L’hyperplasie congénitale des surrénales (HCS) regroupe des maladies autosomiques récessives dues à une carence d’une ou plusieurs enzymes nécessaires à la synthèse par la glande surrénale du cortisol, de l’aldostérone, et des stéroïdes sexuels.
F. Bouferoua, K.N. Benhalla, Service de Pédiatrie A, CHU Issaad Hassani, Béni Messous, Alger.
Date de soumission : 26 février 2021
Résumé : L’hyperplasie congénitale des surrénales (HCS) regroupe des maladies autosomiques récessives dues à une carence d’une ou plusieurs enzymes nécessaires à la synthèse par la glande surrénale du cortisol, de l’aldostérone, et des stéroïdes sexuels. Dans la plupart des cas, cette déficience enzymatique conduit à une accumulation des précurseurs en amont et à l’épuisement des produits en aval. Elle est caractérisée par une insuffisance surrénalienne et divers degrés d’hyper-androgénie (ou hypo-androgénie), selon le type et la sévérité de la maladie. Les filles présentent à la naissance des organes génitaux ambigus. Les formes d’HCS avec perte de sel conduisent à des symptômes de déshydratation au cours des premières semaines de vie, pouvant être fatals. L’HCS non classique n’est généralement pas diagnostiquée avant l’adolescence. D’autres formes rares peuvent se présenter avec une hypertension artérielle, des malformations crâniofaciales et une ambiguïté sexuelle chez les deux sexes. Le diagnostic des déficits enzymatiques responsables de l’hyperplasie congénitale des surrénales (HCS) repose sur le dosage précis des différents stéroïdes. Un traitement hormonal de substitution à vie est nécessaire pour traiter l’insuffisance surrénalienne et diminuer les taux élevés d’androgènes.
Mots clés : HCS, déficit en 21 OH, syndrome de perte de sel, insuffisance surrénale aigüe, 17 OHP, hydrocortisone, fludrocortisone, dépistage néonatal.
Abstract: Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive diseases caused by a deficiency of one or more enzymes necessary for the adrenal gland to synthesize cortisol, aldosterone, and sex steroids. In most cases, this enzyme deficiency leads to an accumulation of upstream precursors and depletion of downstream products, and is characterized by adrenal insufficiency and varying degrees of hyperandrogenism (or hypoandrogenism), depending on the type and severity of the disease. Girls have ambiguous genitalia at birth. The salt loss forms of CAH lead to symptoms of dehydration in the first weeks of life, which can be fatal. Unconventional CAH is usually not diagnosed until adolescence. Other rare forms can present with high blood pressure, craniofacial malformations and sexual ambiguity in both sexes. The diagnosis of enzyme deficiencies responsible for CAH is based on the precise dosage of different steroids. Lifelong hormone replacement therapy is needed to treat adrenal insufficiency and lower high androgen levels.
Key words: CAH, 21 OH deficiency, salt loss syndrome, adrenal insuffiency acute, 17OHP, hydrocortisone, fludrocortisones, new-born screening
Introduction
L’hyperplasie congénitale des surrénales est une maladie génétique à transmission autosomique récessive, qui est due à une anomalie dans la voie de biosynthèse du cortisol, soit un déficit d’une des enzymes de la stéroïdogénèse, ayant pour conséquence un excès d’androgènes.
