S. ALI HALASSA*, N. ZIDOUNI**. Maître de conférences A, Responsable du PNLAT, MSPRH Professeur chef de service, Président du comité d’experts de la tuberculose, MSPRH Service de Pneumo-Phtisiologie Matiben, CHU Béni-Messous
Résumé : L’organisation actuelle de la lutte antituberculeuse en Algérie est le résultat conjoint du développement progressif des services de santé, et des efforts accomplis dans l’application d’un programme national antituberculeux moderne et réaliste.
Depuis 1964, ce programme a évolué en plusieurs étapes, suivant le développement des services de santé et l’évolution des conceptions mondiales de la lutte contre la tuberculose concernant les régimes de chimiothérapie.
L’évolution des différents indicateurs épidémiologiques à révélé :
- Une diminution constante de l’incidence des cas de tuberculose pulmonaire contagieuse,
- Une diminution de la prévalence globale des cas de tuberculose,
- Une diminution de la prévalence de la multi-résistance,
- Une augmentation du nombre des cas de tuberculose extra-pulmonaire.
De nouveaux défis sont encore à relever notamment :
- Maintenir la qualité du diagnostic des cas de tuberculose pulmonaire par la supervision et l’évaluation des services cliniques et des laboratoires,
- Améliorer la qualité de la prise en charge des cas résistants aux antituberculeux dans le domaine de la compliance des cas traités,
- Améliorer la prise en charge des cas de tuberculose extra pulmonaire.
L’innovation thérapeutique marquée par l’utilisation de biothérapie au cours des maladies inflammatoires chroniques, qui pose un problème d’identification des cas de tuberculose pouvant être révélés par l’utilisation de telle thérapeutique
Abstract : The current organization of tuberculosis control in Algeria is a joint outcome of the progressive development of health services and the efforts made to implement a modern and realistic national tuberculosis program.
Since 1964, this program has evolved in several stages, depending on the development of health services and the evolution of global conceptions of the fight against tuberculosis concerning chemotherapy regimens.
The evolution of the various epidemiological indicators revealed:
- A steady decline in the incidence of contagious pulmonary tuberculosis,
- A decrease in the overall prevalence of tuberculosis cases,
- A decrease in the prevalence of multidrug resistance,
- An increase in the number of extra-pulmonary tuberculosis cases
Nowadays new challenges remain:
- Maintain the quality of the diagnosis of pulmonary tuberculosis cases by monitoring and assessing clinical services and laboratories,
- Improving the quality of management of TB resistant cases in the area of compliance of previously treated cases,
- Improve the management of cases of extra-pulmonary tuberculosis.
The therapeutic innovation by the use of biotherapy during the chronic inflammatory diseases, raise a new matter about the identification of tuberculosis cases revealed by the use of such therapeutics.
Introduction :
Dès l’indépendance du pays en 1962, le problème de la tuberculose s’est imposé comme l’une des priorités de santé publique. Sous l’effet conjugué de l’amélioration progressive des conditions de vie et de l’application des mesures techniques du programme national de lutte contre la tuberculose, l’incidence de la tuberculose a régulièrement diminué et l’Algérie est actuellement un pays d’incidence modérée. Cependant, l’application graduelle des mesures de lutte antituberculeuse qui ont permis de réduire la morbidité et la mortalité de cette maladie, ont mis en évidence l’émergence de nouvelles tendances évolutives dont le programme de lutte doit tenir compte.
En effet, une série de mutations démographiques, épidémiologiques et thérapeutiques, sont apparues, créant un impact sur les plans diagnostique et thérapeutique concernant notamment la tuberculose latente, et les formes extra-pulmonaires de la maladie. Cette situation inédite impose de relever de nouveaux défis afin de ne pas réduire ou menacer les actions primordiales du programme de lutte qui sont la détection et la guérison des cas de tuberculose contagieuse afin de diminuer les sources d’infection et de réduire la transmission du bacille tuberculeux dans la collectivité.
