepteurs (nicotiniques). Un processus de dépolarisation de la fibre musculaire s’ensuit, provoquant la contraction musculaire. Enfin, la membrane reprend en quelques millisecondes sa polarité et est prête à une autre contraction. L’ACH est dégradée dans la fente synaptique par l’acétylcholinestérase. L’ACH agit aussi sur les récepteurs muscariniques situés au niveau des ganglions parasympathiques et sont responsables d’une bradycardie, bronchoconstriction et hypersécrétion.
Tous les curares agissent au niveau des récepteurs cholinergiques de la plaque motrice mais diffèrent par le mode d’action, leurs délais et durées d’action et par certains effets adverses.
Les curares dépolarisants (CD) : sont représentés par une seule molécule, la succinylcholine, qui n’est que l’assemblage de 2 ACH. La succinylcholine dépolarise naturellement la fibre musculaire et provoque des myoclonies, mais reste fixée sur son récepteur durant 7 à 8 minutes rendant la fibre musculaire non dépolarisable durant cette période. Ils sont dits ‘’curares dépolarisants ou non compétitifs’’, et agissent rapidement mais pour une courte durée. Leur seule indication est l’estomac plein. Ils sont dégradés par les pseudocholinesthérases au niveau plasmatique.
Les curares non dépolarisants (CND) : Deux familles appartiennent à ce groupe. La molécule se fixe sur le récepteur, par compétition avec l’ACH, rendant imperméable la membrane musculaire et ne permettent donc pas la dépolarisation de la celle-ci. Leurs délais et durées d’action sont variables selon la molécule utilisée. Ils sont dits ‘’curares non dépolarisants ou compétitifs’’. Cette compétition avec l’ACH explique l’efficacité de certains antidotes dans la levée de leur action. Il existe deux sous-groupes : les benzylisoquinolines et les aminostéroides. Leur métabolisme varie en plasmatique non enzymatique (réaction d’Hoffman) ou enzymatique pour certains benzylisoquinolines, ou enfin hépatico-rénal pour les aminostéroides.
Les anti-curares sont de deux types, la néostigmine et le sugammadex (9, 10, 11). Ces deux molécules ont des modes d’action totalement différents. La néostigmine est un anticholinestérasique qui bloque l’action physiologique de l’acétylcholinestérase, augmentant ainsi le taux d’ACH dans la fente synaptique, ce qui permet de déplacer par compétition le CND des sites d’action. Plusieurs inconvénients sont rattachés à cette vieille molécule. D’abord un délai d’action lent, mais aussi la nécessité d’une entame franche d’une décurarisation physiologique, enfin une action concomitante sur les récepteurs muscariniques, imposant l’association d’atropine et la limitation des doses. Elle agit avec tous les CND. Le sugammadex agit au niveau plasmatique par une action directe entre une molécule de sugammadex et une molécule de CND. Cette action est irréversible. La baisse du taux de CND dans le sang est alors rapide entrainant la libération des récepteurs par effets des masses. Les avantages sont nombreux, action rapide en moins de 5, voire 2 minutes ; une action possible quel que soit le niveau de curarisation, une adaptation des doses en fonction du monitorage de la curarisation et enfin l’absence d’action sur les récepteurs muscariniques. L’inconvénient principal est sa spécificité d’action limitée à seulement deux molécules de la famille des aminostéroides, le vécuronium et surtout le rocuronium; mais cependant les plus fréquemment utilisés. Le couple rocuronium-sugammadex a révolutionné la pratique de la curarisation en particulier en chirurgie abdomino-pelvienne, qui nécessite un relâchement parfait, tout au long de la chirurgie. La cœlioscopie en est l’exemple type ; puisqu’un relâchement adéquat induit par le vécuronium permet une bonne visibilité et un espace opératoire avec une pression intra-abdominale la moins élevée possible et avec moins d’effets adverses. Le retard de décurarisation qui s’ensuit est rapidement et facilement levé par le sugammadex, aidé par le monitorage NMT. D’un autre coté, une décurarisation rapide voire vitale, comme dans le cas d’une intubation endotrachéale impossible ou d’une réaction allergique aux aminostéroides, n’est possible qu’avec le sugammadex mais à une dose très élevée.
