Le déficit en GH de l’adulte (GHDA : Growth Hormone Deficiency in Adults) est un syndrome clinico-biologique en rapport avec une baisse de la production de l’hormone de croissance par les cellules somatotropes hypophysaires suite à une anomalie congénitale ou acquise de la région hypothalamo-hypophysaire (HPT-HPP)..
M. Bensalah, S. Ould Kablia, Service d’Endocrinologie, Hôpital Central de l’Armée, Mohamed Nekkache, Aïn Naâdja, Alger.
Date de soumission : 08 Août 2020.
Résumé : Le déficit en GH de l’adulte (GHDA : Growth Hormone Deficiency in Adults) est un syndrome clinico-biologique en rapport avec une baisse de la production de l’hormone de croissance par les cellules somatotropes hypophysaires suite à une anomalie congénitale ou acquise de la région hypothalamo-hypophysaire (HPT-HPP). Ces conséquences cliniques sont dominées par une répartition androïde des graisses, insulinorésistance, une hypertension artérielle, une dyslipémie avec un risque vasculaire élevé. Il est également responsable d’une baisse de la densité osseuse, de troubles cognitifs et d’une altération de la qualité de vie. Le diagnostic de GHDA repose sur la réalisation de tests dynamiques en dehors des déficits congénitaux avec anomalies de la région HPT-HPP, le test oral à la Macimoréline est récemment introduit et reconnu par les sociétés savantes comme bonne alternative au test à l’insuline. Le traitement par hormone de croissance permet d’améliorer les paramètres cliniques, le LDL cholestérol, la densité osseuse et la qualité de vie. Ces effets au long cours n’ont pour le moment pas montré une augmentation du risque de néoplasie.
Mots clés : déficit en GH de l’adulte, diagnostic, Macimoréline, bénéfices du traitement.
Abstract: Growth hormone deficiency in adults (GHDA) is a syndrome due to decrease production of growth hormone by somatroph pituitary cells because of congenital or acquired lesion of the hypothalamic pituitary region. Its clinical consequences are represented by android obesity, insulin resistance, dyslipidaemia, high blood pressure, decrease of brain density, cognitive disorders and alteration of quality of life. Diagnosis of GHDA require the necessity of dynamic stimulation tests except in congenital growth hormone deficiency with abnormalities of the hypothalamic pituitary region. Macimorelin oral test is a recently introduced test recognized by scientific societies as a good alternative to insulin tolerance test. The substitutive treatment with growth hormone improves clinical health, decreases LDL cholesterol levels; improves bone density and quality of life. Long-time safety of growth hormone treatment has been proved without risk of neoplasm.
Keywords: adult GH deficiency, diagnosis, Macimorelin, treatment benefits.
Qu’est-ce que le déficit en hormone de croissance de l’adulte ?
Le déficit en hormone de croissance de l’adulte (GHDA) est un syndrome clinique lié à la baisse de la sécrétion de l’hormone de croissance due à une anomalie organique ou congénitale de la région hypothalamo-hypophysaire. Il est responsable principalement d’anomalies métaboliques qui augmentent le risque cardiométabolique avec répartition androïde des graisses, augmentation du risque d’hypertension artérielle, diminution de la fibrinolyse, intolérance au glucose, insulinorésistance, diminution de la qualité de vie et apparition de troubles cognitifs 1,2,3.
Le déficit somatotrope est à l’origine d’une diminution de la masse maigre et de la densité osseuse, d’une baisse de la capacité à l’exercice, de troubles cardiovasculaires, de dyslipémie et d’une diminution du bien-être 2,4,5,6.
Il engendre par ailleurs des troubles cognitifs dominés par les anomalies de l’attention, de la mémoire, de la fonction exécutive et du langage appréciés par de multiples tests neuropsychologiques 7,8.
Dans le même sens, le déficit somatotrope est à l’origine d’une diminution de la qualité de vie : les troubles mnésiques, la détérioration de l’image de soi, l’anxiété, la dépression, l’apathie, les difficultés de jugement, l’insomnie, la fatigue, les difficultés dans la planification, dans l’organisation, la prise de décision et la communication en sont les principaux signes 39.
Quelles sont les étiologies du déficit en hormone de croissance de l‘adulte ?
Le GHDA peut être en rapport avec un déficit isolé ou combiné hypophysaire congénital connu et traité depuis l’enfance ou encore en rapport avec une cause acquise (adénome hypophysaire, craniopharyngiome, maladie infiltrative, radiothérapie, séquelles de chirurgie de la région hypothalamo-hypophysaire, traumatisme crâniens) 10.
