La maladie de Charcot Marie Tooth (CMT), neuropathie héréditaire la plus fréquente partout dans le monde, est caractérisée par une hétérogénéité clinique et génétique avec plus de 80 gènes pouvant être impliqués dans sa pathogénie.
M. Tazir, S. Nouioua, M. Bellatache, L. Ali Pacha, Laboratoire de Neurosciences, Université Benyoucef Benkhedda, Service de Neurologie CHU Mustapha Bacha, Alger.
Date de soumission : 10 Février 2020.
Résumé : La maladie de Charcot Marie Tooth (CMT), neuropathie héréditaire la plus fréquente partout dans le monde, est caractérisée par une hétérogénéité clinique et génétique avec plus de 80 gènes pouvant être impliqués dans sa pathogénie. La forme classique débutant entre la première et la 3ème décade, marquée par un déficit et une amyotrophie des jambes lentement progressifs, un steppage à la marche, des pieds creux et parfois une scoliose, est compatible avec une espérance de vie quasi normale, et dans plus de 50% des cas, elle est due à la duplication du gène PMP22, caractérisant la forme démyélinisante autosomique dominante. La forme de CMT liée à l’X due à une mutation de la connexine 32 vient en 2ème position dans la fréquence mondiale. En Algérie et dans les pays du Maghreb, les formes autosomiques récessives (AR) de CMT constituent plus de la moitié des cas diagnostiqués. La forme axonale AR causée par une mutation fondatrice du gène LMNA est caractérisée par un phénotype plus sévère avec une progression rapide du déficit moteur des muscles distaux des membres inférieurs aux muscles proximaux aggravant la déambulation et menant à un arrêt précoce de la marche. Le gène GDAP1 dans ses formes AR axonale et démyélinisante est caractérisé par un début précoce, des déformations squelettiques importantes (pieds creux varus et scoliose), confinant le patient rapidement à la chaise roulante. L’avènement des techniques de séquençage des gènes à large échelle (exome sequencing) a permis d’identifier de nombreux nouveaux gènes impliqués dans les CMT et les protéines correspondantes déficientes et ainsi élargir la compréhension des mécanismes physiopathologiques responsables, permettant d’ouvrir de larges avenues aux essais thérapeutiques. La multitude des gènes impliqués a amené les experts de ces pathologies à réfléchir sur une autre manière de classer les différentes formes de CMT, remplaçant le système de classification alphanumérique par un système d’identification du patient basé sur le mode de transmission héréditaire, le type de neuropathie et le gène impliqué.
Mots clés : Maladie de Charcot-Marie-Tooth, ADCMT, ARCMT, XCMT, LMNA, GDAP1, NGS, WES.
Abstract: Charcot Marie Tooth disease (CMT), the most common hereditary neuropathy worldwide, is characterized by clinical and genetic heterogeneity with more than 80 genes that may be involved in its pathogenesis. The classic form beginning between the first and the third decade, marked by a slowly progressive deficit and an amyotrophy of the legs, a steppage gait, pescavus and scoliosis, is compatible with almost a normal life expectancy, and in more than 50% of cases itis due to the duplication of the PMP22 gene, characterizing the autosomal dominant demyelinating form. The X-linked form of CMT due to a Connexin 32 mutation is in 2nd place in the world frequency. In Algeria and the Maghreb countries, the autosomal recessive forms (AR) of CMT constitute more than half of the diagnosed cases. The axonal AR form caused by a founding mutation of the LMNA gene is characterized by a more severe phenotype with a rapid progression of the motor deficit of the distal muscles of the lower limbs to the proximal muscles, aggravating ambulation and leading to early cessation of walking. The GDAP1 gene in its axonal and demyelinating AR forms is characterized by an early onset, significant skeletal deformities (pescavus varus and scoliosis) quickly confining the patient to the wheelchair. The advent of large-scale gene sequencing techniques (exome sequencing) has made it possible to identify many new genes involved in CMTs and corresponding deficient proteins and thus broaden the understanding of the pathophysiological mechanisms, making it possible to open wide avenues for therapeutic trials. The multitude of genes involved has led experts in these pathologies to consider another way of classifying the different forms of CMT, replacing the alphanumeric classification system with a patient identification system based on the mode of inheritance, the type of neuropathy and the involved gene.
Keywords: Charcot- Marie-Tooth disease, ADCMT, ARCMT, XCMT, LMNA, GDAP1, NGS, WES
Introduction
La maladie de Charcot Marie Tooth (CMT) fait partie du vaste chapitre des neuropathies héréditaires qui comprend, en plus des neuropathies héréditaires motrices et sensitives (HMNS) ou CMT, les neuropathies héréditaires motrices (HMN) et les neuropathies héréditaires sensitives et dysautonomiques (HSAN). Ces deux dernières entités, beaucoup plus rares, ne sont pas abordées dans cet article, mais on peut dire que malgré la diversité génétique, il est possible d’établir une distinction entre les HMSN qui comportent des troubles moteurs et sensitifs, les HSAN où prédominent les manifestations sensitives et dysautonomiques et les HMN dans lesquelles seules les fibres motrices sont atteintes (1, 2).