Physiologie – Physiopathologie
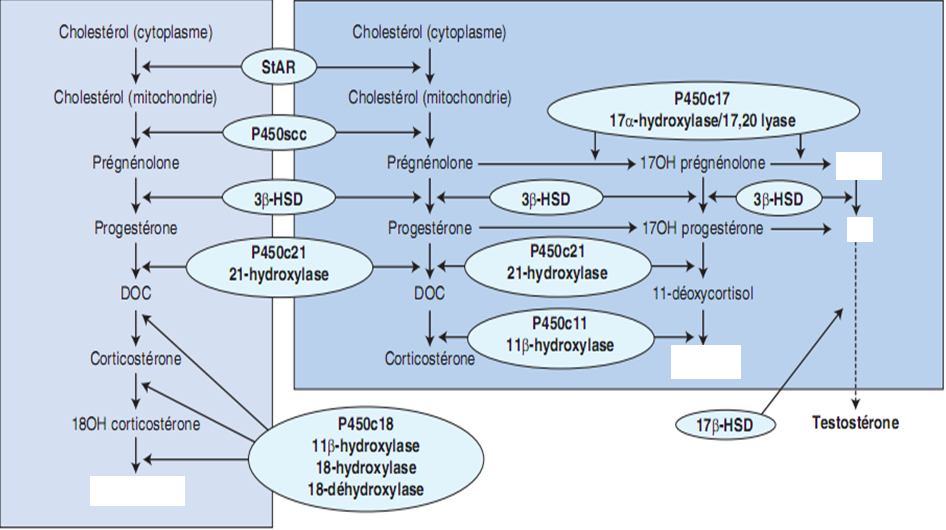
Les stéroïdes surrénaliens sont synthétisés à partir du cholestérol et 80% environ de celui-ci proviennent des lipoprotéines circulantes. Cette synthèse se fait par la succession de réactions enzymatiques (figure 1).
Dans la zone fasciculée sont synthétisés les glucocorticoïdes dont le métabolite actif est le cortisol. L’aldostérone est synthétisée dans la zone glomérulée et les androgènes surrénaliens sont synthétisés dans la zone réticulée (déhydroépiandrostènedione ou DHEA, sulfate de DHEA, ∆ 4-androstènedione).
Le cortisol a un effet sur le métabolisme glucidique, lipidique et protidique, un effet sur le métabolisme phosphocalcique, et une action anti inflammatoire. Sa synthèse se fait sous l’action de la corticotropin releasing hormone (CRH) et l’adrenocorticotophic hormone (ACTH) (hormones hypothalamo-hypophysaires).
La carence en cortisol est à l’origine de la levée du rétrocontrôle négatif hypothalamo-hypophysaire, augmentant la sécrétion de CRH et d’ACTH. Cette élévation est responsable de l’hyperplasie du cortex surrénalien et de l’augmentation de la sécrétion des précurseurs du cortisol en amont du bloc enzymatique.
L’aldostérone favorise la réabsorption du Na+ et l’élimination du K+ H+. Sa synthèse est régulée par le système rénine-angiotensine. Un déficit en aldostérone est donc responsable d’une perte d’eau et de sel dans les urines et donc d’une déshydratation avec hyponatrémie et hyperkaliémie avec un risque de choc hypovolémique.
Dans le bloc en 21-OH et 11-ß-OH, il y a un excès de sécrétion de la synthèse d’androgènes par une déviation du métabolisme des substrats d’amonts. Cet excès est responsable d’une virilisation des fœtus de sexe féminin (46 XX) alors que les fœtus de sexe masculin naissent sans anomalie des organes génitaux externes.
Les blocs enzymatiques à révélation précoce entrainent, quelle que soit l’enzyme déficiente, un défaut de synthèse du cortisol, qui pourrait être à l’origine d’hypoglycémies sévères surtout chez le nouveau-né.
Figure 1 : Synthèse de stéroïdes surrénaliens
Clinique
Déficit en 21-hydroxylase
Son incidence est de 1/15.000 naissances en France. Elle varie selon les régions géographiques et l’appartenance ethnique.
Le gène de la 21-hydroxylase se situe sur le bras court du chromosome 6 (ch6p21.3) dans la région de classe 3 du système majeur d’histocompatibilité HLA. Dans 75% des cas, le déficit est dû à une mutation du gène CYP 21 B. Les patients porteurs de mutations entrainant une activité résiduelle nulle de l’enzyme ont une forme classique avec perte de sel, et ceux porteurs d’une mutation entrainant un déficit incomplet avec une activité résiduelle de 2%, ont une forme sans perte de sel.
L’enzyme 21-hydroxylase permet la transformation de la 17-hydroxyprogestérone (17 OHP) en 11-déoxycortisol sur la voie de synthèse du cortisol et de la progestérone en désoxycorticostérone (DOC) sur la voie de synthèse de l’aldostérone.