Conclusion :
L’anesthésie générale a connu une évolution extraordinaire ces 30 dernières années. Les facteurs ayant contribué à cela sont dominés par le développement des moyens de monitorage, l’apparition de certaines molécules innovantes et une meilleure connaissance du retentissement chirurgical sur le patient. L’anesthésie ‘’discipline’’ a transcendé sa mission initiale pour devenir la ‘’médecine périopératoire’’. En effet, à coté de sa mission d’endormissement du patient, le MAR s’est engagé pleinement dans la mission de prise en charge périoépratoire du patient, dans son volet réhabilitation, traitement des conséquences de la chirurgie et prévention des complications associées aux soins.
Référence :
– L. Cherfi, pratique de l’ALR en Algérie. ….
– E. Viel et col. Chronicisation de la douleur. Le cas des douleurs post-chirurgicales et post-traumatiques. SFAR 2013
– RPC curarisation en anesthésie. AFAR 2000, 19 fi : 34-37
– ASA physical status classification system, 2014
– Terriet and al. BjA 2000
– Thwaites and al. BjA 1997
– Taheri A. Anesth Analg 1999.
– Martini and al. Improved quality of surgical conditions compa- red with moderate block in retroperitoneal laraproscory. BjA 2014 9 – Staehr-Rye AK and al. Optimised surgical space during low pressure laparoscopy with deep neuromuscular blockade. Dan Med J. 2013
Van Wijk and al. DNB reduces intra-abdominal pressure re- quirements during laparoscopic surgery during laparoscopic cho- lecystectomy: a prospective observational study. Acta Anaesthesio- logica Scandinavica 2015.
Annexe 1 :
Classification de l’American Society of anesthesiologists (ASA), permet d’évaluer l’état de santé préopératoire, ainsi que la réserve fonctionnelle des patients avant une intervention chirurgicale
ASA 1 : Absence de maladie
ASA 2 : Présence d’une maladie sans atteinte systémique ni ré- percussion fonctionnelle
ASA 3 : Présence d’une maladie avec atteinte systémique ou ré- percussion fonctionnelle.
ASA 4 : Présence d’une maladie mettant en jeu le pronostic vital.
ASA 5 : Etat moribond avec décès prévisible dans les 24 heures, avec ou sans intervention chirurgicale.
ASA 6 : Patient en état de mort cérébrale, candidat potentiel au don d’organes
NB : ‘’U’’ dans le cas d’une intervention pratiquée en urgence, rajouter la lettre U (Exp. : ASA 3U)
Pr Nadia FELLAH1, Pr Dalila BENMOUSSA2 ,Anesthésie Réanimation, CHU Bab El Oued Alger. Anesthésie Réanimation, CPMC Alger.
Abstract :
For a long time, pain has been neglected. Scientific advances, particularly in its pathophysiology and pharmacology, have enabled us to better understand the mechanisms of pain and thus develop more appropriate therapeutic strategies.
The management of pain should not only concern pain specialists but should be oriented towards the education of all caregivers. The aim of this article is to provide the essential basis for the understanding and management of pain in everyday practice.
Résumé :
Pendant longtemps, la douleur a été négligée, aujourd’hui, les avancées scientifiques, notamment dans sa physiopathologie et sa pharmacologie, nous ont permis de mieux comprendre les mécanismes de la douleur et de développer ainsi des stratégies thérapeutiques plus adéquates.
La prise en charge de la douleur ne doit pas concerner seulement les spécialistes de la douleur mais doit s’orienter vers l’éducation de l’ensemble des soignants. L’objectif de cet article est d’apporter les bases indispensables à la compréhension et à la prise en charge de la douleur en pratique quotidienne.
Introduction :
La douleur est le premier motif de consultation en médecine générale. Selon les études internationales, la prévalence de la douleur chronique varie de 10.01% à 55,2% en population générale [1]. L’International Association for the Study of Pain (IASP), a défini la douleur comme une « Expérience sensorielle, émotionnelle, désagréable, associée à une lésion tissulaire réelle ou potentielle ou décrite dans des termes évoquant une telle lésion. »[2] Lorsqu’elle devient chronique, la douleur est responsable d’un lourd fardeau pour le patient, son entourage familial ainsi que pour la société. Elle a un impact important sur ses activités journalières, son état émotionnel, évoluant progressivement vers un isolement.
