L’atrophie multisystématisée (AMS) est une pathologie neurodégénérative rare, sporadique, survenant le plus souvent dans la sixième décennie avec une prédominance masculine.Atrophie multisystématisée
W.Amer El Khedoud, A. Saadi, A. Benhaddadi, B. Mammeri, S. Lougani, N. Kassouri, F. Ferrat, Service de Neurologie, EHS Ben Aknoun, Alger
Date de soumission : 10 Février 2020.
Résumé : L’atrophie multisystématisée (AMS) est une pathologie neurodégénérative rare, sporadique, survenant le plus souvent dans la sixième décennie avec une prédominance masculine. Sur le plan clinique, l’AMS se caractérise par l’association variable d’un syndrome Parkinsonien peu dopa sensible, d’un syndrome cérébelleux, d’un syndrome pyramidal et de troubles dysautonomiques. La prédominance d’un des deux syndromes, Parkinsonien ou cérébelleux, permet de les classer en deux formes cliniques AMS-P et AMS-C. Elle est considérée comme l’une des deux principales étiologies de syndrome Parkinsonien atypique avec la paralysie supranucléaire progressive (PSP). Elle est classée parmi les “synucléinopathies”, comme la maladie de Parkinson idiopathique (MPI) et la démence à corps de Lewy (DCL). L’AMS est le syndrome Parkinsonien atypique neurodégénératif le plus fréquent. Son pronostic est plus sombre que celui de la maladie de Parkinson idiopathique (MPI). La moitié des patients est en fauteuil roulant après 5 ans, avec une survie médiane inférieure à 10 ans. L’AMS se diagnostique à partir de critères cliniques définissant un niveau « probable » ou « possible » établis par Gilman et al, en 1998 et révisés en 2008. Le diagnostic de certitude repose sur une confirmation anatomopathologique. Nous rapportons l’étude clinique et radiologique de trois cas d’AMS.
Mots clés : Atrophie multisystématisée, syndrome Parkinsonien atypique, ataxie cérébelleuse, dysautonomie, IRM.
Abstract: Multiple system atrophy (MSA) is a rare and sporadic, progressive neurodegenerative disorder which occurs most frequently in the sixth decade with a male predominance, characterized by progressive autonomic failure, parkinsonian features, and cerebellar and pyramidal features in various combinations. Patients are classified as MSA-C or MSA-P depending on the predominance of cerebellar ataxia or parkinsonism. It is considered to be one of the two main etiologies of atypical parkinsonian syndrome with progressive supranuclear palsy (PSP). It is classified among “synucleinopathies” such as idiopathic Parkinson’s disease (MPI) and dementia with Lewy bodies (DCL). AMS is the most common atypical neurodegenerative parkinsonian syndrome. Their prognosis is worse than that of idiopathic Parkinson’s disease (DPI). Half of the patients are in a wheelchair after 5 years, with an average survival of less than 10 years. In a consensus conference on diagnosis held in 1998 and reviewed in 2008, three levels of certainty were established, possible, probable, and definite MSA, with the diagnosis of definite MSA requiring autopsy confirmation. We report the clinical and radiological study of three cases of MSA.
Keywords: multiple system atrophy; Parkinsonism syndrome; cerebellar ataxia; autonomic failure, MRI.
Introduction
L’atrophie multisystématisée (AMS) est une pathologie neurodégénérative rare, sporadique, survenant le plus souvent dans la sixième décennie avec une prédominance masculine [1,2]. Sur le plan clinique, l’AMS se caractérise par l’association variable d’un syndrome Parkinsonien peu dopasensible, d’un syndrome cérébelleux, d’un syndrome pyramidal et de troubles dysautonomiques. La prédominance d’un des deux syndromes, Parkinsonien ou cérébelleux, permet de les classer en deux formes cliniques AMS-P et AMS-C [3,4]. Elle est considérée comme l’une des deux principales étiologies de syndrome Parkinsonien atypique avec la paralysie supranucléaire progressive (PSP). Elle est classée parmi les “synucléinopathies”, comme la maladie de Parkinson idiopathique (MPI) et la démence à corps de Lewy (DCL). L’AMS est le syndrome Parkinsonien atypique neurodégénératif le plus fréquent. Elle peut être parfois confondue avec la maladie de Parkinson idiopathique (MPI) notamment parce que certaines formes d’AMS sont dopasensibles. La prévalence de l’AMS est faible, 10 fois moindre que la MPI.