Le syndrome de perte de sel est la conséquence d’un déficit complet de l’enzyme (activité résiduelle nulle), la surrénale ne peut pas synthétiser le cortisol et l’aldostérone.
On distingue la forme classique avec ou sans perte de sel et la forme non classique à révélation tardive.
Dans la forme classique, nous avons :
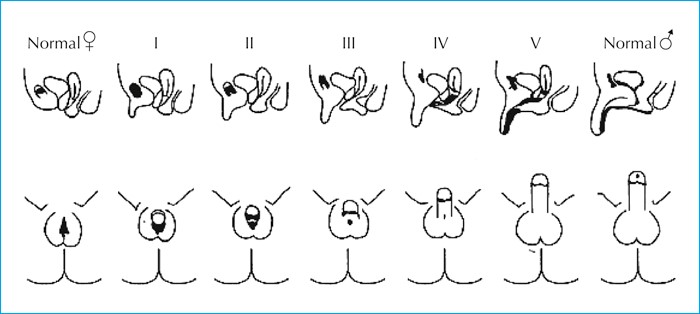
La forme virilisante pure qui se révèle à la naissance par une virilisation des organes génitaux externes de la petite fille, qui est côtée selon le stade de Prader de 1 à 5 (figure 2), allant de l’hypertrophie clitoridienne jusqu’à l’aspect masculin des organes génitaux externes mais sans palpation des gonades avec des organes génitaux internes (utérus et ovaires) normaux.
Figure 2 : classification de Prader.
En absence de dépistage, le diagnostic chez le garçon est difficile car les organes génitaux sont normaux, il est souvent posé vers l’âge de 2 à 4 ans devant l’apparition de signes cliniques de pseudo puberté, à savoir une augmentation de la taille de la verge sans augmentation du volume des testicules ainsi qu’une accélération de la vitesse de croissance et de la maturation osseuse.
La forme avec perte de sel qui se manifeste entre le 8ème et le 15ème jour de vie par l’apparition de troubles digestifs, une mauvaise prise pondérale, des vomissements puis déshydratation qui peut être sévère pouvant se compliquer d’un état de choc. Le tableau clinique peut se compliquer d’hypoglycémie en rapport avec le déficit en glucocorticoïdes.
Le diagnostic de l’hyperplasie congénitale des surrénales est facilement posé chez le nouveau-né de sexe féminin devant la présence d’une ambiguïté sexuelle, mais reste difficile lorsqu’il s’agit d’un garçon ou une fille au stade 5 de Prader ; il doit être évoqué devant les épisodes de déshydratation répétés, les signes cliniques déjà suscités, ou perturbation de l’ionogramme afin de pouvoir réaliser une prise en charge précoce et améliorer le pronostic vital.
La forme non classique est de révélation tardive, elle peut se manifester soit à la période pubertaire dans un tableau d’hyper-androgénie avec hirsutisme, acné, trouble du cycle ou stérilité, soit plus précocement dans un tableau de pseudo puberté précoce.
Examens complémentaires
En présence de syndrome de perte de sel, l’ionogramme objective une hyponatrémie avec natriurèse conservée et une hyperkaliémie à kaliurèse basse.
Les autres androgènes, ∆4-androsténédione (∆4) et sulfate de déhydroépiandrosténédione (SDHEA) sont élevés, la testostérone est élevée, elle trouve tout son intérêt, en particulier chez la fille.
Le dosage du cortisol chez le nouveau-né n’a pas d’intérêt car il est physiologiquement bas. Dans le syndrome de perte de sel, le taux d’aldostérone est très bas avec une réninémie élevée. L’échographie pelvienne montre la présence de structures féminines, un utérus et des ovaires
La génitographie n’est pas utile pour le diagnostic, mais surtout pour la prise en charge chirurgicale, elle permet d’opacifier la cavité müllerienne postérieure lorsqu’elle existe, et montrer le niveau de son implantation sur la face postérieure de l’urètre, ce qui permet de préciser la technique chirurgicale.