La douleur est un phénomène subjectif et individuel ; chacun d’entre nous a sa propre sensibilité face à la douleur. La douleur chronique est une expérience multidimensionnelle et pour mieux la comprendre, il est important de distinguer ses différentes composantes [3] :
La composante sensori-discriminative Elle représente les mécanismes neurophysiologiques permettant le décodage des caractéristiques de la douleur (localisation, intensité et qualité).
– La composante affectivo-émotionnelleElle correspond à la tonalité désagréable (pénible, insupportable), au niveau de détresse psychologique, d’anxiété, de dépression et de la tolérance à la douleur.
– La composante cognitive
Elle consiste en l’ensemble des processus mentaux susceptibles d’influencer la perception de la douleur et les réactions comportementales qu’elle détermine (processus d’attention et de diversion de l’attention ; interprétations et valeurs attribuées à la douleur ; anticipation, références à des expériences douloureuses antérieures ; décisions sur le comportement à adopter). Cette composante est influencée par des facteurs socio-culturels et l’histoire personnelle du patient.
– La composante comportementale
Elle inclue l’ensemble des manifestations verbales ou non verbales observables (plaintes, gémissements, position antalgique). Elle représente un bon moyen de communication, notamment chez les patients non communicants.
La douleur chronique est donc un phénomène complexe, subjectif, multidimensionnel, soumis à de nombreuses influences culturelles, sociales, économiques et religieuses. Cet aspect intrinsèque et subjectif, fait que nous ne sommes pas tous égaux devant la douleur.
Cependant, il est possible aujourd’hui, de distinguer les douleurs selon leurs mécanismes physiopathologiques et leurs caractéristiques, ce qui permettra de proposer une thérapeutique adaptée à chacun de nos patients. Il est clair que la prise en charge de la douleur doit donc être individuelle et personnalisée.
Par opposition à la douleur aiguë, qui est un symptôme attirant l’attention sur un problème d’intégrité physique, la douleur chronique perd son caractère protecteur ; en plus de son impact physique, elle affecte tous les aspects de la vie quotidienne comme le simple fait de marcher, se laver et s’habiller ; elle est aussi responsable de troubles du sommeil avec une somnolence diurne affectant ainsi l’activité sociale et professionnelle.
La persistance de la douleur a des répercussions émotionnelles aussi préjudiciables que les répercussions physiques. Aujourd’hui, la douleur chronique est considérée comme une réelle maladie évoluant pour son propre compte. Il s’agit d’un modèle bio-psycho-social dont la prise en charge doit être globale.
Évaluation de la douleur :
L’évaluation de la douleur est impérative, elle contribue au diagnostic, et oriente le choix du traitement antalgique. Elle permet également d’apprécier l’efficacité du traitement.
La douleur est souvent sous-évaluée par les soignants, et de leur côté, certains patients peuvent avoir des difficultés à exprimer l’intensité de la douleur qu’ils ressentent. C’est pour cela que sa recherche doit être systématique et son évaluation structurée, recherchant :
Topographie (par ex. territoire nerveux)
Caractéristiques (par ex. aiguë, chronique, composantes neuropathiques et nociceptive)
Intensité (EVA, EN, EVS)
Bilan psycho-social approfondi lors d’évolution prolongée (retentissement sur sommeil, humeur, travail, entourage et ressources propres du patient).
1- Topographie
Le siège et l’irradiation de la douleur doivent être précisés grâce à l’utilisation du schéma corporel sur lequel il faut indiquer où se trouve votre douleur habituelle (depuis les 8 derniers jours) en hachurant la zone et en mettant sur le schéma un « S » pour une douleur près de la surface douloureuse, un « P » pour une douleur plus profonde et un « I » à l’endroit où la douleur est la plus intense.
2- Type de douleur
La douleur peut être classée selon sa durée d’évolution et son mécanisme physiopathologique :
2-1 Classification selon sa durée d’évolution [4]
On distingue :
La douleur aiguë, dont la durée est inférieure à 1 mois ; c’est une douleur symptôme dont la lésion tissulaire est habituellement évidente ; elle est liée à une hyper activation du système nerveux. Elle a une fonction de protection et disparaît après un traitement curatif.