Son pronostic est plus sombre que celui de la maladie de Parkinson idiopathique (MPI), la moitié des patients est en fauteuil roulant après 5 ans, avec une survie médiane inférieure à 10 ans [2].
Les facteurs de mauvais pronostic seraient l’apparition précoce de troubles urinaires, la présence de signes dysautonomiques sévères ou d’un stridor [5]. Les causes de décès les plus fréquentes sont les infections broncho-pulmonaires et les morts subites [6].
L’AMS se diagnostique à partir de critères cliniques définissant un niveau « probable » ou « possible » établis par Gilman et al, en 1998 et révisés en 2008 [3,4]. Le diagnostic de certitude repose sur une confirmation anatomopathologique retrouvant notamment des inclusions oligodendrogliales intracytoplasmiques (ou glial cytoplasmic inclusion [GCI]) majoritairement constituées d’alpha-synucléine [7] présentes dans l’ensemble du système nerveux central avec une forte densité dans les régions appartenant au système olivo-ponto-cérébelleux et aux boucles motrices cortico-striato-corticales [8].
Cas Cliniques
Figure 01
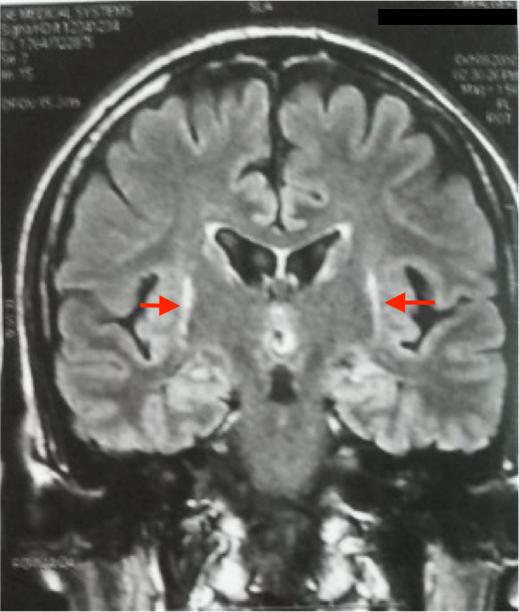
Figure 02
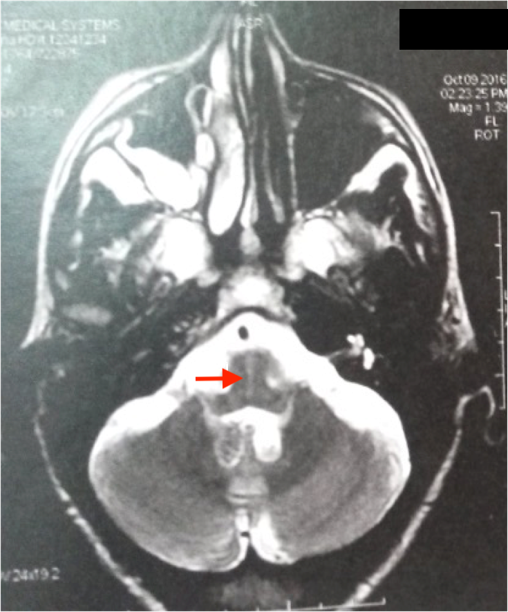
Figure 03
Cas 1 : Mme G.H.F. âgée de 50 ans, sans antécédents particuliers, a présenté une lourdeur des membres inférieurs, suivie un an après par l’installation d’un déséquilibre à la marche avec chutes fréquentes, incontinence urinaire et lenteur à l’élocution. L’évolution s’est faite vers l’aggravation progressive avec arrêt de la marche au bout de deux ans confinant la patiente au fauteuil roulant.