Les causes congénitales du déficit en GH peuvent être en rapport avec des anomalies génétiques avec ou sans anomalies de dévalement de la glande hypophysaire. Il s’agit des anomalies des gènes LHX3, LHX4, Hesx1, Prop 1, Pit 1, ATRX1 ; qui associent des déficits hypophysaires combinés. Le déficit en GH congénital peut être également isolé par anomalie du gène de la GH. Les causes acquises sont représentées par les adénomes hypophysaires, causes les plus fréquentes du GHDA acquis, les craniopharyngiomes, la radiothérapie de la région hypothalamo-hypophysaire, les maladies infiltratives de la région hypothalamo-hypophysaire et les traumatismes crâniens dont le déficit en GH est l’atteinte la plus fréquente parmi les axes endocriniens 10.
Figure 1 : Étiologies du déficit en GH de l’adulte 10
Quelles sont les conséquences cliniques du déficit en GH de l’adulte ?
Le déficit en GH de l’adulte associe un profil clinico-biologique particulier avec répartition androïde des graisses, hypertension artérielle, insulinorésistance, modification des paramètres lipidiques avec augmentation du cholestérol LDL, diminution du cholestérol HDL et profil pro-inflammatoire confirmé par certaines études. Ce profil pro-inflammatoire correspond à l’augmentation de la Protéine C Réactive (CRP), certaines cytokines, la pregnancy associated plasma protein, du stress oxydatif avec dysfonction endothéliale 3,4,10.
Toutes ces modifications augmentent le risque cardiovasculaire des patients atteints de GHDA. Ces patients ont donc une augmentation du risque de maladie coronaire comme l’a démontré l’étude Framingham 11.
Rosen et Bengtsson12, ont été les premiers à démontrer que l’espérance de vie des patients déficitaires en hormone de croissance, était réduite. Le taux de mortalité élevé était essentiellement dû à la survenue de la maladie cardiovasculaire. Une étude rétrospective portant sur 333 patients estimait que le risque de mortalité doublait par maladie cardio-vasculaire avec 58% de décès. Dans une autre étude menée au royaume uni sur 1.014 patients hypo-pituitaires, la mortalité était liée aux causes vasculaires 13.
Les accidents vasculaires cérébraux et la cardiopathie ischémique sont plus fréquents chez les femmes que chez les hommes.
Les patients déficitaires en GH ont un nombre élevé de plaques d’athérome au niveau des artères carotides et fémorales par rapport aux sujets contrôles et une réduction de la distension artérielle. Ces données sont retrouvées aussi bien chez les adultes jeunes que ceux plus âgés. Les performances cardiaques sont réduites avec une diminution de la masse ventriculaire gauche, une diminution de la fraction d’éjection et des troubles de la fonction diastolique droite. A cela s’ajoute également une diminution de l’épaisseur du septum interventriculaire du VD et de la fraction d’éjection du VD de 14% 5,14,15,16.
Par ailleurs, la majorité des études ont démontré une augmentation de l’épaisseur intima-média (EIM) chez les patients hypo-pituitaires. Cette augmentation est corrélée aux valeurs basses d’IGF117.
Le déficit en GH de l’adulte entraîne une diminution de la masse et de la densité osseuses avec augmentation de l’incidence des fractures et de l’ostéopénie18.
Ces modifications peuvent apparaître si le déficit somatotrope est isolé ou associé à d’autres déficits antéhypophysaires et même en absence du déficit gonadotrope 19.
Certains GHD adultes, et GHD enfants devenus adultes, (20%, 30% respectivement), ont une ostéoporose avec un T score <2,5. Les patients de moins de 30 ans ont une ostéoporose plus sévère que les contrôles sains. Les patients âgés de plus de 60 ans ne diffèrent pas des contrôles. Ceux âgés entre 30-45 ans ont une sévérité intermédiaire 19.
Holmes et collaborateurs démontrent que les hommes et les femmes qui ont un déficit en GH durant plus de deux ans ont une diminution significative de la densité osseuse au niveau des vertèbres lombaires et ceci même en absence de déficit gonadotrope 18.
Dans le même sens, Colao et collaborateurs 4, retrouvent une baisse significative de la densité osseuse avec un risque fracturaire 2 à 3 fois plus élevé chez les patients atteints de déficit en GH sévère et très sévère, qu’il soit isolé ou associé à d’autres déficits anté-hypophysaires.
Sur le plan histologique, les patients GHD ont une augmentation du volume de l’os trabéculaire, une augmentation de la résorption osseuse, une augmentation de l’épaisseur des cellules ostéoïdes suggérant un retard à la minéralisation osseuse 20.