Les outils d’analyse de l’évolution du programme de lutte antituberculeuse (PNLAT)
- Sources de données L’analyse épidémiologique de cette évolution se réfère :
- Avant l’année 1982, aux résultats de deux ensembles de travaux :
- Les enquêtes tuberculiniques entreprises après l’indépendance et qui renseignent sur le risque d’infection tuberculeuse et sur les tendances évolutives de la tuberculose.
- Les enquêtes régionales de huit wilayates du centre du pays de 1975 à 1981 dans le cadre de l’application du programme national de lutte contre la tuberculose.
- Après l’année 1982, au recueil du système d’information installé à partir de cette année avec l’analyse de la prévalence et l’incidence de morbidité tuberculeuse couvrant tout le pays.
2. Données de population
Les taux de déclaration pour 100.000 habitants (/105 h) ont été calculés à partir des estimations de population de la Direction de la Population du Ministère de la Santé pour l’année correspondant à l’année de déclaration des cas.
Résultats
1. Avant l’indépendance
Avant de commencer de traiter l’évolution de la tuberculose après l’indépendance, il est primordial de rap- peler des résultats d’enquêtes réalisées avant 1962 et qui fournissent des renseignements intéressants sur la morbidité tuberculeuse de cette époque. Ainsi, A. LEVI
- VALENSI rapporte les résultats d’une étude de Pancrazi, réalisée entre 1954 et 1956, portant sur 230.000 assurés sociaux de caisses diverses. Le nombre de cas de tuberculose pulmonaire bacillifère a été le suivant :
- 235 cas pour 100.000 assurés en 1952
- 276 cas pour 100.000 assurés en 1953
- 280 cas pour 100.000 assurés en 1954
Les auteurs estiment que « ces chiffres sont probablement inférieurs à la réalité, car les ouvriers agricoles musulmans, qui de toute évidence fournissent le plus gros contingent de tuberculeux ne sont pas compris dans nos statistiques ».
Après 1954, il est permis d’avancer sans grand risque d’erreur que la dégradation des conditions de vie pendant la guerre de libération, les arrestations massives, les déplacements et les regroupements de population ont certainement introduit des facteurs d’aggravation de la situation de la tuberculose en Algérie et qu’à l’indépendance en 1962, le problème posé par cette affection était probablement plus grave qu’en 1954.
2. Après l’indépendance
- Les enquêtes tuberculiniques
- Risque Annuel d’Infection (RAI) : l’incidence annuelle de l’infection ou Risque Annuel de l’Infection dépend du nombre des sources d’infection présentes dans la collectivité, c’est-à-dire du nombre de cas de tuberculose pulmonaire à frottis positif (TPM+). Le RAI est exprimé en pourcentage. Le RAI peut être mesuré par les enquêtes tuberculiniques qui apprécient le taux de sujets présentant une réaction positive à l’intradermo-réaction à la tuberculine. Plusieurs enquêtes ont montré que les paramètres de contagiosité qui relient le RAI et la prévalence des sources d’infection, c’est -à-dire les cas de tuberculose pulmonaire à microscopie positive est de 10, ce qui signifie que 10 personnes en moyenne sont infectées (ou réinfectées) au cours d’une année par chaque cas non connu de tuberculose pulmonaire. On estime qu’un RAI de 1 % correspond à la présence de 50 à 55 cas de tuberculose pulmonaire contagieuse (TPM+) pour 100.000 habitants. Cependant, depuis la généralisation de la vaccination BCG, cette corrélation n’est plus aussi précise, et sujette à des erreurs d’interprétation. Pour étudier l’ampleur du problème de la tuberculose dans le pays et faute d’un système de déclaration de la tuberculose au lendemain de l’indépendance, de nombreuses enquêtes tuberculiniques ont été réalisées. Bien qu’elles ne soient pas comparables, chacune apporte des informations essentielles sur le risque annuel d’infection (RAI) et sur la tendance évolutive de la morbidité tuberculeuse pour la période étudiée.