L’examen neurologique retrouve un syndrome Parkinsonien akineto-rigide bilatéral et symétrique, (amimie, rareté du clignement, hypokinésie, hypertonie plastique), une dysarthrie mixte cérébelleuse et extrapyramidale, des réflexes ostéo-tendineux exagérés aux 4 membres et un syndrome dysautonomique fait d’incontinence urinaire et d’une hypotension orthostatique.
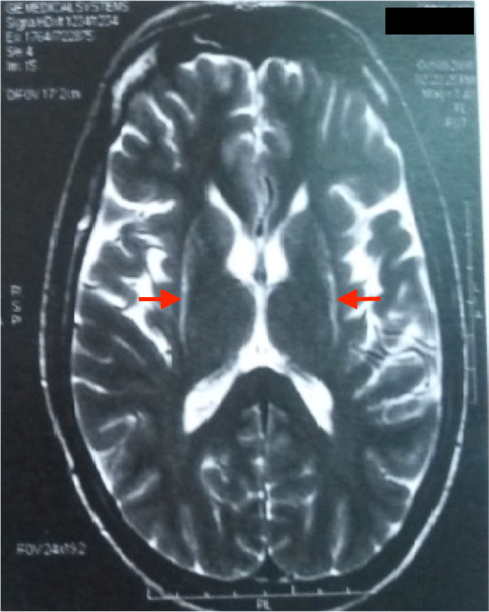
L’IRM cérébrale a montré des anomalies typiques d’une MSA. Sur les séquences T2 et FLAIR, on note un hypersignal de la bordure postérolatérale du putamen (pointe des flèches rouges) (figure 1,2) et un hypersignal au niveau du pont : début du « signe de la croix » dû à une atrophie des fibres pontiques, qui débute habituellement par l’hypersignal vertical (figure 3).
Cas 2 : Mr D.S. âgé de 62 ans sans antécédents particuliers qui a présenté un déséquilibre à la marche entrainant des chutes, suivi un an plus tard d’une lourdeur aux membres inferieurs associée à des troubles de l’élocution, troubles sexuels et incontinence urinaire.
L’examen neurologique a objectivé un syndrome cérébelleux stato-cinétique très important, un syndrome dysautonomique (troubles mictionnels et hypotension orthostatique : chute de la pression artérielle systolique de 30 mmHg lors du passage de la position couchée à debout), un discret syndrome Parkinsonien akinéto-rigide et des réflexes ostéo-tendineux exagérés aux 4 membres. L’IRM cérébrale est sans anomalie.
Cas 3 : Mr A. A. âgé de 52 ans, a présenté à l’âge de 48 ans des mictions impérieuses associées à une impuissance sexuelle suivie en quelques mois de pertes de connaissance brèves lors du passage rapide de la position assise à la position debout (le bilan cardiaque est sans anomalies), quelques mois après son hospitalisation ; apparition d’une lourdeur aux MI avec déséquilibre à la marche.
Examen neurologique : Syndrome dysautonomique sévère fait d’une hypotension orthostatique symptomatique, une impériosité mictionnelle avec un résidu post mictionnel à l’échographie vésicale, un discret syndrome Parkinsonien akinéto-rigide et des réflexes exagérés aux 4 membres. L’IRM cérébrale est sans anomalies.
Le bilan thyroïdien, carentiel (Vitamine B12, E), sérologies infectieuses, le bilan auto-immun et paranéoplasique étaient négatifs chez les trois patients.
Le bilan neuropsychologique : MMSE et BREF n’ont pas trouvé de déclin cognitif.
Le traitement par la L-Dopa n’a entrainé aucune amélioration sauf pour le cas 3 où la réponse était partielle au début.