Les patients atteints de déficit en hormone de croissance sont atteints de troubles psychologiques, d’une diminution de la qualité de vie et de troubles cognitifs 21.
Ces troubles sont dominés par les difficultés de l’attention, par des troubles mnésiques, des troubles du langage, de la concentration et de la compréhension engendrant une diminution du bien-être et une tendance dépressive 8,22.
Les récepteurs de l’hormone de croissance sont présents dans de nombreuses parties du cerveau : l’hippocampe, région impliquée dans la compréhension et la mémoire, le cortex cérébral, les amygdales et l’hypothalamus. Les sites de liaison de la GH sont particulièrement abondants au niveau des plexus choroïdes 23.
L’axe GH/IGF1 joue un rôle important dans la réparation des cellules neuronales du SNC. En effet, l’IGF1 intervient dans la myélinisation, remyélinisation et la protection des oligodendrocytes 23.
La GH et l’IGF1 permettent aussi de maintenir le tonus et la réactivité vasculaire et participent à la réparation du SNC en cas d’hypoxie cérébrale.
L’altération de la qualité de vie dans le déficit en GH est évaluée par un outil pratique et simple comprenant 25 questions avec des réponses dichotomiques QOL AGHDA (quality of life growth hormone deficiency in adults, un score élevé atteste d’une altération de la qualité de vie.

Figure 2 : profil clinico-biologique du GHDA.
Comment poser le diagnostic de déficit en GH chez l’adulte ?
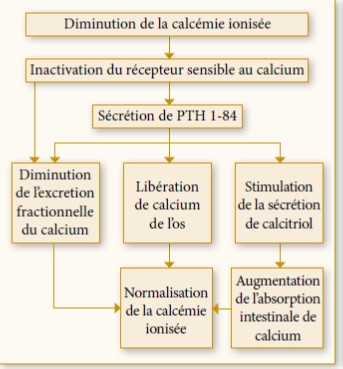
Les valeurs basales de GH et d’IGF1 n’ont pratiquement pas d’intérêt, en dehors des situations de lésions organiques de la région hypothalamo-hypophysaire avec déficits multiples >3. Un taux d’IGF1 <-2DS ou une valeur <84 ng/ml suffit pour poser le diagnostic de déficit en GH. Dans les autres situations, la réalisation de tests de stimulation dynamique est nécessaire. Le test à l’insuline/GH reste le test de référence pour poser le diagnostic de GHDA en présence d’une hypoglycémie <2,2 mmol/l avec un cut-off reconnu de 5 µg/L pour le déficit partiel et 3 µg/L pour le déficit total. En présence de contre-indications, le test au glucagon est une bonne alternative à condition qu’il soit interprété en fonction de l’IMC (indice de masse corporelle). Le cut-off de 3 µg/L est admis pour les patients avec IMC <25 Kg/m2 et ceux dont le IMC est entre 25 et 30 Kg/m2. Il est de 1 µg/L pour les patients dont l’IMC est >30 Kg/m2. Le test à la GH RH arginine représente une très bonne alternative au test à l’insuline, cependant, la GHRH n’étant plus commercialisée, ce test est donc irréalisable 1,10.
Un nouveau test a révolutionné l’exploration de l’axe somatotrope de l’adulte, il s’agit d’un nouveau test oral à la Macimoréline qui est un agoniste de la ghreline qui stimule l’hormone de croissance. La FDA a autorisé l’utilisation de ce test pour la recherche du déficit en GH de l’adulte en Décembre 2017 pour un cut-off de 2,8 µg/L24.
Le test de stimulation de l’hormone de croissance n’est pas indiqué en cas d’atteinte organique avec déficits hypophysaires multiples ≥3 et des taux d’IGF1 <-2 DS ainsi que chez les patients avec déficit en GH d’origine génétique. Les patients ayant ≤2 déficits anté-hypophysaires avec des taux d’IGF1 <-2 DS doivent bénéficier d’un test de stimulation de l’hormone de croissance pour confirmer le déficit en GH 10.