- Ainsi SUTHERLAND et coll. estiment que le risque annuel d’infection tuberculeuse lors d’une enquête réalisée en 1969 à Sidi Bel Abbès et dans l’agglomération oranaise, a diminué de façon importante en comparaison aux résultats d’une enquête réalisée en 1951.
- Les enquêtes tuberculiniques de 1966-1969 : De nombreuses enquêtes ont été réalisées lors des années 1966 et 1969. Elles ont permis d’estimer le Risque Annuel d’Infection tuberculeuse moyen : il varie de 2,5% à 4% selon les régions.
- M. KOUIDRI, lors d’une enquête réalisée dans l’agglomération algéroise, a estimé le Risque Annuel d’Infection à 3,5% en 1966.Les enquêtes de Blida, réalisées à cinq ans d’intervalle en 1976 et 1981, ont donné les résultats suivants : le RAI est de 1,42% en 1976 et de 0,75% en 1981. La diminution annuelle du risque a été estimée à – 12,4% dans cette région au cours de la période de l’enquête.
- Enquêtes des huit wilayates du centre :Les enquêtes de Blida, réalisées à cinq ans d’intervalle en 1976 et 1981, ont donné les résultats suivants : le RAI est de 1,42% en 1976 et de 0,75% en 1981. La diminution annuelle du risque a été estimée à – 12,4% dans cette région au cours de la période de l’enquête.
Enquêtes des huit wilayates du centre :
Ces enquêtes régionales ont concerné 8 wilayates du centre du pays, sur la base des déclarations annuelles des cas de tuberculose faites régulièrement depuis 1975 jusqu’à l’année 1981 par les secteurs sanitaires. L’incidence de la tuberculose a diminué régulièrement passant de 78 cas de tuberculose pulmonaire bacillifère pour 100.000 habitants en 1975 à 60 cas en 1981 et de 150 à 120 cas pour la tuberculose toutes formes. Le meilleur fonctionnement des services de santé dans cette région a permis d‘améliorer le dépistage (Tableau 1).
| Année | Incidences pour 100.000 Habences | Incidences pour 100.000 Hab | Incidences pour 100.000 Hab |
| TPM+ | TEP | @ | |
| 1975 | 78 | 45 | 200 |
| 1976 | 74 | 40 | 180 |
| 1977 | 79 | 29 | 150 |
| 1978 | 75 | 18 | 120 |
| 1981 | 60 | 17 | 100 |
Surveillance de la morbidité tuberculeuse
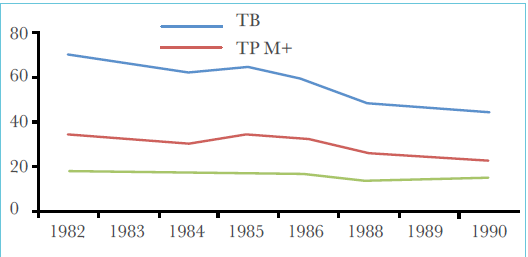
Depuis 1982, la déclaration des cas de tuberculose est centralisée au niveau national et l’analyse de l’évolution de la situation s’effectue de façon plus précise.
La figure 1 montre l’évolution de l’incidence enregistrée annuellement dans notre pays depuis l’année 82. L’incidence décroît régulièrement jusqu’en 1990 puis après avoir stagné durant la première moitié de cette décennie, elle augmente ensuite lentement durant la deuxième moitié (Figure 2).