Discussion
Le diagnostic de MSA probable selon les critères de Gilman a été retenu chez nos 3 patients devant la présence constante de signes dysautonomiques associés à un syndrome Parkinsonien bilatéral et symétrique ainsi que des réflexes vifs avec des anomalies à l’IRM cérébrale en faveur d’une (MSA-P) chez le 1er cas, et à un syndrome cérébelleux stato-cinétique dominant le tableau clinique avec un discret syndrome Parkinsonien et reflexes vifs chez le 2ème cas, (MSA-C).
Le 3ème cas a présenté des signes dysautonomiques, une hypotension orthostatique symptomatique parfois syncopale, isolée pendant cinq ans entrainant un retard important au diagnostic.
Les critères diagnostiques de l’AMS établis en 1998 (4, 5) ont été récemment revus lors d’une nouvelle conférence de consensus (3, 7). Ces nouveaux critères sont destinés à affiner le diagnostic et à inclure des signes additionnels ainsi que le résultat d’explorations paracliniques.
Le diagnostic d’AMS repose avant tout sur des critères cliniques consensuels [3] au sein desquels des anomalies spécifiques ont été identifiées à l’IRM.
Les critères diagnostiques définissent deux types d’AMS : l’AMS-P où le syndrome Parkinsonien prédomine comme dans notre cas 1 et l’AMS-C où le syndrome cérébelleux est au premier plan comme chez notre patient cas 2.
Par ailleurs, on classe la maladie selon trois niveaux de certitude : AMS « possible » (Tableaux II et III), « probable » (Tableau I) et « certaine » [3,4]. La certitude diagnostique est obtenue à l’examen neuropathologique post-mortem, montrant une dégénérescence des structures olivo-ponto-cérébelleuses et de la voie nigrostriée associée à d’abondantes inclusions gliales intracytoplasmiques d’alpha-synucléine (8).
Nos 3 patients répondent aux critères d’AMS probable. (Tableau I).
Tableau I. Critères pour le diagnostic d’AMS probable.
|
Maladie de début sporadique, progressive, chez un adulte (> 30 ans), caractérisée par : – une dysautonomie avec incontinence urinaire (associée à une dysfonction érectile chez l’homme) ou hypotension orthostatique (HO) survenant dans les 3 minutes du lever avec chute de pression artérielle (PA) d’au moins 30 mmHg pour la systolique (PAS) ou de 15 mmHg pour la diastolique (PAD) • et un syndrome parkinsonien (bradykinésie avec rigidité, tremblement ou instabilité posturale) peu dopasensible • ou un syndrome cérébelleux (ataxie à la marche avec dysarthrie cérébelleuse, ataxie des membres ou dysfonction oculomotrice cérébelleuse). |
Tableau II. Critères pour le diagnostic d’AMS possible.
|
Maladie de début sporadique, progressive, chez un adulte (> 30 ans), caractérisée par : – un signe suggérant une dysautonomie : mictions impérieuses sans autres explications, vidange vésicale incomplète, dysfonction érectile chez les hommes, ou HO n’ayant pas les critères exigés dans l’AMS probable • et un syndrome parkinsonien (bradykinésie avec rigidité, tremblement ou instabilité posturale) • ou un syndrome cérébelleux (ataxie à la marche avec dysarthrie cérébelleuse, ataxie des membres ou dysfonction oculomotrice cérébelleuse) • et au moins un des critères additionnels du tableau III |
Les signes cliniques principaux
Le syndrome Parkinsonien, plus fréquemment de forme akinéto-rigide, est dit « atypique » car il ne répond pas ou peu au traitement dopaminergique, et est associé à d’autres signes qui sont inhabituels dans la maladie de Parkinson idiopathique (MPI) comme l’instabilité posturale et les chutes précoces chez nos 3 patients ; la progression rapide vers le fauteuil roulant pour le cas 1 et le confinement au lit pour le cas 3 ainsi que la dysarthrie pour les cas 1 et 2 (signes considérés comme des drapeaux rouges dans la MPI et en faveur d’une AMS-P) (tableau 3). Néanmoins, une dopasensibilité est retrouvée chez environ un tiers des patients avec une excellente dopasensibilité en début de maladie pour 10 % d’entre eux comme chez notre patient, cas 3, qui a répondu partiellement à la L-dopa.