Déficit en 11-β-hydroxylase (11-βOH)
Il réalise 5 à 8% des hyperplasies congénitales des surrénales, son incidence est de 1/200.000 dans la population générale. Le déficit de cette enzyme est responsable d’un défaut de synthèse du cortisol et d’aldostérone. La mutation la plus fréquente est le R448H se situant sur l’exon 8, et la transmission est autosomique récessive.
Clinique
Le tableau clinique à la période néonatale est identique à celui de la 21-OH, avec en plus, le développement d’une hypertension artérielle dans 2/3 des cas les premières années de vie, cette dernière est due à l’accumulation du DOC. Le syndrome de perte de sel est beaucoup plus rare.
Biologie
Le diagnostic est fondé sur l’élévation du composé S et de la DOC, qui malheureusement ne sont pas de pratique courante par manque de disponibilité dans notre pays, le plus souvent on se base sur le dosage de la 17 OHP qui est modérément élevée, avec élévation de la testostérone et de la ∆4, et contrairement au déficit en 21-OH, la réninémie est basse.
Bloc en 3-β-hydroxystéroïde déshydrogénase
Le déficit en 3-β-hydroxystéroïde déshydrogénase (3-βHSD) est responsable d’environ 1 à 10% des hyperplasies congénitales des surrénales. Son déficit est responsable d’un déficit en cortisol, aldostérone et androgènes surrénaliens. Le gène est situé sur le chromosome 1 en p13.1, et une trentaine de mutations ont été décrites.
Clinique
Les anomalies des organes génitaux externes du fœtus de sexe masculin, sont de degré variable, elles sont dues à un défaut de synthèse des androgènes, il peut s’agir d’un hypospadias périnéo-scrotal ou périnéal, un micropénis, gonades ectopiques ou scrotum bifide, avec dans tous les cas des organes génitaux internes normaux. Le syndrome de perte de sel se voit dans la moitié des cas.
Les filles ne présentent pas d’anomalies des organes génitaux, et le diagnostic ne peut être porté les premiers mois de la vie en l’absence de perte de sel ; mais de discrets signes de virilisation peuvent se voir et s’accentuer dans l’enfance et l’adolescence pouvant être à l’origine d’une prémature pubarche, acné, accélération de la vitesse de croissance et de la maturation osseuse.
Biologie
17 OHP/17OH prégnénolone et 17 OHP/cortisol élevés. Dans les formes avec perte de sel, la réninémie est élevée.
Autres déficits
Les autres déficits sont beaucoup plus rares, nous ne ferons que les citer : bloc en 17-α-hydroxylase, 450 oxydoréductase et le Star.
Assignation du sexe
Elle doit être la plus rapide possible, mais après consensus pluridisciplinaire qui doit tenir compte de l’âge de diagnostic, des possibilités chirurgicales en fonction de la sévérité de l’androgénéisation (Prader 5), et le souhait des parents qui est souvent influencé par leur vécu socioculturel ; le gros problème se pose pour les enfants nés avec un morphotype masculin, un bourgeon génital complètement masculin, la palpation des gonades n’étant pas faite à la naissance et l’enfant est déclaré d’emblée garçon, et ce n’est que quelques mois ou années plus tard que les parents consultent pour absence de gonades, le plus souvent devant cette situation, même si le caryotype réalisé déclare le sexe féminin, les parents gardent l’enfant dans le sexe déclaré à la naissance, c’est-à-dire garçon, d’où la nécessité d’un examen systématique des organes génitaux externes à la naissance, expliquer la pathologie aux parents ainsi que les possibilités thérapeutiques et le devenir à long terme en cas de choix erroné du sexe.
Traitement
Déficit en 21-OH et 11-β-OH
Traitement médical : Le traitement doit être entrepris immédiatement après avoir posé le diagnostic.
Si le tableau clinique est celui d’un syndrome de perte de sel, l’urgence est à la réhydratation intraveineuse, l’apport de sel et l’apport intraveineux en glucocorticoïdes, et ce, selon un protocole bien codifié ; le relai per os est entrepris dès que l’état clinique s’améliore ainsi que la normalisation de l’ionogramme.