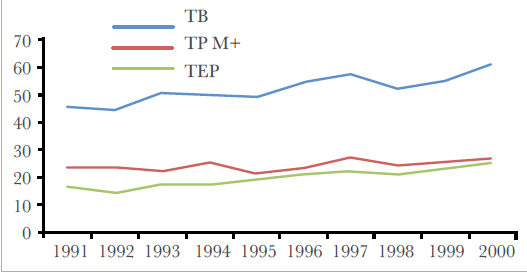
Durant le début des années 2000 et après un plan de relance du Programme National de lutte contre la tuberculose, on constate une augmentation plus marquée des incidences déclarées notamment celle de la tuberculose extra-pulmonaire. L’augmentation se poursuit sauf pour l’incidence de la tuberculose pulmonaire, qui régresse à partir de l’année 2005 et décline pour la première fois sous le seuil de 17 cas pour 100.000 habitants en 2015. Durant cette période, l’incidence annuelle des cas de tuberculose extrapulmonaire déclarés augmente, rejoignant en 2000 la courbe d’incidence des cas de tuberculose pulmonaire à microscopie positive puis la dépasse pour atteindre une incidence considérable de 38,4 cas pour 100.000 habitants en 2015. (Figure 3)
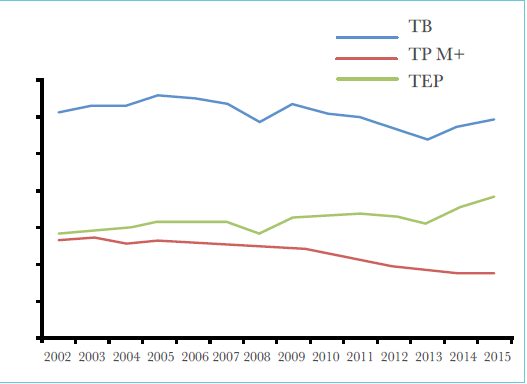
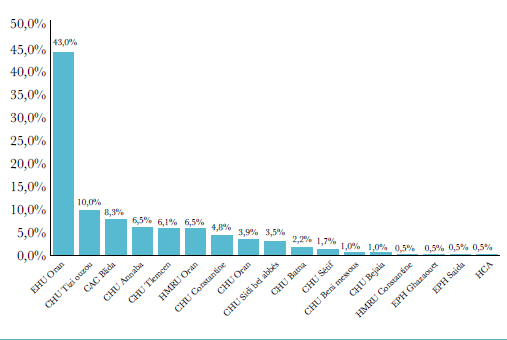
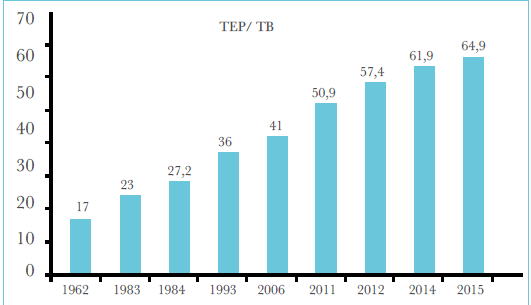
En conséquence, les tuberculoses extra-pulmonaires représentent un pourcentage croissant de toutes les formes de tuberculose atteignant près de 65 % d’entre elles alors qu’elles ne franchissaient pas les 17% en 1962, 23% en 1982 et 38% en 2002 (figure 4).
La résistance bactérienne
La surveillance régulière de la résistance de Mycobactérium tuberculosis aux antibiotiques a fait l’objet de plusieurs enquêtes nationales. Les enquêtes menées respectivement en 1988 et en 2002 sur la prévalence de la résistance bactérienne parmi les nouveaux cas de tuberculose ont permis de prouver la diminution de la résistance de 8.6 % en 1988 à 5,9 % en 2002 et une stabilisation de la multi résistance de 1,2% en 1988 à 1,4% en 2002 (tableau II).
| Cas testés (nombre) | Souches résistantes | ||||
| A un médica- ment au moins | Multi-résistantes | ||||
| Nombre | % | Nombre | % | ||
| 1988 | 243 | 21 | 8.6 | 3 | 1.2 |
| 2002 | 574 | 34 | 5.9 | 8 | 1.4 |
Au cours de cette année 2002, une première enquête nationale sur la séroprévalence du virus de l’immuno-déficience humaine (VIH) chez les tuberculeux, choisi comme groupe sentinelle pour la surveillance de l’épidémie de VIH, a révélé une séroprévalence de 0.18% de sujets porteurs du virus dans ce groupe de malades. Cette enquête est venue confirmer le faible impact en Algérie, du VIH, sur l’incidence de la tuberculose et sur les résultats du traitement.