Le syndrome cérébelleux est au premier plan dans l’AMS-C, plus tardif et inconstant dans l’AMS-P où il est parfois difficile à distinguer des troubles posturaux du syndrome Parkinsonien. Notre patient cas 2 a présenté un syndrome cérébelleux au 1er plan associé à un discret syndrome Parkinsonien en faveur d’une AMS-C.
La dysautonomie est quasi constante, précoce et sévère contrairement à la MPI ou à la paralysie supra nucléaire progressive (PSP). Elle est un pilier central pour le diagnostic, son dépistage est donc primordial.
La dysautonomie, définie dans les critères consensuels, comprend une hypotension orthostatique (HO) et/ou des troubles vésico-sphinctériens.
- L’Hypotension orthostatique est définie par une chute de la pression artérielle systolique d’au moins 20 mm d’Hg ou une baisse de la pression artérielle diastolique d’au moins 10 mm d’Hg, enregistrée dans les 3 minutes suivant le passage à l’orthostatisme comme ce fut le cas chez nos patients 1 et 2 alors qu’elle était symptomatique pour le cas 3.
- Les troubles vésicosphinctériens sont souvent inauguraux dans l’AMS et précèdent généralement l’HO (10, 11). Les manifestations cliniques d’hyperactivité vésicale sont fréquentes au début de la maladie (urgences mictionnelles, impériosités, pollakiurie, fuites par impériosités) [12-15]. L’incontinence urinaire est le symptôme le plus fréquemment rapporté (13, 16, 18, 19) comme ce fut le cas chez nos trois patients.
- L’impuissance est souvent précoce, et elle est quasi constante chez l’homme (16, 18).
- Les autres signes de dysautonomie sont la dysfonction érectile, retrouvée chez nos 2 patients cas 2 et 3 et les troubles gastro-intestinaux dominés par la constipation.
- Il existe aussi des troubles de la thermorégulation avec une intolérance à la chaleur ou la présence de mains froides et violacées.
La présence d’un syndrome pyramidal est retrouvée chez environ 50 % des patients mais il n’est jamais au premier plan du tableau clinique. Chez nos 3 patients on a retrouvé des réflexes ostéo- tendineux vifs aux 4 membres.
La dysarthrie est précoce, mixte (parkinsonienne et cérébelleuse), retrouvée chez nos patients 1 et 2 ; elle s’associe souvent à des troubles de la déglutition et une dysphonie. Ces troubles sont pourvoyeurs de pneumopathie de déglutition qui sont une des principales causes de décès [6].
Les troubles cognitifs sont le plus souvent modérés, sous la forme d’anomalies de nature sous-cortico-frontale touchant les tâches attentionnelles, l’exécution, la planification et la mémoire de travail. Ils se rapprochent plus de ceux rencontrés dans la MPI et semblent plus sévères dans les AMS-P.
Pour nos 3 patients ; il n’a pas été retrouvé de troubles cognitifs.
L’imagerie est actuellement incluse dans le diagnostic d’AMS possible (3-20).
L’IRM cérébrale et l’imagerie fonctionnelle peuvent apporter des arguments supplémentaires pour le diagnostic d’AMS. L’IRM cérébrale peut montrer une atrophie putaminale, pontique et des pédoncules cérébelleux moyens. Sur les séquences pondérées en T2, on retrouve souvent un hyposignal de la partie postérieure du putamen, parfois associé à un hypersignal de la bordure postérolatérale du putamen, un hypersignal en forme de « croix » pontique et parfois des hypersignaux floconneux des pédoncules cérébelleux moyens [21] comme retrouvé chez notre patient cas 1.