En l’absence de signes aigus d’insuffisance surrénale, le traitement est entrepris par voie orale à base d’hydrocortisone à une dose d’attaque de 50 mg/m2/j chez le nouveau-né pendant 10 à 15 jours puis on passe à une dose d’entretien de 20 à 25 mg/m2/j chez le nouveau-né et de 10 à 15 mg/m2/j pour l’enfant plus grand (la dose d’attaque chez le nourrisson est de 20-25 mg/m2/j).
Le traitement minéralocorticoïde à base de Fludrocortisone est associé à la dose de 50 à 100 µg/j, lorsque ce dernier est disponible ; le plus souvent dans notre pays, on se contente de la supplémentation en sel à raison de 1 à 2 g/j, et qui doit être maintenue les deux premières années ; au-delà, la supplémentation en sel se fera dans l’alimentation.
Le médecin informera les parents des différentes situations de stress pouvant engendrer une décompensation et la conduite à tenir devant chaque situation, le patient sera muni d’une carte où sera mentionné le diagnostic d’insuffisant surrénalien, le nom du médecin à contacter et la conduite à réaliser en urgence.
Traitement chirurgical : Le but du traitement chirurgical est d’obtenir un aspect féminin des organes génitaux externes, avec des voies urinaires normales sans obstruction ni infections à répétition et une vie sexuelle normale.
La chirurgie doit se faire dans un centre spécialisé ayant l’expérience de la pathologie, avec une équipe chirurgicale compétente.
L’acte chirurgical consiste en une vaginoplastie associée à une clitoridoplastie ; le plus grand défi est la préservation du pédicule vasculo-nerveux du gland, permettant ainsi une vie sexuelle normale.
Il faut savoir que c’est une chirurgie lourde qui n’est pas dénuée de complications à type d’atrophie du clitoris, sténose vaginale, troubles mictionnels avec incontinence urinaire, infection, fistules urétrovaginales ayant pour conséquence une anomalie du flux menstruel et un retentissement sur la vie sexuelle. L’indication chirurgicale ne se pose pas pour le Prader 1 et 2.
L’âge auquel doit être faite la chirurgie, fait actuellement l’objet de controverses, les équipes qui préconisent un acte chirurgical dans l’enfance, le conseillent vers l’âge de 6 mois, ayant pour arguments que la peau phallique est un bon matériel de reconstruction vaginale en période néonatale, du fait que les tissus sont imprégnés en œstrogènes placentaires ; et sur le plan psychosocial, ça permet de construire précocement une identité de l’enfant et son insertion psychosociale en fonction du sexe choisi. Les équipes qui préconisent un acte chirurgical tardif, soit à la période pubertaire, ont pour argument de diminuer le risque de sténose vaginale et le recours aux dilatations.
Déficit en 3-βHSD
L’hydrocortisone (HC) est associée à la fludrocortisone (FC) en cas de syndrome de perte de sel. Le traitement chirurgical consiste en la génitoplastie masculinisante qui dérive de la chirurgie de l’hypospadias et consiste à désenfouir et découder la verge et une uréthroplastie. Des complications peuvent se voir à type de fistules ou sténose de l’urètre.
Évolution
Le suivi à la consultation est très rapproché à la période néonatale puis se fait trois à quatre fois par an. La surveillance se base sur la croissance staturo-pondérale et l’état d’hydratation. Le bilan biologique comportera l’ionogramme sanguin, 17 OHP, ∆4 et testostérone. Le bilan radiologique comportera l’âge osseux.
L’objectif du traitement est de maintenir un équilibre hormonal satisfaisant à des doses minimales efficaces d’hydrocortisone et de fludrocortisone afin d’éviter leur retentissement sur la croissance staturo-pondérale (HC) et sur la tension artérielle (FC).
La croissance de ces patients n’est pas optimale, et ceci est dû à l’excès d’androgènes surrénaliens d’une part et le traitement par les glucocorticoïdes d’autre part, entrainant une fusion précoce du cartilage de conjugaison avec comme conséquence une taille finale inférieure à la taille cible.
Les recommandations actuelles selon le consensus international des hyperplasies congénitales des surrénales est de ne pas dépasser la dose de 10 à 15 mg/m2/j.