La douleur chronique, est une douleur qui persiste au delà de 3 à 6 mois ; il s’agit d’une douleur maladie qui persiste au-delà de la durée prévisible de guérison. Elle n’a pas de fonction de protection, bien au contraire, elle est responsable d’une dégradation de la santé et de la fonction de l’individu. Au stade de chronicité, le traitement de la douleur est réadaptatif avec comme corollaire une amélioration de la qualité de vie.
2-2 Classification selon le mécanisme physiopathologique[5], [6]
Différents types de douleurs sont observés selon le mécanisme physiopathologique :
La douleur nociceptive
Lorsqu’une partie du corps est endommagée, les nocicepteurs envoient des messages de douleur au cerveau le long des nerfs périphériques et de la moelle épinière.
La douleur est ressentie comme constante, localisée et souvent comme persistante ou pulsatile.
Elle peut être mécanique (maximum en fin de journée, calmée par le repos, ne réveillant pas la nuit, provoquée par la mobilisation) ou inflammatoire (nocturne, articulaire, raideur matinale, diminuant avec la mobilisation). En général, elle disparait à la cicatrisation ; l’examen neurologique (tests de la sensibilité) est normal. Sur le plan thérapeutique, la douleur nociceptive est bien soulagée par les analgésiques classiques.
La douleur neuropathique
Elle est causée par une lésion ou une affection du système somato-sensoriel, souvent séquellaire. Sa topographie évocatrice ; elle est située en territoire déficitaire. La douleur est spontanée ou provoquée ; les tests de la sensibilité sont anormaux (chaud/froid, toucher, piqûre).
Le diagnostic de douleur neuropathique est complexe, il se fait sur un faisceau d’arguments cliniques caractérisés par la présence de symptômes sensoriels positifs et négatifs[8].
Les anomalies sensorielles et la douleur sont souvent concomitantes[1,3]et sont susceptibles d’évoluer dans le temps.[9]
Symptômes sensoriels positifs
Symptômes sensoriels négatifs
L’intérêt de cette distinction entre douleur nociceptive et douleur neuropathique, est que sur le plan thérapeutique, cette dernière répond mal aux analgésiques classiques et nécessite des antalgiques spécifiques tels que les antidépresseurs et les antiépileptiques.
Intensité de la douleur
Elle doit être appréciée à l’aide d’échelles standardisées, qui permettent une appréciation globale de la douleur.
4-1- Echelles unidimensionnelles :
Elles permettent une appréciation quantitative de l’intensité de la douleur.
rectement au patient le niveau de sa douleur. Elle nécessite évidemment une coopération du patient et une bonne compréhension.
Classiquement, on distingue 3 échelles d’auto-évaluation : L’Échelle Visuelle Analogique (EVA), l’Échelle Numérique (EN) et l’Échelle Verbale Simple (EVS). Ces 3 échelles doivent être présentées au patient, qui choisira celle qui lui convient.
Échelle Visuelle Analogique (EVA) :
Elle est considérée comme étant la plus sensible, reproductible, fiable, doit être utilisée en priorité, lorsque c’est possible. Elle se présente comme une réglette graduée, avec 2 faces.
La face côté patient : il doit mobiliser le curseur le long d’une ligne droite représentant sa douleur, dont l’une des extrémités correspond à « Absence de douleur », et l’autre « Douleur maximale imaginable ».
La face côté praticien : il doit mettre la note en face du curseur.
Pas de douleur
Douleur faible
Douleur modérée
Douleur sévère
Douleur très sévère
Pire douleur possible
0
1
4
6
8
10
Échelle numérique (EN) :
Outil le plus simple et le plus couramment utilisé.
Il suffit de demander au patient de noter sa douleur de 0 à 10. En sachant que la note 0 représente la douleur maximale imaginable.
Échelle Verbale Simple (EVS) :
Il suffit de demander au patient d’entourer le chiffre qui représente le mieux sa douleur.