Elle peut cependant être normale au début de la maladie.
Les études en tomographie par émission de positons (TEP) avec le 18 F-fluorodésoxyglucose (FDG) ont montré un hypométabolisme au niveau du putamen et du cervelet [22].
Chez un patient présentant un syndrome Parkinsonien atypique sans syndrome cérébelleux, la démonstration en TEP-FDG d’un hypométabolisme cérébelleux permet le diagnostic d’une AMS-P « possible » (Tableau II).
De même, chez un patient présentant un syndrome cérébelleux sans syndrome Parkinsonien, l’observation d’une dénervation dopaminergique nigrostriée par TEP ou tomographie d’émission monophotonique (TEMP) (DATSCAN)), est en faveur d’un diagnostic d’AMS-C « possible » [23]. Ces examens ne sont cependant pas des examens de routine.
Un tiers des patients AMS répond au traitement par lévodopa mais souvent de manière transitoire. Une réponse excellente à la lévodopa est néanmoins retrouvée chez 10 % des patients en début de maladie [24]. Les effets secondaires à type d’hypotension orthostatique sont des facteurs limitant à l’augmentation du traitement.
Un traitement à base de L-dopa a été institué chez nos 3 patients, il n’y a pas eu de réponse pour les cas 1 et 2, et une réponse partielle pour le cas 3.
Il n’existe pas de traitements validés pour agir sur le syndrome cérébelleux.
Une prise en charge rééducative est indispensable au décours de l’AMS afin de faciliter les déplacements et de prévenir les chutes.
Une prise en charge multidisciplinaire (cardiovasculaire et réadaptative) de l’hypotension orthostatique et des troubles vésicosphinctériens (24, 25) permet d’améliorer le quotidien des patients. Pour l’hypotension orthostatique, nos patients ont bénéficié de conseils hygiéno-diététiques : une bonne hydratation, un régime riche en sel, le port de bas de contention dès le lever et dormir avec une surélévation de la tête de 30°. Ils ont été adressés en cardiologie pour prise en charge de leur hypotension orthostatique et en rééducation fonctionnelle pour exploration et prise en charge de leurs troubles vésicosphinctériens.
Conclusion
L’AMS est une affection neurodégénérative dont le diagnostic reste essentiellement clinique. Les examens d’imagerie morphologique et fonctionnelle peuvent toutefois permettre un diagnostic un peu plus précoce.
Les ressources thérapeutiques de cette affection restent limitées sur le plan médicamenteux notamment pour la prise en charge des troubles moteurs. Le traitement symptomatique de la dysautonomie à la fois cardiovasculaire et vésicosphinctérienne ainsi que le traitement dopaminergique permettent d’améliorer les conditions de vie du patient.
Une prise en charge multidisciplinaire semble indispensable à l’accompagnement de ces patients et de leur famille.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Photos : Service de Neurologie de l’EHS Ben Aknoun.
Références
- Bjornsdottir A, Gudmundsson G, Blondal H, Olafsson E. Incidence and prevalence of multiple system atrophy: a nation-wide study in Iceland. J Neurol Neurosurg Psychiatry 2013;84 (2):136–40.
- Wenning GK, Geser F, Krismer F, Seppi K, Duerr S, Boesch S, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013; 12(3):264–74.
- Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008; 71(9): 670–6.
- Gilman S, Low PA, Quinn N, Albanese A, Ben-Shlomo Y, Fowler CJ, et al. Consensus statement on the diagnosis of multiple system atrophy. J Auton Nerv Syst 1998; 74(2–3):189–92.
- Kollensperger M, Geser F, Ndayisaba JP, Boesch S, Seppi K, Ostergaard K, et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: final analysis of the European multiple system atrophy registry. Mov Disord 2010; 25(15): 2604–12.