Les progrès récents dans les domaines de la génétique moléculaire et la biologie cellulaire ont révolutionné nos connaissances sur les neuropathies héréditaires, sachant que plus de 80 gènes associés aux CMT ont été identifiés et plus d’une vingtaine d’anomalies génétiques sont en relation avec les HMN et HSAN.
Les HMSN ou CMT sont les maladies neurodégénératives du système nerveux périphérique les plus courantes, avec une fréquence variable dans les différentes populations, et dont la caractéristique est une hétérogénéité clinique, neuropathologie et génétique.
Cliniquement, la maladie de Charcot Marie Tooth est caractérisée par une perte progressive de la motricité distale des membres inférieurs avec amyotrophie, et de la sensibilité distale associée à des déformations des pieds : pieds creux et orteils déformés en marteau.
L’étude électrophysiologique demeure le premier examen à réaliser pour le diagnostic et la classification des HMSN.
CMT1 et CMT2 sont classiquement transmis selon le mode autosomique dominant, cependant des formes autosomiques récessives ou liées à l’X sont de plus en plus décrites. Les formes dominantes sont plus fréquentes aux États Unis, en Europe de l’ouest et au Japon, alors que dans d’autres pays comme ceux du bassin méditerranéen et notamment d’Afrique du Nord, où la prévalence des mariages consanguins est élevée, les formes autosomiques récessives peuvent représenter plus de 50% des cas (3-4).
L’étude génétique des CMT a débuté en 1991 avec l’identification d’une duplication de 1,4Mb dans le chromosome 17 contenant le gène PMP22 (protéine myélinique périphérique 22) qui est à l’origine, pour une grande part, des formes CMT1 (5, 6).
Depuis ce temps, il y a eu des progrès supplémentaires importants ayant permis la compréhension des bases moléculaires de nombreuses formes de CMT. En effet, de très nombreux gènes sont actuellement connus comme étant impliqués dans les CMT, ce qui a conduit à une meilleure connaissance de la physiopathologie de ces affections et de la biologie cellulaire du système nerveux périphérique.
L’étude neuropathologique qui s’effectue sur des biopsies nerveuses d’un nerf sensitif comme le sural ou le péronier superficiel, plus récemment sur des biopsies de peau, et qui peut montrer les lésions de neurodégénération des fibres nerveuses, a contribué grandement à la compréhension des mécanismes pathophysiologiques des mutations génétiques à l’origine des CMT. Cependant, le développement des techniques génétiques modernes permettant l’identification de mutations de gènes connus ou de gènes candidats, a modifié l’approche diagnostique des CMT. Actuellement, la biopsie nerveuse est rarement indiquée pour le diagnostic des neuropathies héréditaires. Néanmoins, la biopsie nerveuse et l’analyse des fibres nerveuses des biopsies cutanées sont utiles dans une perspective de recherche et d’études de fonctionnalité couplée à une corrélation avec les altérations de gènes découverts dans l’exome ou le génome de patients, grâce aux nouvelles méthodes de séquençage de l’ADN à large échelle.
Phénotype classique des CMT et classification
Comme indiqué précédemment, les CMT sont un groupe hétérogène cliniquement et génétiquement, l’âge de début, l’évolution clinique et les résultats électrophysiologiques étant variables selon les différentes formes clinico-génétiques. Dans la majorité des cas, l’évolution des troubles est lente avec un âge de début habituellement dans la première ou deuxième décade.
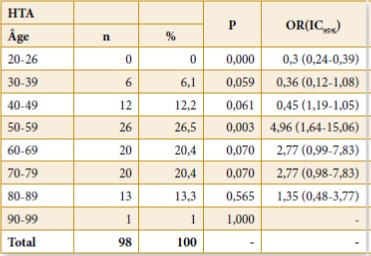
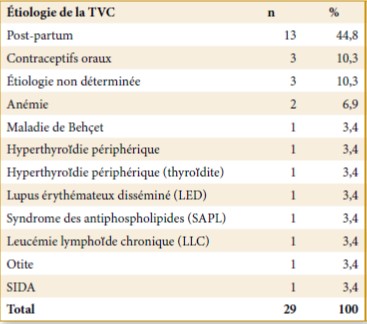
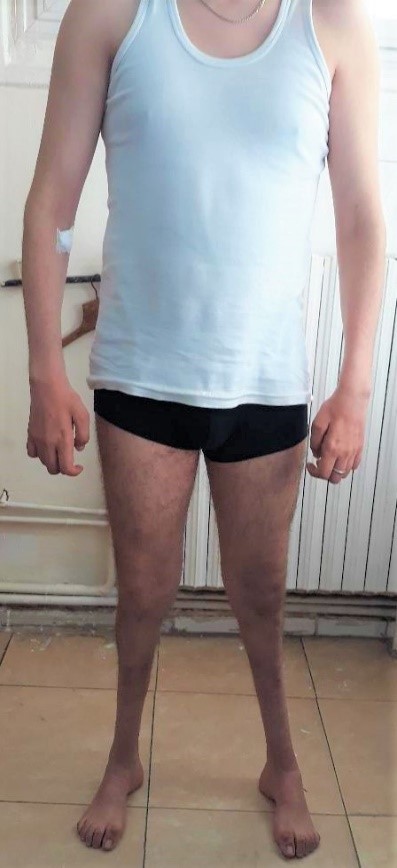
Le phénotype commun comprend un steppage à la marche, des pieds creux, une hypoesthésie distale et une amyotrophie distale des membres inférieurs donnant l’aspect de « jambes de coq ». Les membres supérieurs sont atteints plus tardivement avec une amyotrophie progressive pouvant aboutir à un aspect de « mains en griffe ». (Photo 1 et 2)
Photo1 : Amyotrophie des jambes avec aspect en « jambes de coq ». Amyotrophie des mains et rétraction réductible des doigts.