Commentaires
Selon ces résultats présentés, il est permis de constater que le Programme National de Lutte contre la Tuberculose (PNLAT) en Algérie a évolué en plusieurs étapes, suivant le développement des conditions socio-économiques générales et des services de santé ainsi que l’évolution des conceptions mondiales de la lutte contre la tuberculose.
1. 1964-1990 : Les années phares du Programme National de Lutte Anti Tuberculeuse
Au cours de cette période, il y a eu une réduction considérable du Risque Annuel d’Infection, de la morbidité liée à cette maladie et la régression spectaculaire de la tuberculose de l’enfant,
- La gratuité des soins médicaux dans les services de santé publique, appliquée dès le 1er janvier 1974, a facilité l’accès aux soins à une large couche de la population,
- La vaccination BCG des nouveau-nés a été généralisée,
- La mise en place d’un réseau de laboratoires de microscopie, permettant de faire la preuve de la tuberculose dans près de 85% des nouveaux cas de tuberculose pulmonaire a permis d’atteindre les objectifs fixés par l’Organisation Mondiale de la Santé (OMS),
- La mise à la disposition, dans tous les services de santé publique et à tous les cas de tuberculose (pulmonaire et extra-pulmonaire), de la chimiothérapie de courte durée de six mois fondée sur l’association Rifampicine-Isoniazide à partir de 1980,
- La supervision étroite des services de santé et les séminaires annuels de supervision et d’évaluation ont permis d’assurer la formation continue des personnels et d’entretenir leur motivation.
2. 1990-1999 : Ralentissement des activités du Programme National
À partir de 1990, la courbe de l’évolution épidémiologique connait une stagnation puis une élévation de l’incidence des cas de tuberculose à cause de nombreux problèmes notamment :
- La dégradation des conditions socio-économiques générales
- L’instabilité politique et de l’insécurité qui règne dans certaines zones du pays, poussant les populations de ces zones à migrer vers les agglomérations urbaines rendent difficile l’adaptation des services de santé aux nouveaux besoins exprimés,
- Les supervisions et les séminaires d’évaluation sont interrompus, les équipes des secteurs sanitaires se retrouvent isolées, sans support technique ni contrôle des procédures de diagnostic avec comme conséquence le non-respect des critères de diagnostic recommandés,
- Cette situation a entraîné une augmentation des cas de tuberculose pulmonaire à microscopie négative dans certains secteurs, et surtout l’augmentation considérable des cas présumés de tuberculose extra-pulmonaire.
1. 2000 – 2015 : La relance du Programme National et les nouveaux défis ;
La relance des activités du programme antituberculeux a permis d’entreprendre des mesures telles :
- Les séminaires d’information qui ont eu lieu en 2001 et 2002,
- L’adoption et la généralisation des associations de médicaments antituberculeux en proportions fixes, pour le traitement de tous les malades tuberculeux (adultes et enfants) selon les recommandations de l’OMS.
- Le lancement de « l’approche pratique de la santé respiratoire », nouvelle stratégie proposée par l’OMS pour intensifier la détection des cas de tuberculose,
- L’installation des Laboratoires Régionaux de Référence et l’équipement du Laboratoire National de Référence en moyens de diagnostic rapide de la résistance,
- La mise en place de nouvelles structures : Services de contrôle de la tuberculose et des maladies respiratoires (SCTMR) qui remplacent désormais les anciens Dispensaires Anti Tuberculeux (DAT). Les SCTMR sont organisés pour être des services de première référence dans la prise en charge de la tuberculose et des maladies respiratoires du territoire couvert par ces services,
- La reprise des séminaires annuels de supervision et d’évaluation qui permettent chaque année d’établir des plans d’action en fonction des problèmes rencontrés dans chaque wilaya,
- La formation continue des personnels de la lutte contre la tuberculose (laborantins et les médecins des SCTMR, les coordinateurs du programme au niveau des wilayas).