- Papapetropoulos S, Tuchman A, Laufer D, Papatsoris AG, Papapetropoulos N, Mash DC. Causes of death in multiple system atrophy. J Neurol Neurosurg Psychiatry 2007; 78(3):327–9.
- rojanowski JQ, Revesz T. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol 2007;33(6):615–20.
- Jellinger KA. Neuropathological spectrum of synucleinopathies. Mov Disord 2003; 18(Suppl. 6):S2–12.
- Kollensperger M, Geser F, Seppi K, Stampfer-Kountchev M, Sawires M, Scherfler C, et al. Red flags for multiple system atrophy. Mov Disord 2008;23(8):1093–9.
- Sakakibara R, Hattori T, Uchiyama T et al. Urinary dysfunction and orthostatic hypotension in multiple system atrophy: which is the more common and earlier manifestation? J Neurol Neurosurg Psychiatry 2000; 68(1):65-9.
- Mabuchi N, Hirayama M, Koike Y et al. Progression and prognosis in pure autonomic failure (PAF): comparison with multiple system atrophy. J Neurol Neurosurg Psychiatry 2005;76(7):947-52.
- Wenning GK, Tison F, Ben Shlomo Y et al. Multiple system atrophy: a review of 203 pathologically proven cases. Mov Disord 1997;12(2):133-47.
- Wenning GK, Scherfler C, Granata R et al. Time course of symptomatic orthostatic hypotension and urinary incontinence in patients with postmortem confirmed parkinsonian syndromes: a clinicopathological study. J Neurol Neurosurg Psychiatry 1999; 67(5):620-3.
- Joseph PA, Arné P, Barat M et al. Troubles vésicosphinctériens et explorations du sytème urinaire. In: Les syndromes parkinsoniens atypiques et secondaires. Paris : Masson (ed) 2006:106-12.
- Bonnet AM, Pichon J, Vidailhet M et al. Urinary disturbances in striatonigral degeneration and Parkinson’s disease: clinical and urodynamic aspects. Mov Disord 1997; 12(4):509-13.
- Wenning GK, Ben Shlomo Y, Magalhaes M et al. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain 1994;117(Pt 4):835-45.
- Wenning GK, Ben Shlomo Y, Magalhaes M et al. Clinicopathological study of 35 cases of multiple system atrophy. J Neurol Neurosurg Psychiatry 1995; 58(2):160-6.
- Kirchhof K, Apostolidis AN, Mathias CJ et al. Erectile and urinary dysfunction may be the presenting features in patients with multiple system atrophy: a retrospective study. Int J Impot Res 2003; 15(4):293-8.
- Kollensperger M, Stampfer-Kountchev M, Seppi K et al. Progression of dysautonomia in multiple system atrophy: a prospective study of self-perceived impairment. Eur J Neurol 2007; 14(1):66-72.
- Brooks DJ, Seppi K. Proposed neuro-imaging criteria for the diagnosis of multiple system atrophy. Mov Disord 2009; 24(7):949-64.
- Schrag A, Good CD, Miszkiel K, Morris HR, Mathias CJ, Lees AJ, et al. Differentiation of atypical parkinsonian syndromes with routine MRI. Neurology 2000; 54(3):697–702.
- Eckert T, Barnes A, Dhawan V, Frucht S, Gordon MF, Feigin AS, et al. FDG PET in the differential diagnosis of parkinsonian disorders. Neuroimage 2005; 26(3):912–21.
- Munoz E, Iranzo A, Rauek S, Lomeña F, Gallego J, Ros D, et al. Subclinical nigrostriatal dopaminergic denervation in the cerebellar subtype of multiple
- Wenning GK, Geser F, Poewe W. Therapeutic strategies in multiple system atrophy. Mov Disord 2005; 20(Suppl. 12):S67–76.
- Freeman R. Clinical practice. Neurogenic orthostatic hypotension. N Engl J Med 2008; 358(6):615–24.