Photo 2 : Pieds creux avec aspect des orteils en marteau.
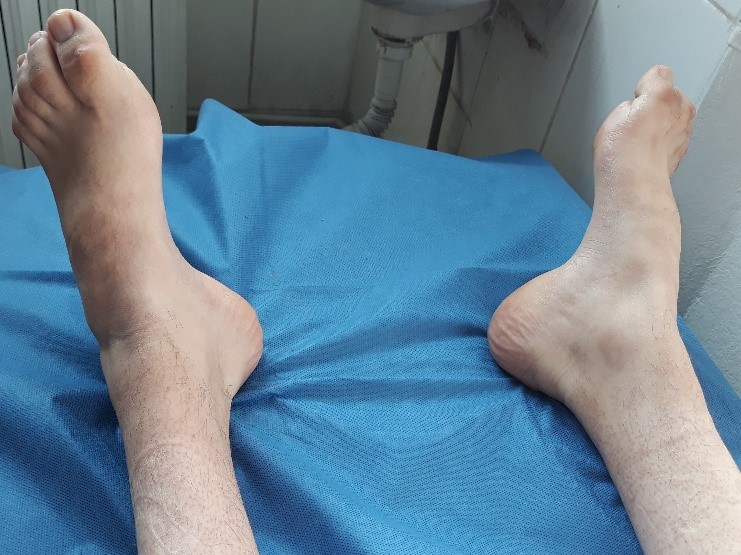
L’examen neurologique révèle une diminution ou une abolition des réflexes avec une atteinte motrice et sensitive distale symétrique. La marche et la stabilité sont habituellement affectées en raison des troubles proprioceptifs et des déformations squelettiques (pieds creux et orteils en marteau, voir photo 2).
L’étude des conductions nerveuses (VCN) permet de classer les CMT en formes démyélinisantes, intermédiaires ou axonales. Les VCN des membres supérieurs et les amplitudes des potentiels d’action musculaire (CMAP) ainsi que les potentiels sensitifs sont nécessaires alors que les nerfs des membres inférieurs sont souvent inexcitables.
Quand la vitesse de conduction motrice (VCM) du médian est <38m/s, la forme démyélinisante ou CMT1 est diagnostiquée, alors qu’une VCM du nerf médian >38m/s associée à des CMAP réduites avec absence des potentiels d’action sensitifs conduisent au diagnostic de la forme axonale ou CMT2. La forme intermédiaire ou CMT I est diagnostiquée quand la VCM du médian se situe entre 38 et 45m/s (en pratique, ces chiffres pouvant aller au-delà de ces limites). L’électromyographie de détection met en évidence des signes de dénervation chronique diffus aux quatre membres.
Certains enfants atteints de CMT ont un tableau clinique sévère avec un retard de la marche et des déformations orthopédiques des pieds et souvent du rachis et une VCM du médian très réduite (15m/s). Ces formes précoces ont été classées CMT3 et décrites auparavant comme syndrome de Déjerine-Sottas (DSS) ou neuropathie congénitale avec hypomyélination (CHN).
Actuellement, la sévérité de la neuropathie peut être évaluée par le score CMT comportant 9 paramètres cliniques et électrophysiologiques (7).
La classification des CMT est également basée sur le mode de transmission génétique. CMT1 et CMT2 sont de transmission autosomique dominante, CMTX est liée à l’X. Les formes autosomiques récessives sont classées CMT4 ou ARCMT1 pour les formes démyélinisantes et ARCMT2 pour les formes axonales ; sachant qu’une transmission autosomique récessive est fortement suspectée lorsqu’il y a au moins deux enfants atteints d’une même fratrie issue de parents consanguins indemnes.
Ces différentes formes de CMT sont subdivisées selon l’anomalie génétique constatée. Actuellement plus de 80 gènes, dont la mutation peut entrainer un phénotype CMT, sont identifiés. Cependant, des études récentes ont montré qu’environ 90% des diagnostics moléculaires dans les populations occidentales concernaient des mutations ou réarrangements dans les gènes PMP22 (CMT1A), GJB1 (CMTX1), MPZ (CMT1B) et MFN2 (CMT2A) dans l’ordre décroissant de fréquence, alors que les mutations des autres gènes sont relativement beaucoup plus rares (8-9). Dans les formes axonales, les mutations des gènes LMNA, GDAP1et SH3TC1 se sont révélées relativement fréquentes dans les populations du bassin méditerranéen et Maghrébines (10).
Certains patients avec des tableaux de neuropathies congénitales (DSS ou CHN) peuvent avoir des mutations dans les gènes dominants ou récessifs tels que MPZ, PMP22, EGR2, GDAP1, MTPR2 et PRX (11).