2. Les nouveaux défis
Devant ces problèmes émergents qui ont surgi sur la scène de la lutte antituberculeuse, de nouveaux défis sont encore à relever notamment :
- Maintenir la qualité du diagnostic des cas de tuberculose pulmonaire par :
- La supervision et l’évaluation des activités du personnel médical notamment dans l’application des critères de diagnostic des cas de tuberculose pulmonaire à microscopie négative, et identifier le besoin de sessions de formation pour les médecins qui ne respectent pas ces critères,
- Le contrôle de qualité des laboratoires de microscopie pour contrôler les performances des microscopistes afin d’identifier leur besoin en formation et recyclage,
- L’élargissement du réseau de laboratoires de culture à toutes les wilayas pour augmenter le taux de tuberculose confirmé par la culture dont le taux ne dépasse pas 4% sur l’échelle nationale depuis quelques années.
- Améliorer la qualité de la prise en charge des cas résistants aux antituberculeux dans le domaine de la compliance des cas traités ; pour atteindre ce défi, l’application des directives du programme national concernant ce volet doit être renforcée.
- Le dépistage précoce des cas par la réalisation systématique de test de sensibilité chez tous les patients déjà traités avant d’instaurer le traitement de 2ème ligne,
- La réalisation chaque fois que possible des tests rapides de diagnostic de la résistance type Xpert MTB/RIF en particulier chez les nouveaux cas contaminés par un cas multi-résistant,
- La centralisation de la prise en charge des cas à bacilles multi-résistants (MDR) dans des centres de référence au niveau des services de pneumo-phtisiologie hospitalo-universitaires,
- L’approvisionnement régulier des centres de référence en médicaments de 3ème ligne de qualité contrôlée,
- L’enregistrement des cas chroniques et des cas de tuberculose à bacilles multi-résistants sur un registre spécial au niveau de chaque service de pneumo-phtisiologie hospitalo-universitaires et une fiche de déclaration individualisée.
- Améliorer la prise en charge des cas de tuberculose extra pulmonaire (TEP) :
- Améliorer la qualité de diagnostic des cas de tuberculose extra pulmonaire par la multiplication des laboratoires d’anatomo-pathologie et de culture du bacille tuberculeux (BK) pour diminuer la proportion des cas présumés de TEP,
- Organiser des séminaires d’information et sensibilisation sur le problème émergent des TEP, destinés aux praticiens spécialistes d’organe qui prennent en charge les cas de TEP,
- Editer un consensus validé par les spécialistes d’organes sur la prise en charge thérapeutique de chaque localisation : schémas thérapeutiques, modalités de surveillance, critères de guérison,
- Actualiser le système d’information de la tuberculose pour une surveillance rigoureuse du devenir des cas de TEP mis sous traitement antituberculeux,
- Installer des équipes de recherche opérationnelle pluridisciplinaires pour fournir des solutions rationnelles concernant les situations conflictuelles notamment la durée de traitement de certaines localisations.
- Tuberculose au cours de l’utilisation des traitements de biothérapie
L’utilisation de biothérapies en particulier les anti-TNF alpha sont indiqués dans les maladies inflammatoires chroniques, graves et invalidantes comme la polyarthrite rhumatoïde, la spondylarthrite ankylosante, le psoriasis sévère et sa forme rhumatismale, l’arthrite juvénile idiopathique ainsi que certaines maladies inflammatoires chroniques de l’intestin (MICI) comme la rectocolite hémorragique et la maladie de Crohn. Ces médicaments diminuent le TNFα sérique, ce qui va permettre de contrôler l’inflammation et donc l’évolution de la maladie mais exposent les patients à d’importants risques d’infection, en particulier à la tuberculose maladie. Ainsi, compte tenu des risques graves encourus par les patients, il est impératif, avant d’initier un traitement par les anti-TNFα, de procéder de façon systématique à la recherche d’une tuberculose maladie ou d’une infection tuberculeuse, pour éviter le risque d’une réactivation tuberculeuse.