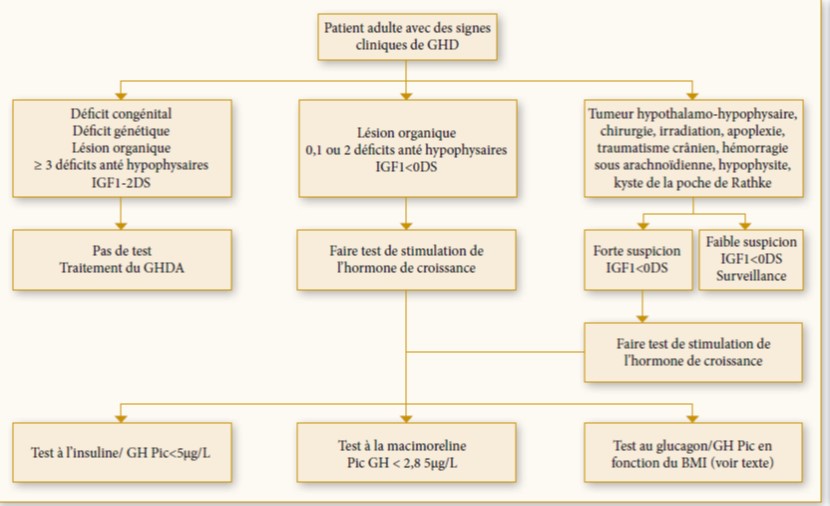
Figure 3 : Protocole d’évaluation du déficit en GH de l’adulte selon les recommandations de l’AACE – Growth hormone task force 2019
Comment évaluer les patients atteints de déficits en hormone de croissance congénitaux en phase de transition ?
La phase de transition concerne les adolescents âgés entre 15 et 18 ans atteints de déficit congénital en hormone de croissance sous traitement par GH recombinante ayant atteint leur taille finale. Le maintien de l’hormone de croissance chez les adolescents chez lesquels le déficit persiste, permet de normaliser la composition corporelle, le profil lipidique et la maturation osseuse ainsi que la qualité de vie. La transition du service d’endocrinologie pédiatrique vers un service d’endocrinologie de l’adulte doit se faire en étroite collaboration entre l’endocrinologue et le pédiatre pour assurer une prise en charge adéquate en cette période vulnérable 10,25.
La réalisation de test de stimulation en phase de transition n’est nécessaire que chez une catégorie de patients. Ne seront pas re-testés les patients atteints de déficit congénital en hormone de croissance avec déficits multiples >3, les patients porteurs de mutations génétiques type PRPOP1, PIT1, LHX3, LHX4, HESEX1, PITX2, les patients atteints de déficit en GH par anomalie de la GH ou de son récepteur, les patients atteints de déficit congénital en GH avec anomalies structurelles ou acquises périnatales10 25.
Par contre, les adolescents atteints de déficit en GH idiopathique isolé avec des taux d’IGF1 entre 0 et -2 DS ou <-2 DS, les patients atteints de déficit en GH avec un ou deux autres déficits anté-hypophysaires, les patients avec déficit isolé en GH avec hypoplasie hypophysaire ou post-hypophyse ectopique ou d’une irradiation crânienne, doivent bénéficier d’un test de stimulation de l’hormone de croissance après un mois d’interruption du traitement par hormone de croissance 10.
Le nombre de tests à réaliser dépend du degré de présomption de la persistance du déficit en GH. Si la suspicion de persistance de déficit est élevée comme en cas de déficit isolé en GH avec hypoplasie hypophysaire ou post-hypophyse ectopique ou d’une irradiation crânienne, un seul test de stimulation est nécessaire. Si la suspicion de GHD est faible en cas d’absence d’anomalies à l’IRM hypothalamo-hypophysaire, d’absence de déficits hypophysaires associés ou de taux d’IGF1 normal bas <0 DS ; deux tests de stimulation de l’hormone de croissance sont nécessaires pour infirmer ou confirmer la persistance du déficit 10.
Le test à l’insuline sur GH est le test de référence en phase de transition. En cas de contre-indications, le test au glucagon et le test à la Macimoréline sont une bonne alternative. Le seuil en phase de transition pour le test à l’insuline est de 5,6 µg/L avec une sensibilité de 77% et une spécificité de 93% 25. Pour les nouvelles recommandations de l’AACE, ce seuil est de 5 µg/L.
Il est important à souligner que la réalisation des tests de stimulation doit se faire en équilibre hormonal des autres déficits anté-hypophysaires.