Le diagnostic de neuropathie héréditaire est plus difficile à établir en l’absence d’histoire familiale ; néanmoins ce diagnostic est évoqué devant des troubles symétriques à début infantile ou juvénile d’une neuropathie distale lentement progressive associée à des pieds creux. L’électrodiagnostic contribue également au diagnostic en montrant une réduction uniforme de toutes les vitesses de conduction nerveuse, par opposition aux neuropathies acquises où les VCM sont réduites de façon asymétrique avec des signes de dispersion temporelle ou des blocs de conduction.
La biopsie nerveuse n’est pas recommandée dans les formes ADCMT1 et les CMT liés à l’X mais peut être considérée comme un moyen nécessaire pour mieux caractériser la neuropathie dans certaines formes non communes (CMT2 et ARCMT) afin d’orienter les tests moléculaires. En effet, les anomalies histologiques peuvent être suffisamment caractéristiques de l’anomalie génétique sous-jacente, comme celles rencontrées dans les mutations MPZ, GJB1, PRX, FGD4 et LMNA.
En définitive, l’orientation des tests génétiques est avant tout basée sur le phénotype clinique, l’électrodiagnostic et le mode de transmission héréditaire. Un algorithme de diagnostic CMT devrait être basé en premier lieu sur l’étude électrophysiologique avec les vitesses de conduction nerveuses et l’électromyographie pour établir si la neuropathie est démyélinisante ou axonale, en second lieu sur l’histoire familiale et l’arbre généalogique de façon à déterminer si le mode de transmission héréditaire est AD, AR ou lié à l’X. Ces données permettent de planifier ensuite l’étude génétique par la recherche de mutations des gènes correspondants les plus fréquents, à savoir PMP22, GJB1, MPZ, MFN2 et SH3TC2, et pour notre région, LMNA et GDAP1, et de rechercher les plus rares, éventuellement, dans un deuxième temps. En fait, nous rencontrons dans nos régions toutes les formes de CMT, que ce soit les dominantes, liées à l’X ou intermédiaires. Cependant, les formes AR y sont aussi fréquentes, sinon plus, que les autres formes, alors qu’elles sont très rares voire inexistantes dans les pays où la consanguinité est rarissime. Dans cet article nous détaillerons les formes prévalentes au bassin méditerranéen et notamment en Afrique du Nord.
Actuellement, les équipes de diagnostic génétique les plus performantes utilisent les méthodes de séquençage à grande échelle comme le séquençage haut débit appelé next generation sequencing (NGS), et le séquençage à haut débit de l’exome. En effet, l’exome (qui désigne tous les exons ou séquences codantes) de plusieurs patients peut être séquencé simultanément. Plusieurs gènes ciblés ou panel de gènes pour de nombreux patients peuvent être séquencés en une fois. Alors qu’auparavant le séquençage était fastidieux et « time consuming », se faisant gène par gène à la recherche de mutation. Ces nouvelles techniques, y compris celle de bio-informatique, considérées maintenant comme étant les meilleures méthodes d’exploration génétique des CMT et des autres affections neurogénétiques, ont permis la découverte de nombreux nouveaux gènes ces dernières années, permettant d’accroitre la connaissance de nouveaux mécanismes physiopathologiques de ces affections qui conduiront certainement à de nouvelles approches thérapeutiques.
Par ailleurs, ces nouvelles techniques permettent de donner un résultat génétique en un temps record. Pour en apprendre plus sur elles le lecteur peut consulter le site : https://www.ncbi.nlm.nih.gov/books/NBK279899/#app5.Multigene_Panels
Des mécanismes et voies moléculaires très variés ont été mis en évidence dans les neuropathies héréditaires grâce au nombre élevé des gènes et protéines altérés identifiés. Les structures myéliniques, la dynamique mitochondriale, la régulation transcriptionnelle, le turnover protéique ainsi que le transport axonal sont ainsi impliqués dans les différentes formes de CMT, permettant d’explorer de nouvelles stratégies thérapeutiques, malgré leur très grande complexité physiopathologique.
Caractéristiques cliniques et pathologiques des différentes formes de CMT :
- Les CMT autosomiques dominants
Les CMT dominants sont les formes les plus fréquentes en général, notamment dans les pays occidentaux.