Douleur absente
Douleur faible
Douleur modérée
Douleur intense
Douleur extrêmement intense
4-2 Les échelles d’hétéro évaluation :
Elles sont réalisées par un observateur, si le patient est incapable d’évaluer lui même l’intensité de sa douleur (enfant, sujet très âgé, non communicant). Elle est basée sur le comportement verbal et non verbal, cependant, cette méthode sous évalue toujours la douleur du patient. Pour le sujet âgé et le patient non communiquant, on fera appel au questionnaire DOLOPLUS. Pour l’enfant, il existe des questionnaires en fonction de l’âge
ECHELLE DOLOPUSÉvaluation complémentaire de la douleur chez la personne âgée
Nom : prénom : Service : Observation comportementale
DATES
RETENTISSEMENT SOMATIQUE
1 * Plaintes Somatique
* pas de plainte……………………………………………………………………………………………………………………………………….. * plaintes uniquement à la sollicitation………………………………………………………………………………………………………… * plaintes spontanées occasionnelles……………………………………………………………………………………………………………. * plaintes spontanées continues…………………………………………………………………………………………………………………..
0 1 2 3
0 1 2 3
0 1 2 3
0 1 2 3
2 * Positions Antalgique au repos
* pas de position antalgique………………………………………………………………………………………………………………………. * le sujet évite certaines positions de façon occasionnelle………………………………………………………………………………. * position antalgique permanente et efficace……………………………………………………………………………………………….. * position antalgique permanente infficace…………………………………………………………………………………………………..
0 1 2 3
0 1 2 3
0 1 2 3
0 1 2 3
3 * Protection de zones douloureuses
* pas de protection…………………………………………………………………………………………………………………………………… * protection à la sollicitation n’empêchant pas la poursuite de l’examen ou des soins……………………………………….. * protection à la sollicitation empêchant tout examen ou soins………………………………………………………………………. * protection au repos, en l’absence de toute sollicitation………………………………………………………………………………..
0 1 2 3
0 1 2 3
0 1 2 3
0 1 2 3
4 * Mimique
* mimique habituelle……………………………………………………………………………………………………………………………….. * mimique semblant exprimer la douleur à la sollicitation…………………………………………………………………………….. mimique semblant exprimer la douleur en l’absence de toute sollicitation…………………………………………………….mimique inexpressive en permanence et de manière inhabituelle (atone, figée, regard vide)……………………………
0 1 2 3
0 1 2 3
0 1 2 3
0 1 2 3
5 * Sommeil
* sommeil habituel…………………………………………………………………………………………………………………………………… * difficultés d’endormissement………………………………………………………………………………………………………………….. * réveils fréquents (agitation motrice)…………………………………………………………………………………………………………. * insomnie avec retentissement sur les phases d’éveil…………………………………………………………………………………….
0 1 2 3
0 1 2 3
0 1 2 3
0 1 2 3
RETENTISSEMENT PSYCHOMOTEUR
6 * Toilette Et/ou habillage
* possibilités habituelles inchangées……………………………………………………………………………………………………………. * possibilités habituelles peu diminuées (précautionneux mais complet)………………………………………………………….. possibilités habituelles très diminuées, toilette et/ou habillage étant difficiles et partiels………………………………….toilette et/ou habillage impossible, le malade exprimant son opposition à toute tentative……………………………….
0 1 2 3
0 1 2 3
0 1 2 3
0 1 2 3
7 * Mouvements
* possibilités habituelles inchangées……………………………………………………………………………………………………………. possibilités habituelles actives limitées (le malade évite certains mouvements, diminue son périmètre de marche)possibilités habituelles actives limitées (même aidé, le malade diminue ses mouvements)………………………………… * mouvement impossible, toute mobilisation entrainant une opposition…………………………………………………………..
0 1 2 3
0 1 2 3
0 1 2 3
0 1 2 3
RETENTISSEMENT PSYCHOSOCIAL
8 * Communication
* inchangée…………………………………………………………………………………………………………………………………………….. * intensifiée (la personne attire l’attention de manière inhabituelle)………………………………………………………………… * diminuée (la personne s’isole)………………………………………………………………………………………………………………….. * absence ou refus de toute communication………………………………………………………………………………………………..