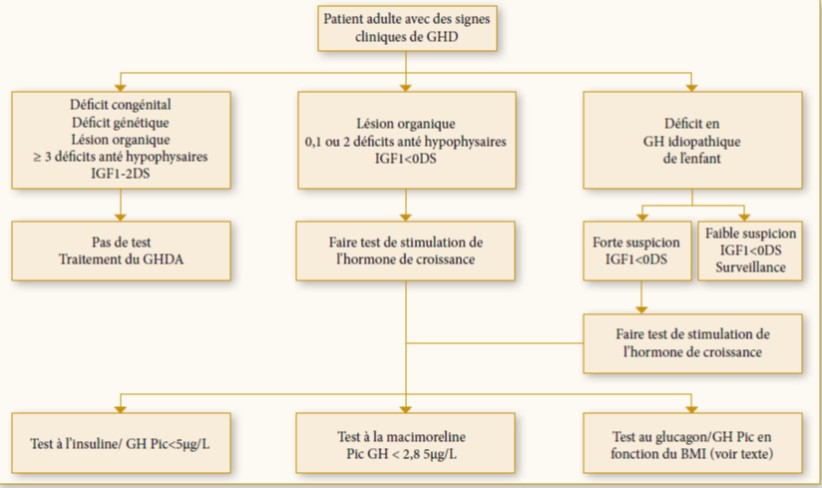
Figure 4 : Protocole d’évaluation du déficit en GH en phase de transition selon les recommandations de l’AACE Growth hormone task force 2019
Comment traiter le déficit en GH de l’adulte ?
L’hormone de croissance humaine recombinante est utilisée pour traiter les patients adultes atteints de déficit en GH. Il est recommandé d’initier le traitement par de faibles doses, en dehors des patientes sous œstrogènes et des patients en phase de transition. Il est recommandé de débuter le traitement à la dose de 0.3-0.4 mg/j chez les patients de moins de 30 ans, 0.2-0.3 mg pour les patients âgés entre 30-60 ans et 0.1-0.2 mg pour les patients de plus de 60 ans, les patients obèses, diabétiques ou en intolérance au glucose 2,3,10.
La titration des doses sera ensuite guidée par les taux d’IGF1 qui doivent être dans les normes pour l’âge et le sexe entre -2 DS et +2 DS. La surveillance de la titration se fera tous les 1 à 2 mois initialement puis tous les 6 mois une fois l’équilibre thérapeutique atteint.
Chez les patients en transition d’un GHD congénital traités pendant l’enfance, les doses administrées sont réduites de 50% par rapport à la dose utilisée pendant l’enfance avec titration en fonction des taux d’IGF1 qui ne doivent pas dépasser +2 DS 10.
La surveillance du traitement se basera sur les données cliniques tous les 6-12 mois avec évaluation de l’IMC, de la pression artérielle, du tour de taille, le profil lipidique, l’électrocardiogramme et l’échocardiographie ainsi que l’échodoppler des troncs supra-aortiques. L’ostéodensitométrie est réalisée avant le traitement puis 2 à 3 années après son initiation 2,3,10.
L’évaluation des autres axes anté-hypophysaires est également nécessaires notamment pour les fonctions corticotrope et thyréotrope, le dosage de la FT4 guidera une éventuelle nécessité de majoration des doses de Lévothyroxine 2,3,10.
Quels sont les bénéfices du traitement par hormone de croissance chez les patients GHDA ?
Le traitement par GH améliore les chiffres tensionnels, augmente la vasodilatation et diminue la rigidité artérielle. La GH améliore la fonction endothéliale qui contribue au changement du tonus vasculaire.
Les marqueurs de l’inflammation sont élevés chez les patients GHD et l’administration de GH diminue la CRP.
La majorité des études ont démontré une augmentation du HDL cholestérol et une diminution du LDL cholestérol et du cholestérol total après institution du traitement par hormone de croissance. Une large étude observationnelle (n=1.206) rapporte une réduction de 7% du LDL cholestérol et du cholestérol total après deux ans de traitement. Mais aucune étude n’a démontré l’effet additif de la GH par rapport aux statines 26.
La majorité des études ont démontré que l’épaisseur intima média est plus importante chez les patients GHD par rapport aux contrôles. Son augmentation est corrélée aux valeurs basses d’IGF1 et l’administration de GH aux patients GHD diminue l’EIM 19.
Par ailleurs, le traitement par hormone de croissance améliore les performances cardiaques, la masse et le volume diastolique du VG comme l’a démontré une étude portant sur dix patients GHD traités par hormone de croissance. 19
Le traitement par hormone de croissance a un effet anabolique sur l’os, les effets sont complexes et diphasiques la GH stimule à la fois la formation et la résorption osseuses.
Après 18-24 mois, la majorité des études objectivaient une augmentation de 4-10% de la densité osseuse avec des effets plus importants au niveau vertébral que fémoral. 27,28,29.
Les patients dont la perte osseuse est plus importante (Z score <-2) ont une meilleure réponse au traitement, particulièrement les hommes avec une efficacité pendant près de 10 ans, mais les effets sur le col restent en plateau après 5 ans de traitement.
Elbornsson et al., ont évalué la densité osseuse chez 109 patients atteints de GHD sur une période de 15 ans et ont mis en évidence une augmentation de la densité osseuse au niveau vertébral et du col du fémur pendant 7 ans. Au delà, elle rejoignait les valeurs basales30.