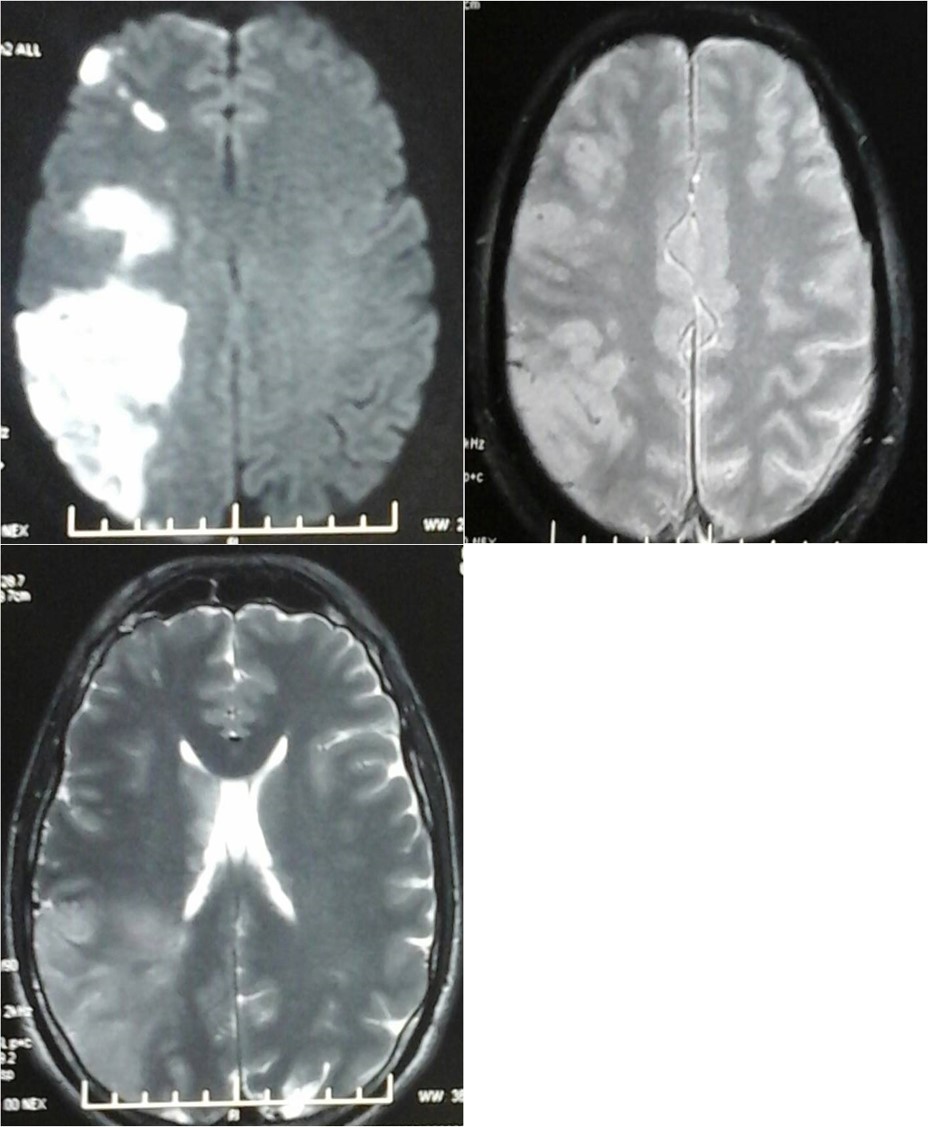
- Les formes AD-CMT1 (démyélinisantes) sont les plus communes (80% des cas). La plupart des patients ont un phénotype CMT classique avec une VCM sur le nerf médian inférieure à 38m/s, des signes neuropathologiques comprenant une réduction importante des axones myélinisés et un aspect en « bulbes d’oignon » des cellules de Schwann (neuropathie hypertrophique) recouvrant des axones entourés d’une myéline fine (Figure). Cliniquement, cette neuropathie hypertrophique peut être diagnostiquée en palpant un gros nerf comme le cubital au coude. À l’heure actuelle, les sous-types CMT1 sont classés de CMT1A à CMT1G. De nouveaux gènes CMT1 sont apparus récemment : FBLN5 et c1orf194 . (Voir le lien https://neuromuscular.wustl.edu/time/hmsn.html pour les détails cliniques correspondant aux CMT1).
Une nouvelle classification proposant de remplacer les lettres par le nom du gène impliqué et prenant en compte la forme démyélinisante (De), axonale (Ax) ou intermédiaire (In), ainsi que l’hérédité (AD, liée à l’X ou AR) a été publiée récemment par Mathis et al (12), et reprise par d’autres auteurs experts dans le domaine (13, 14). L’argument principal pour changer la classification étant que les lettres de l’alphabet ne suffisent plus pour le nombre de gènes impliqués sans cesse croissant. Ainsi, CMT1A devient pour ce système de classification : AD-CMTDe-PMP22. L’avantage d’une telle classification c’est la suppression des lettres et des chiffres remplacés par les noms des gènes et la pathologie (démyélinisante ou axonale).
La plupart des patients AD-CMT1 (ou AD-CMTDe) ont un handicap bénin à modéré interférant peu avec une vie active, peu d’entre eux évoluant vers la chaise roulante. Rarement le phénotype est sévère avec un tableau de neuropathie congénitale type DSS.
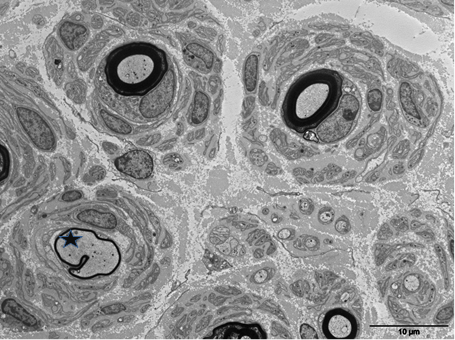
Figure : Microscopie électronique du nerf sural, section transverse. Les axones myélinisés sont entourés de proliférations de cellules de Schwann en bulbes d’oignons. (Etoile : un axone entouré de myéline anormalement fine). Photo JM Vallat, ref. 18.