La puberté survient en général dans un délai normal, le gros problème durant cette période est l’installation de l’obésité et le défaut de compliance ayant pour conséquence un déséquilibre hormonal avec l’installation des signes d’hyper-androgénie.
Diagnostic anténatal et prise en charge
Il a pour but de dépister les enfants atteints d’une forme classique, et ceci concerne les familles où existe un cas index. La détermination du sexe du fœtus de sexe féminin (SRY-) est réalisée à la 6ème SA, il consiste à traiter les mamans porteuses d’un fœtus atteint, au plus tard vers la 9ème SA, par la dexaméthasone, qui a pour rôle de freiner l’hyper-androgénie et donner naissance à un nouveau-né de sexe féminin de morphotype normal, sans signes de virilisation.
Pour ce faire, cela nécessitera l’information des parents de la maladie et des possibilités thérapeutiques, l’étude préalable du gène CYP21 chez les différents membres de la famille, le diagnostic précoce de la grossesse (avant la 6ème SA) et la collaboration du pédiatre, gynécologue et généticien.
Cette prise en charge est possible pour le déficit en 21- OH et 11-β-OH. Cette conduite a tendance à être abandonnée, selon les données des dernières publications qui ont montré l’effet néfaste des corticoïdes sur la mère.
Dépistage néonatal
Il est systématique dans plusieurs pays. Il est surtout utile dans les formes sans ambiguïté sexuelle, car c’est la seule façon de faire un diagnostic précoce, et dans le Prader 5 en l’absence de syndrome de perte de sel. Il est peu couteux, se fait au 3ème jour à l’aide d’une goutte de sang prélevée au talon du nouveau-né, pour dosage de la 17-OHP.
Devenir à l’âge adulte
L’effet néfaste de l’hydrocortisone a été décrit avec des conséquences sur le métabolisme, l’installation de l’obésité, ainsi que sur la minéralisation osseuse. Il a été également décrit des anomalies dans la sexualité, et la fertilité.
Conclusion
La constatation d’une ambiguïté sexuelle à la naissance doit faire déclarer l’enfant de sexe indéterminé. Le choix du sexe définitif nécessite un ensemble d’explorations pour décider de l’orientation finale et donc de la génitoplastie à réaliser, celle-ci doit être assez précoce pour permettre à l’enfant d’avoir une identité sexuelle et pour que la famille puisse mieux vivre le problème qui reste toujours difficile nécessitant un soutien psychologique.
L’examen des organes génitaux externes doit être systématique et minutieux chez le nouveau-né, le nourrisson et l’enfant.
Devant toute déshydratation répétée avec hyponatrémie et hyperkaliémie chez un garçon, penser au diagnostic d’hyperplasie congénitale des surrénales.
Le premier diagnostic à évoquer devant une anomalie de la différenciation sexuelle (DSD) avec des gonades non palpables est l’hyperplasie congénitale des surrénales par bloc en 21-OH.
Devant une cryptorchidie bilatérale, il faut faire un caryotype, il peut s’agir soit d’un 46 XX, stade 5 de Prader d’un bloc en 21-OH ou 11-βOH, soit d’un DSD 46 XY.
La prise en charge est multidisciplinaire nécessitant la collaboration du pédiatre, du chirurgien et du psychologue.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Bibliographie
- Speiser PW, White PC.- Congenital adrenal hyperplasia. N Engl J Med 2003; 349:376-88
- Consensus statement on 21-hydroxylase deficiency from the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. J Clin Endocrinol Metab 2002; 87:4048-53.
- Hyperplasie congénitale des surrénales par déficit en 21-hydroxylase. HAS 2011
- Hughes IA, Houk C, Ahmed SF, Lee PA. Consensus statement on management of intersex disorders. Arch Dis Child 2006; 91:554-63.
- Bachelot A, Plu-Bureau G, Thibaud E, Laborde K, Pinto G, Samara D, et al. Long-term outcome of patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Horm Res 2007; 67:268-76.
- Martine Cools and Birgit Kohler. Disorders of Sex Development. In : Mehul T. Dattani, Charles GD. Brooks, eds Brook’s Clinical Pediatric Endocrinology. London, UK, John Wiley; 2020: 105-31