L’amélioration de la qualité de vie n’est pas toujours corrélée à l’augmentation des taux d’IGF1. Cette amélioration commence les trois premiers mois pour certaines études et au bout d’une année pour d’autres.
Le traitement au long cours permet de maintenir une qualité de vie adéquate chez les patients traités 31.
Elbornsson et al.32, ont évalué la mortalité chez 156 patients avec GHD traités par hormone de croissance pendant plus de 15 ans ; 21 patients sont décédés ; le décès n’était pas d’origine cardiovasculaire. Par ailleurs, 38% des patients avaient bénéficié d’une radiothérapie cérébrale probablement source de mortalité dans cette population. La mortalité liée aux causes cardiovasculaires avait par contre baissé sous traitement.
Ces données sont confirmées par celles rapportées dans la KIMS data base, rapportée par Gaillard et al.33, qui retrouvent une augmentation de la mortalité globale dans le groupe traité par GH (n=13.983) ; elle était corrélée à l’âge jeune au diagnostic, au sexe féminin, aux étiologies du GHD (Craniopharyngiome, tumeurs agressives, diabète insipide, radiothérapie hypophysaire).
Par contre, l’étude danoise retrouve une augmentation de la mortalité cardiovasculaire chez les femmes après analyse du registre national des patients traités par hormone de croissance 34.
Une étude plus récente réalisée par Kokshoorn et al., portant sur 534 patients traités pour GHDA âgés entre 60 et 80 ans, a montré une baisse du LDL et une amélioration de la qualité de vie.
Ainsi, si le traitement par hormone de croissance semble améliorer la mortalité cardiovasculaire chez les patients GHD, la mortalité en général est plus élevée que dans la population générale.
Le traitement par hormone de croissance peut-il être prescrit au long cours avec sécurité ?
La sécurité d’emploi de l’hormone de croissance est favorisée par le respect des indications thérapeutiques, de la posologie ainsi que la surveillance du traitement. Il est clairement établi que le risque de cancer, de tumeur cérébrale, de leucémie d’hémorragie cérébrale n’augmente pas en absence de facteurs de risque 10. Cependant, le risque de second cancer a augmenté chez les patients ayant bénéficié d’une radiothérapie 10. Il est évidemment contre-indiqué d’utiliser l’hormone de croissance chez les patients ayant une néoplasie active. L’étude SAGhE publiée en 2012 avait montré une augmentation de la mortalité chez les adultes traités pendant l’enfance pour GHD. Ces données n’ont cependant pas été confirmées par l’étude européenne ni sur la mortalité ni sur le risque de cancer. Dans le même sens, des données portant sur 150.000 patients traités par GH thérapie avec suivi au long cours sont rassurantes sur le plan de la sécurité au long cours 37.
Par ailleurs, les effets secondaires sont représentés par les œdèmes, les arthralgies, les paresthésies et l’HIC bénigne. La survenue de diabète ou de troubles de la tolérance glucosée est possible chez les patients traités par hormone de croissance notamment dans les populations à risque (antécédents familiaux de diabète de type 2, diabète gestationnel). Il est donc recommandé de doser la glycémie et/ou l’hémoglobine glyquée tous les 6-12 mois.
Quelles sont les perspectives thérapeutiques ?
Le traitement par hormone de croissance est administré par voie sous cutanée quotidiennement. Des essais thérapeutiques visent à développer des formes retards de l’hormone de croissance pour améliorer l’observance thérapeutique des patients. Cinq formes de GH retard ont été développées, cependant plusieurs questions restent posées 10 :
- Quels sont les effets de la GH circulante de façon prolongée sur le risque de cancer et sur le plan métabolique ?
- Les molécules porteuses de la GH permettent-elles une bonne diffusion de l’hormone de croissance vers les tissus cibles ?
- Les effets de la forme retard sont-ils durables ?
- Comment se fera le monitoring de l’IGF1 pour les formes prolongées de GH ?
Conclusion
Le déficit en GH de l’adulte doit être bien documenté et prouvé avant toute initiation d’un traitement substitutifs dont les bénéfices sur le plan métabolique, cardiovasculaire, osseux et sur la qualité de vie ont été prouvés. Bien que le risque de cancer et de diabète, soit faible, le traitement par hormone de croissance doit être individualisé et surveillé pour chaque patient traité. La phase de transition chez les patients atteints de déficit en GH pendant l’enfance, doit être évaluée en collaboration entre pédiatre et endocrinologue afin de maintenir l’effet bénéfique du traitement substitutif chez les patients déficitaires confirmés. Les formes-retard de l’hormone de croissance faciliteront certainement l’observance thérapeutique, des essais au longs cours sont encore nécessaires pour valider leur utilisation.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
- Glynn, N. & Agha, A. Diagnosing growth hormone deficiency in adults. Int. J. Endocrinol. 2012, (2012).