- Les formes AD-CMT2 (ou AD-CMTAx) sont relativement fréquentes pouvant représenter le tiers des formes dominantes. La plus fréquente est CMT2A (AD-CMTAx-MFN2), liée au gène de la mitofusine (MFN2), protéine mitochondriale, dans laquelle on peut observer un phénotype CMT classique avec des VCM du nerf médian supérieures à 45m/s, des potentiels sensitifs souvent absents, mais avec parfois des signes d’atteinte du système nerveux central comme des réflexes vifs, un signe de Babinski ou une atrophie optique. La biopsie nerveuse montre une raréfaction neuronale avec des signes de régénération (axonal sprouting), sans signe de démyélinisation. La plupart des CMT2A sont sévèrement atteints et perdent la marche à l’âge adulte.
En dehors des AD-CMTAx-MFN2 ou CMT2A, les autres sous-types sont très rares, décrits dans une ou deux familles. Elles figurent néanmoins dans la classification actuelle jusqu’à CMT2Z lié au gène MORC2, prenant la dernière lettre de l’alphabet, montant ainsi les limites de cette classification. (Voir le lien https://neuromuscular.wustl.edu/time/hmsn.htmlpour les détails cliniques correspondant à chaque forme de CMT2)
Certaines formes de CMT2 comme CMT2C (TRPV4), CMT2D (GARS), CMT2F (HSPB1) et CMT2L (HSPB8) sont alléliques à certaines formes de neuropathies motrices héréditaires distales, confortant l’hypothèse d’un continuum entre les CMT axonales et les HMN distales (15).
- Les CMT liés à l’X
Un CMT lié à l’X est suspecté lorsque la transmission père-fils est absente sur les arbres généalogiques et quand les patients males sont plus sévèrement atteints (16). Quatre formes dominantes et récessives de CMT liés à l’X ont été décrites. CMTX1 lié à une mutation du gène GJB1 encodant la connexine 32 (Cx32), est la forme la plus fréquente après CMT1A dans les cohortes américaines et européennes.
Les autres formes de CMT liés à l’X sont beaucoup plus rares. (Voir tableau des CMT liés à l’X dans : https://neuromuscular.wustl.edu/time/hmsn.html)
- Les CMT intermédiaires
Les CMT dominants intermédiaires (DI-CMT) sont caractérisés par un phénotype bénin à modérément sévère et des VCM du médian allant de 25 à 50m/s (17). Les formes intermédiaires affectent la myéline et l’axone, ce qui met en exergue la relation étroite entre ces deux structures du nerf périphérique. Six formes cliniques ont été identifiées (Voir classification des CMT intermédiaires dans le lien : https://neuromuscular.wustl.edu/time/hmsn.html)
- Les CMT autosomiques récessifs.
Les CMT autosomiques récessifs (AR-CMT) sont décrits dans les pays où les mariages consanguins sont fréquents (pays du pourtour méditerranéen, du Moyen-Orient et les populations Roms d’Europe).
La plupart des cas AR-CMT sont caractérisés par un début précoce de la maladie et une progression clinique rapide conduisant à des déformations orthopédiques (pieds varus équin, mains déformées Aran-Duchenne et déformations vertébrales majeures) et un arrêt précoce de la marche.
Les CMT autosomiques récessifs comprennent des formes démyélinisantes (AR-CMT1 ou CMT4) avec des VCM <38m/s, et les formes axonales (AR-CMT2) avec des VCM >45m/s aux membres supérieurs, des amplitudes réduites des potentiels moteurs ainsi que des potentiels sensitifs réduits ou absents aux quatre membres. La biopsie nerveuse dans les AR-CMT1 montre une démyélinisation importante avec parfois des anomalies spécifiques de la myéline et une perte axonale secondaire, alors que dans les AR-CMT2 les fibres myélinisées sont très réduites en nombre sans signes évidents de démyélinisation et remyélinisation ou de régénération axonale active.
Au plan génétique, plusieurs types de ARCMT ont été identifiés, avec des caractéristiques cliniques, pathologiques et ethniques propres (18). On peut consulter des formes rares nouvellement identifiées dans le site : https://neuromuscular.wustl.edu/time/hmsn.html.
Le diagnostic moléculaire des AR-CMT est plus complexe du fait de la faible fréquence des gènes identifiés et de leur grande hétérogénéité. Cependant, ce diagnostic génétique peut être orienté par les caractéristiques épidémiologiques, cliniques et pathologiques. Certaines formes de AR-CMT comme celles associées aux gènes récessifs NGRD-1 et LMNA sont dues à des mutations fondatrices dans des populations spécifiques, Roms (Gitans) en Europe et du Maghreb, respectivement ; alors que d’autres formes liées aux gènes GDAP1 et SH3TC2 sont décrites dans tous les pays méditerranéens (19-20), y compris en Algérie : voir la thèse de DESM de Ameur El Khoudoud, Université d’Alger.
AR-CMT1A (CMT4A) correspondant au premier locus AR décrit, lié au gène GDAP1 codant pour une protéine de la membrane mitochondriale, est caractérisé par un début précoce et une neuropathie grave et des déformations distales des membres conduisant à un arrêt de la marche à l’adolescence. Cette forme de CMT liée au gène GDAP1 peut avoir aussi une transmission AD avec un phénotype moins sévère (21).