Conclusion :
En Algérie depuis le plan de relance de 2000, la morbidité tuberculeuse commence à s’améliorer après la recrudescence qu’elle a connu dans les années 90. Elle décroît pour les cas de tuberculose pulmonaire alors qu’elle augmente très fortement pour les cas de tuberculose extra-pulmonaires, plus souvent présumés que prouvés. La lutte antituberculeuse doit prendre en compte ce phénomène et mettre en œuvre des dispositifs pour améliorer le diagnostic et le traitement de ces cas.
L’identification des cas de tuberculose chez les sujets soumis à des biothérapies doit être maitrisée et faire l’objet d’une adoption consensuelle chez les prescripteurs de telles thérapeutiques.
References bibliographiques :
- Les résultats du traitement de la tuberculose pulmonaire en Algé- rie (Participation à l’enquête coopérative de l’Union Internationale contre la tuberculose. P. CHAULET et coll. Bull. Un. Int. Tuberc. 1979, 54, 44-48
- L’enquête Nationale 1997-78. Les caractéristiques de l’échantillon national de malades. R.AMRANE., N. ZIDOUNI Revue Algérienne de Médecine 1980, 3 : 37-39
- Les résultats pratiques du traitement de la tuberculose pulmonaire en 1979-1980. N. ZIDOUNI, P. CHAULET. Revue Algérienne de Médecine 1981, 4 : 93-97
- Résultats comparés des activités de dépistage et de traitement de la tuberculose en Algérie avant et après généralisation de la chimiothé- rapie de courte durée. N. ZIDOUNI, L. OUAR, N. CHAKER, N. KEITA., P. CHAULET. Maghreb Medical, 1982, 49: 37 – 45.
- La déclaration des cas de tuberculose en Algérie. Expérience d’un an de centralisation des données. N. ZIDOUNI, N. BELKAALOUL, L. OUAR, N. CHAKER, R. AMRANE, P. CHAULET. Revue Algé- rienne de Pneumo-Phtisiologie, 1983, 1,2 : 34 – 38.
- Evolution de la Tuberculose en Algérie de 1962 à 1983, Bulletin épi- démiologique. No. 9, 1985 ; Division d’épidémiologie, R. AMRANE 7.Relevé Epidémiologique Hebdomadaire No. 36 – 5 septembre 1986, Organisation Mondiale de la Santé
- L’utilisation d’un réseau hiérarchisé de soins primaires dans la prise en charge des malades tuberculeux au cours d’une enquête commu- nautaire réalisée dans 45 secteurs (districts) sanitaires d’Algérie. N. ZIDOUNI. Bull. Un. Int. Tub. Mal. Resp. 1989, 64: 37 – 39.
- Methodological problems in evaluating tuberculosis treatment pro- grammes. N. ZIDOUNI, P. CHAULET. Tuberculosis Surveillance Research Unit of the IUATLD. Progress Report 1989, 1 : 66-78.
- Comment traiter les tuberculeux ? P. CHAULET, N. ZIDOUNI Journal Algérien de Médecine 1992, 2 : 248-254.
- Treatment of failure and relapse cases of pulmonary tuberculosis within a National programme based on short – course chemotherapy. (Preliminary Report). L. MAZOUNI, N. ZIDOUNI, F. BOULAH- BAL., P. CHAULET. Tuberculosis Surveillance Research Unit of the IUATLD. Progress Report 1992, 1: 36-42.
- Evaluation of applied strategies of tuberculosis control in Deve- loping World.
- CHAULET P., ZIDOUNI N. Tuberculosis . A comprehensive Inter- national Approach. edited by LEE B. REICHMAN, and EARL. S. HERSHFIELD., MARCEL DEKKER, Inc. 1993, New York, 27 . pp 601 – 627.