0 1 2 3
0 1 2 3
0 1 2 3
0 1 2 3
9 * Vie sociale
* participation habituelle aux différentesactivités (repas, animation, ateliers thérapeutiques…)………………………….. * participation aux différentes activités uniquement à la sollicitation…………………………………………………………….. * refus partiel de participation aux différentes activités…………………………………………………………………………………. * refus de toute vie sociale…………………………………………………………………………………………………………………………
0 1 2 3
0 1 2 3
0 1 2 3
0 1 2 3
10 * Troubles du comportement
* comportement habituel………………………………………………………………………………………………………………………….. * troubles du comportement à la sollicitation et itératif…………………………………………………………………………………. * troubles du comportement à la sollicitation et permanent…………………………………………………………………………… * troubles du comportement permanent (en dehors de toute sollicitation)………………………………………………………..
0 1 2 3
0 1 2 3
0 1 2 3
0 1 2 3
SCORE
4-3- Echelles pluridimensionnelles : Elles apprécient la douleur de façon quantitative et qualitative. Elles sont essentiellement utilisées dans le cadre de la douleur chronique, elles permettent également de mesurer l’efficacité thérapeutique sur chacune des dimensions de la douleur (sensorielle et émotionnelle). Il existe plusieurs questionnaires comme le questionnaire de Saint Antoine (QDSA), le Brief Pain inventory (BPI), etc.
Pour exemple, le BPI, en plus de l’intensité de la douleur, il évalue ses répercussions sur la qualité de vie du patient.
Concernant la douleur neuropathique, devant la difficulté du diagnostic, depuis quelques années, il existe des questionnaires spécifiques d’aide au diagnostic comme le DN4 Questionnaire DN4 : Permet d’estimer la probabilité d’une douleur neuropathique. Il s’agit de poser 4 questions avec 10 items, le médecin comptabilise les réponses, en comptant 1 point pour chaque « oui », et 0 point pour chaque « non ». Le test est considéré comme étant positif si le score ≥ 4/10. La sensibilité de ce questionnaire est estimée à 82,9 %, et sa spécificité à 89,9 %.
Traitement de la douleur :
À la fin de l’évaluation, le praticien doit être en mesure de distinguer le type de douleur, car celui-ci va dicter la conduite thérapeutique. En effet, la douleur nociceptive va nécessiter des antalgiques classiques, comme les AINS, le paracétamol et les opiacés. Alors que la douleur neuropathique va nécessiter des antalgiques spécifiques, tel que les antidépresseurs et les antiépileptiques.
La prescription des antalgiques doit également répondre à certains principes généraux :
Principes généraux du traitement de la douleur Traitement étiologique de la maladie Traitement antalgique selon les mécanismes et l’intensité de la douleur Voie d’administration simple: la voie orale doit être privilégiée Intervalles réguliers Traiter les accès douloureux par des interdoses Prévenir les effets secondairesTraiter l’anxiété et la dépression Association de moyens non médicamenteux Réévaluation régulière de la réponse au traitement
Le traitement de la douleur par excès de nociception :
Il s’appuie principalement sur les recommandations de l’OMS (Organisation Mondiale de la Santé). Celles-ci éditées en 1996 concernent initialement les douleurs d’origines cancéreuses et nociceptives. L’échelle de l’OMS à trois paliers, représente une méthode efficace et simple pour assurer une prise en charge médicamenteuse de la douleur. L’utilisation de co-antalgiques doit être envisagée à chacun des paliers. Elle met en parallèle l’intensité de la douleur et le niveau de l’antalgique requis. Cette règle a souvent été mise en défaut, mais elle reste très utile, en pratique quotidienne, pour guider les prescriptions d’antalgiques dans le cadre de la composante nociceptive de la douleur.
Le traitement de la douleur neuropathique :
Pour ce type de douleur, plus difficile à soulager, les objectifs thérapeutiques recherchent rarement la guérison, mais surtout une amélioration de la qualité de vie du patient, par une baisse de l’intensité de la douleur.
La prise en charge thérapeutique de la douleur neuropathique repose essentiellement sur certains anti-épileptiques et antidépresseurs.
Ces 2 classes thérapeutiques agissent de façon synergique du fait d’un mécanisme d’action différent.