- Thomas, J. D. J. & Monson, J. P. Adult GH deficiency throughout lifetime. Eur. J. Endocrinol. 161, 97–106 (2009).
- Reed, M. L., Merriam, G. R. & Kargi, A. Y. Adult growth hormone deficiency – benefits, side effects, and risks of growth hormone replacement. Front. Endocrinol. (Lausanne). 4, 1–14 (2013).
- Colao, a. Bone Loss Is Correlated to the Severity of Growth Hormone Deficiency in Adult Patients with Hypopituitarism. J. Clin. Endocrinol. Metab. 84, 1919–1924 (1999).
- Colao, A. Di Somma, C, Pivonello, Ret al. The cardiovascular risk of adult GH deficiency (GHD) improved after GH replacement and worsened in untreated GHD: A 12-month prospective study. J. Clin. Endocrinol. Metab. 87, 1088–1093 (2002).
- McGauley, G. The psychological consequences and quality of life in adults with growth hormone deficiency. Growth Horm. IGF Res. 10 Suppl B, S63–S68 (2000).
- Erlanger, D. M., Kutner, K. C. & Jacobs, a R. Hormones and cognition: current concepts and issues in neuropsychology. Neuropsychol. Rev. 9, 175–207 (1999).
- Falleti, M. G., Maruff, P., Burman, P. & Harris, A. The effects of growth hormone (GH) deficiency and GH replacement on cognitive performance in adults: A meta-analysis of the current literature. Psychoneuroendocrinology31, 681–691 (2006).
- McMillan, C. V, Bradley, C., Gibney, J., Russell-Jones, D. L. & Sönksen, P. H. Psychometric properties of two measures of psychological well-being in adult growth hormone deficiency. Health Qual. Life Outcomes4, 16 (2006).
- Kevin. C. J. Yuen, Beverly. M. K. Biller, Sally. Radovick et al. American association of clinical endocrinologists and American college of endocrinology guidelines for management of growth hormone deficiency in adults and patients transitioning from pediatric to adult care. Endocrine Practice. Vol 25 n°11. November 2019.
- Hodis HN, Mack WJ, LaBree, Selzer RH, Liu CR, Liu CH, A. S. The role of carotid arterial intima-media thickness in predicting clinical coronary events. Ann Intern Med128, 262–269 (1998).
- Rosen T, B. B. Premature mortality due to cardiovascular disease in hypopituitarism. Lancet336, 285–288 (1990)
- Tomlinson, J. W. Holden, N Hills, R Ket al. Association between premature mortality and hypopituitarism. West Midlands Prospective Hypopituitary Study Group. Lancet357, 425–31 (2001).
- Jallad RS, Lieberman B, Vianna CB, Vieira ML, Ramieres JA, K. M. Effects of growth hormone replacement therapy on metabolic and cardiac parameters, in adult patients with childhood onset growth hormone deficiency. Growth Horm. IGF Res. 13, 81–88 (2003).
- Capalbo D, Lo Vecchio A, Spinelli L, Palladino A, Tiano C, Lettiero Y, Lombardi G, Calao A, S. M. Subtle alterations of cardiac performance in children with growth hormone deficiency: results of a two years prospective case-control study. J. Clin. Endocrinol. Metab. 94, 3347–3355 (2009).
- Maison P, C. P. Cardiac effects of growth hormone in adults with growth hormone deficiency: a meta-analysis. Circulation108, 2648–2652 (2003).
- Colao A, Di Somma C, Filipella M, Rota F, Pivonello R, Orio F, Vitale G, L. G. Insulin-like growth factor -1 deficiency determines increased intima-media thickness at common carotid arteries in adult patients with growth hormone deficiency. Clin. Endocrinol. (Oxf). 61, 360–366 (2003).
- Holmes SJ, Economou G, Whitehouse RW, Adams JI, S. S. Reduced bone mineral density in patients with adult onset growth hormones deficiency. J. Clin. Endocrinol. Metab. 78, 669–674 (1996).
- Molitch, M. E., Clemmons, D. R., Malozowski, S., Merriam, G. R. & Vance, M. L. Evaluation and treatment of adult growth hormone deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 96, 1587–1609 (2011).