- L’introduction des programmes de santé respiratoire dans le cur- sus des études de spécialité en Pneumo-Phtisiologie. ZIDOUNI N., NAFTI S. Revue des Maladies Respiratoires, 10, 2: 1993 , R.123. Journal Algérien de Médecine 1994, 4 : 62-74.
- Le traitement curatif de la tuberculose dans le monde. P. CHAU- LET, N. ZIDOUNI. Rev. Pneumol. Clin. 1994, 50, 247-255.
- Le dilemme des cas de tuberculose pulmonaire à microscopie né- gative. Comment les différencier ? N. ZIDOUNI. Tubercle and Lung Diseases, 1996, 77, 2.
- Les cas de tuberculose pulmonaire à microscopie négative obser- vés dans un secteur d’Alger. N. ZIDOUNI, L. BAOUGH, P. CHAU- LET. Tubercle and Lung Diseases, 1996, 77,69.
- Evaluation des activités de lutte antituberculeuse d’un dispensaire spécialisé en milieu urbain à Alger. N. ZIDOUNI, L. BAOUGH, P. CHAULET. Tubercle and Lung Diseases, 1996, 77,71.
- N. CHAULET P. , ZIDOUNI. Failures in Tuberculosis Chemo- therapy. MYCOBACTERIA. Vol II ch12, edited by P.R.J. GAN- GADARHAM, P.A. JENKINS. CHAPMAN and HALL , Medical Microbiology series, NewYork,1997, pp 313-334.
- Le traitement des cas de tuberculose à bacilles multi-résistants. L. MAZOUNI, N. ZIDOUNI et coll. Tubercle and Lung Diseases, 1997, 5, S49.
- Le traitement des cas d’échec-rechute de tuberculose pulmonaire. D. MEKIDECHE, N. ZIDOUNI, D. YALA. Tubercle and Lung Diseases, 1997, 5, S78.
- Epidémiologie de la tuberculose en Algérie. N. ZIDOUNI. Ca- hiers de la Santé, 1998, 2, 46-50.
- Les cas de tuberculose pulmonaire multi-résistants observés dans un service de pneumophtisiologie d’Alger par L BAOUGH, Y KES- SAS, A BESSAH, S KHEDER, Y HAMOU, S ALI HALASSA, N ZIDOUNI, O YALA, F BOULAHBAL. The Int. Journ. Tub. Lung. Dis. 2001,5(11), S16
- L’infection à VIH/SIDA en Algérie: le point sur la surveillance par R BELKAID et N ZIDOUNI. ALGERIE SANTE, 2001, 07:28-30.
- La contribution des écoles de médecine à la prise en charge de la tuberculose et des maladies respiratoires par N. ZIDOUNI. The Int. Journ. Tub. Lung. Dis. 2006, (10),11 S14
- The responsibilities of medical and nursing schools in tubercu- losis care and control in countries with medium and high tuberculo- sis incidence par P. CHAULET, N.ZIDOUNI. TUBERCULOSIS. A Comprehensive International Approach. Third edition. edited by M.C RAVIGLIONE, L.B. REICHMAN, E.S. HERSHFIELD, Mar- cel Dekker Inc. New York, 2006, 219, 44: 1083- 1095
- L’apport de l’APSR dans l’identification des problèmes priori- taires de santé respiratoire par N. Zidouni. The Int. Journ. Tub. Lung. Dis. 2008, (12),11 S 89
- L’approche pratique de la santé respiratoire en Algérie par N. ZI- DOUNI, L. BAOUGH, Y. LAID, P. CHAULET. The Int. J. Tuberc. Lung Dis., 2009, 13(8): 1029-1037
- Manuel de la lute antituberculeuse à l’usage des personnels médi- caux. Programme national de lutte contre la tuberculose, MSPRH 2011, 256p
- ALI HALASSA. Impact de l’approche pratique de la santé respi- ratoire sur le fonctionnement des services de contrôle de la tubercu- lose et les maladies respiratoires. Thèse DESM, Alger, 2014