Dans ce type de douleur, le choix thérapeutique se porte sur des médicaments ayant une AMM pour les douleurs neuropathiques et selon les recommandations internationales qui ont pour but lorsqu’il existe plusieurs options de hiérarchiser ces options thérapeutiques en fonction du niveau de preuve.
La plupart des recommandations internationales, préconisent en 1ère intention l’administration d’un antidépresseur tricyclique et/ou un anticonvulsivant.
Le recours à la morphine est préconisé en 2ème intention, car même si la morphine fait partie des molécules susceptibles de moduler de façon favorable la douleur neuropathique, sa gestion au long cours peut être problématique. Depuis 2011, nous disposons de recommandations pour le traitement de la douleur neuropathique périphérique dans le Maghreb francophone. [10]
Le traitement des douleurs mixtes :
Certaines douleurs présentent les 2 mécanismes (nociceptif et neuropathique), comme c’est le cas de la douleur cancéreuse, les radiculopathies lombaires, etc. … Ces douleurs nécessitent une approche thérapeutique visant les 2 mécanismes (antalgiques classiques et antalgiques spécifiques).
Traitement non pharmacologique :
Il peut être une alternative ou un complément efficace au traitement pharmacologique. Il s’agit de thérapeutiques physiques ou réadaptatives, et psychologiques.
Leur objectif est de limiter les conséquences néfastes de la douleur sur l’organisme en tant qu’agent de stress. Un tel état de relaxation peut être obtenu de plusieurs manières (massages, bains chauds, détente musculaire, méditation, etc.), Cependant, dans la prise en charge globale de la douleur, les thérapies cognitivo-comportementales, sont des techniques plus particulièrement utilisées comme la sophrologie et l’hypnose.
Conclusion :
La perception et la verbalisation de la douleur sont très variables d’un individu à un autre, sa prise en charge doit être personnalisée. L’évaluation de la douleur est un des actes médicaux les plus difficiles, il s’agit d’un phénomène subjectif avec plusieurs dimensions, plusieurs mécanismes et un profil évolutif.
Cette évaluation ne se résume pas simplement à l’intensité, mais également à de nombreuses autres variables tel que le mécanisme de la douleur, le niveau de détresse psychologique, d’incapacités … ; d’où la nécessité de la considérer comme une véritable maladie, en rechechant sa cause chaque fois que cela est possible, en recherchant son impact physique, psychologique, sans négliger sa répercussion familiale et socio professionnelle. Pour une prise en charge optimale, il ne faut pas oublier de procéder à une réévaluation régulière de la douleur ainsi que l’efficacité du traitement antalgique. Des objectifs thérapeutiques réalisables doivent être fixés dès la première consultation car le soulagement complet de la douleur chronique n’est pas toujours possible ; le but est donc l’amélioration de la qualité de vie avec une limitation du handicap, et ceci grâce l’utilisation combinée de thérapeutiques pharmacologiques diverses, associée à des thérapeutiques non pharmacologiques.
Du temps et de la disponibilité sont nécessaires à l’établissement de ce bilan global, indispensable pour une bonne prise en charge de la douleur chronique.
Revue bibliographique
(Verhaak et al, 1998; Elliot et al, 1999; Rustoen et al, 2004)
Definition de l’International Association for the Study of Pain (IASP) – 1979.
Fédération Nationale des Centres de Lutte contre le Cancer. SOR Evaluation de la douleur chez l’adulte et l’enfant atteints d’un cancer sept. 2 3 P.32
Cole. Hosp Physician. 2002;38:23-30.
Serrie A, Thurel C. In La douleur en pratique quotidienne. Dia- gnostic et traitements. Ed Arnette 2002. pp.34-35.
Le livre de l’externe – ECN/65. Bases neurophysiologiques et évaluation d’une douleur aiguë et d’une douleur chronique. www.s- editions.com.
Baron R. Clin j Pain. 2000;16:S12-S20..
[8](Eds) In: Classification of Chronic Pain: Descriptions of Chronic Pain Syndromes and Definitions of Pain Terms. 1994:209-212. [9]Gilron I et al. CMAj 2006;175:265-75.
[10]Griene B, et al. Pharmacological treatment of peripheral neuropathic pain: expert panel recommendations for the French-speaking maghrebian region. Douleur et Analgésie 2011