- Bravenboer N, Holzman P, de Boer H, Blok GJ, L. P. Histomorphometric analysis of bone mass and bone metabolism in growth hormone deficient adult men No Title. B
- Baum, H. B. Katznelson, LSherman, J C a et al. Effects of Physiological Growth Hormone (GH) Therapy on Cognition and Quality of Life in Patients with Adult- Onset GH Deficiency. Endocrinol. Metab. 83, 3184–3189 (1998).
- Pavlovic, D. Pekic, S. Stojanovic, M. et al. Chronic cognitive sequelae after traumatic brain injury are not related to growth hormone deficiency in adults. Eur. J. Neurol. 17, 696–702 (2010).
- High, W. M. Briones-Galang, M Clark, Jet al. Effect of growth hormone replacement therapy on cognition after traumatic brain injury. J. Neurotrauma27, 1565–1575 (2010).
- Jose M Garcia, Beverly M K Biller, Márta Korbonits et al. Macimorelin as a Diagnostic Test for Adult GH Deficiency. J Clin Endocrinol Metab 2018 Aug 1;103(8): 3083-3093.
- Adda Grimberg, Sara A DiVall, Constantin Polychronakos et al. Guidelines for Growth Hormone and Insulin-Like Growth Factor-I Treatment in Children and Adolescents: Growth Hormone Deficiency, Idiopathic Short Stature, and Primary Insulin-Like Growth Factor-I Deficiency. Horm Res Paediatr. 2016;86(6): 361-397.
- Abs, R. Feldt-Rasmussen, U. Mattsson, A F. et al. Determinants of cardiovascular risk in 2589 hypopituitary GH-deficient adults – A KIMS database analysis. Eur. J. Endocrinol. 155, 79–90 (2006).
- Ueland, T. Götherström, G. Bosæus, I et al. Effects of 12 months of GH treatment on cortical and trabecular bone content of IGFs and OPG in adults with acquired GH deficiency: A double-blind, randomized, placebo-controlled study. J. Clin. Endocrinol. Metab. 87, 2760–2763 (2002).
- Nilsson, a G. Effects of growth hormone replacement therapy on bone markers and bone mineral density in growth hormone-deficient adults. Horm. Res. 54 Suppl 1, 52–57 (2000).
- Appelman-Dijkstra, N. M., Claessen, K. M. J. a, Roelfsema, F., Pereira, A. M. & Biermasz, N. R. Long-term effects of recombinant human GH replacement in adults with GH deficiency: A systematic review. Eur. J. Endocrinol. 169, (2013).
- Elbornsson, M., Gotherstrom, G., Bosaeus, I., Bengtsson, B. -A., Johannsson, G., & Svensson, J. (2012). Fifteen years of GH replacement increases bone mineral density in hypopituitary patients with adult-onset GH deficiency. European Journal of Endocrinology, 166(5), 787–795.
- Kołtowska-Häggström, M., Jonsson, B., Isacson, D. & Bingefors, K. Using EQ-5D to derive general population-based utilities for the Quality of Life Assessment of Growth Hormone Deficiency in Adults (QoL-AGHDA). Value Heal. 10, 73–81 (2007).
- Elbornsson, M. Götherström, G. Bosæus, I. et al. Fifteen years of GH replacement improves body composition and cardiovascular risk factors. Eur. J. Endocrinol. 168, 745–753 (2013).
- Gaillard, R. C. Mattsson, A F. Åkerblad, Ann Cet al. Overall and cause-specific mortality in GH-deficient adults on GH replacement. Eur. J. Endocrinol. 166, 1069–1077 (2012).
- Van Bunderen, C. C van Nieuwpoort, I Caroline A, Lucia I et al. Does growth hormone replacement therapy reduce mortality in adults with growth hormone deficiency? Data from the Dutch National Registry of Growth Hormone Treatment in adults. J. Clin. Endocrinol. Metab. 96, 3151–9 (2011).
- Jean-Claude Carel, Emmanuel Ecosse, Fabienne Landier, Djamila Meguellati-Hakkas, Florentia Kaguelidou, Grégoire Rey, Joël Coste. Long-term mortality after recombinant growth hormone treatment for isolated growth hormone deficiency or childhood short stature: preliminary report of the French SAGhE study. J Clin Endocrinol Metab. 2012 Feb;97(2): 416-25.
- Anthony J Swerdlow, Rosie Cooke, Dominique Beckers et al. Cancer Risks in Patients Treated with Growth Hormone in Childhood: The SAGhE European Cohort Study. J Clin Endocrinol Metab. 2017 May 1;102(5): 1661-1672
- Kirstine Stochholm, Wieland Kiess. Long-term safety of growth hormone-A combined registry analysis. Clinical Endocrinol (oxf). 2018 Apr;88(4): 515-528
.
Télécharger le PDF de cet article