D’autres formes de AR-CMT telles que celles associées à des anomalies de la myotubularine (MTMR2 et MTMR13) donnent des tableaux de CMT sévères, caractérisées par une myéline redondante avec des outfoldings et infoldings à la biopsie nerveuse, pouvant orienter le diagnostic moléculaire, lorsqu’elle est possible, ce qui n’est pas évident. Autrement, dans certaines formes, des particularités cliniques comme une cyphoscoliose au premier plan et une hypoacousie, (AR-CMT1C, SH3TC2), une parésie des cordes vocales avec une respiration stertoreuse (AR-CMT1A, GDAP1), une ataxie sensitive avec des troubles moteurs discrets et des VCM très réduites (AR-CMT1F, PRX), permettent d’orienter le diagnostic génétique.
Parmi les formes axonales de ARCMT, la forme ARCMT2A, ou AR-CMTAx-LMNA (selon la classification récemment proposée mettant en évidence le gène impliqué à la place des lettres de l’alphabet), identifiée dans une famille marocaine de neuf membres et dont la mutation fondatrice c.892C>T (p. Arg298Cys) du gène LMNA a été par la suite caractérisée chez des familles algériennes, est une forme de CMT axonale qui débute entre 11 et 14 ans et qui s’aggrave assez rapidement avec une atteinte précoce des muscles proximaux des membres inférieurs rendant la marche de plus en plus difficile avec steppage et dandinement (22-23). Certains patients atteignent le stade de la chaise roulante après 15 à 20 ans d’évolution mais d’autres peuvent rester ambulants plus longtemps (23). Cette mutation fondatrice prédomine dans une région du Maghreb située entre le Centre et l’Ouest de l’Algérie et le Maroc. L’ancêtre commun le plus récent aurait vécu il y a 800 à 900 ans (24).
Mesures thérapeutiques
Actuellement, aucun traitement spécifique n’est disponible pour guérir ou arrêter la progression des neuropathies héréditaires, en sachant que des essais thérapeutiques divers notamment de thérapies géniques, sont en cours d’étude ; l’un des plus récents essais médicamenteux étant l’essai au PXT3003, une combinaison de trois médications déjà connues (baclofene, naltrexone et D-sorbitol) (25, 26).
Cependant, il est bien reconnu que les traitements symptomatiques sont nécessaires, entre autres pour corriger des déformations squelettiques et ainsi améliorer la qualité de vie du patient. La physiothérapie et les mesures de réadaptation demeurent les meilleures approches thérapeutiques pour les patients qui présentent un phénotype classique de CMT. La kinésithérapie est indispensable pour lutter contre l’atrophie musculaire et surtout les rétractions capsulo-tendineuses, permettant ainsi d’éviter, ou du moins retarder et limiter, les déformations articulaires. La prescription de chaussures orthopédiques et/ou des orthèses spécifiques (releveurs du pied le plus souvent) est fortement recommandée, ainsi que des équipements disponibles adaptés aux déformations des mains.
La prise en charge des déformations orthopédiques des pieds et de la colonne vertébrale nécessite parfois un recours à la chirurgie.
La paralysie des cordes vocales que l’on peut observer dans certaines formes de ARCMT (GDAP1, MTMR2) peut bénéficier d’un traitement au laser efficace, réduisant de façon satisfaisante les troubles de la phonation et de la respiration (27).
La douleur et la dépression peuvent être présentes dans certaines formes de CMT, pouvant nuire à la qualité de vie du patient. Les douleurs chroniques sont souvent associées à ces neuropathies et peuvent être liées à une origine musculo-squelettique ou neuropathique. Dans ces cas des traitements symptomatiques comme la thérapie physique, les interventions orthopédiques, le soutien psychologique et les médicaments s’avèrent souvent très utiles.
Conclusion
Le diagnostic positif des CMT est relativement aisé, reposant sur le tableau clinique et l’électrophysiologie ainsi que l’évolution lentement progressive. Le diagnostic des différentes formes cliniques repose sur l’électrodiagnostic mais aussi sur le type de transmission héréditaire et le testing moléculaire. Celui-ci est nettement facilité par les méthodes modernes de diagnostic génétique comme le NGS, le séquençage d’exome et l’analyse bio-informatique.
Dans la littérature européenne et nord-américaine les formes dominantes et liées à l’X représentent plus de 80% des cas, alors que dans les pays comme l’Algérie et les autres pays du Maghreb où le taux de consanguinité est encore important, les formes autosomiques récessives représentent plus de 50% des cas.
La prise en charge des patients et des familles atteintes de neuropathies héréditaires consiste à effectuer un conseil génétique, et surtout à programmer une physiothérapie au long cours avec des spécialistes en médecine physique pour prévenir et traiter les complications orthopédiques de ces affections, en attendant les thérapies curatrices spécifiques.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Photos : Originales. Collection personnelle de l’auteure.
Références
- Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular Neurologic, genetic, and electrophysiological findings in hereditary polyneuropathies. Arch Neurol 1968; 18:603–8.
- Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980; 103:259–80.
- Harding AE, Thomas PK. Autosomal recessive forms of hereditary motor and sensory neuropathy. J NeurolNeurosurgPsychiatry1980; 43:669–78.
- Dubourg O, Azzedine H, Verny C, Durosier G, Birouk N, Gouider R, et al. Autosomal recessive forms of demyelinating Charcot–Marie–Tooth disease. Neuro molecular Med 2006; 8:75–86.
- Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, et al. DNA duplication associated with Charcot–Marie–Tooth disease type 1A. Cell 1991;66:219–32.
- Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, Hoogendijk JE, Baas F, et al. Duplication in chromosome 17p11.2 in Charcot–Marie–Tooth neuropathy type 1a (CMT 1a). Neuromuscul Disord 1991; 1:93–7.
- Murphy SM, Herrmann DN, McDermott MP, Scherer SS, Shy ME, Reilly MM, et al. Reliability of the CMT neuropathy score (second version) in Charcot–Marie–Tooth disease. J Peripher Nerv Syst 2011; 16:191–8.
- Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot–Marie–Tooth disease subtypes and genetic testing strategies. Ann Neurol 2011; 69:22–33.
- Gess B, Schirmacher A, Boentert M, Young P. Charcot–Marie–Tooth disease: frequency of genetic subtypes in a German neuromuscular center population. Neuromuscular Disorder 2013; 23:647–51.
- Tazir M, Bellatache M, Nouioua S, Vallat JM. Autosomal recessive Charcot Marie Tooth disease: from genes to phenotypes. J Peripher NervSyst. 2013;18(2):113–129.
- El-Abassi R, England JD, Carter GT. Charcot–Marie–Tooth disease: an overview of genotypes, phenotypes, and clinical management strategies. PMR 2014; 6:342–55.
- Mathis S, Goizet C, Tazir M, Magdelaine C, Lia AS, Magy L, et al. Charcot Marie Tooth diseases: an update and some new proposals for the classification. J Med Genet 2015;52(10):681–90
- Magy L, Mathis S, Le Masson G, Goizet C, Tazir M, Vallat JM. Updating the classification of inherited neuropathies: results of an international survey. Neurology. 2018;90:e870–6
- Bird TD. Charcot Marie Tooth (CMT) Hereditary Neuropathy 1998 Sep 28 [updated 2020 Jan 2. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. Gene Reviews. Seattle (WA): University of Washington, Seattle; 1993-2020
- Solla P, Vannelli A, Bolino A, Marrosu G, Coviello S, Murru MR, et al. Heat shock protein 27 R127W mutation: evidence of a continuum between axonal Charcot–Marie–Tooth and distal hereditary motor neuropathy. J Neurol Neurosurg Psychiatry 2010; 81:958–62.
- Boerkoel CF, Takashima H, Garcia CA, Olney RK, Johnson J, Berry K, et al. Charcot–Marie–Tooth disease and related neuropathies: mutation distribution and genotype–phenotype correlation. Ann Neurol 2002; 51:190–201.
- Nicholson G, Myers S. Intermediate forms of Charcot–Marie–Tooth neuropathy: a review. Neuromolecular Med 2006; 8:123–30.
- Tazir M, Hamadouche T, Nouioua S, Mathis S, Vallat JM. Hereditary motor and sensory neuropathies or Charcot Marie Tooth diseases: an update. J Neurol Sci. 2014;347(1-2):14–22
- Kalaydjieva L, Gresham D, Gooding R, Heather L, Baas F, de Jonge R, et al. N-myc downstream-regulated gene 1 is mutated in hereditary motor and sensory neuropathy-Lom. Am J Hum Genet 2000; 67:47–58.
- Gosselin I, Thiffault I, Tétreault M, Chau V, Dicaire MJ, Loisel L, et al. Founder SH3TC mutations are responsible for a CMT4C French-Canadians cluster. Neuromuscul Disord 2008; 18:483–92.
- Sivera R, Frasquet M, Lupo V, et al. Distribution and genotype-phenotype correlation of GDAP1 mutations in Spain. Sci Rep. 2017;7(1):6677.)
- Bouhouche A, Benomar A, Birouk N, et al. A locus for an axonal form of autosomal recessive Charcot Marie Tooth disease maps to chromosome 1q21.2-q21.3. Am J Hum Genet 1999; 65: 722-727.
- Tazir M, Azzedine H, Assami S, et al. Phenotypic variability in autosomal recessive axonal Charcot Marie Tooth disease due to the R298C mutation in lamin A/C. Brain. 2004;127(Pt 1):154–163.
- Hamadouche T, Poitelon Y, Genin E, et al. Founder effect and estimation of the age of the c.892C>T (p.Arg298Cys) mutation in LMNA associated to Charcot Marie Tooth subtype CMT2B1 in families from North Western Africa. Ann Hum Genet 2008; 72: 590-597.
- Attarian S, Dubourg O, Funalot B, Gonnaud PM, Lacour A, Magy L, et al. A phase II randomized, placebo-controlled multicenter clinical trial of three doses of PXT3003 in 80adult patients with CMT1A treated for 1 year. J Peripher Nerv Syst 2013; 18(Suppl.): S7-8.
- Morena J, Gupta A, Hoyle JC. Charcot-Marie-Tooth: From Molecules to Therapy. Int J Mol Sci. 2019;20(14):3419.
- Nouioua S, Hamadouche T, Funalot B, et al. Novel mutations in the PRX and the MTMR2 genes are responsible for unusual Charcot Marie Tooth disease phenotypes. Neuromuscul Disord. 2011;21(8):543–550.
Télécharger le PDF de cet article