Le syndrome de Chediak-Higashi (SCH) est une maladie génétique autosomique récessive rare, caractérisée par un albinisme oculo-cutané, une immunodéficience de l’activité cytotoxique des lymphocytes T et des cellules NK, responsables d’infections récurrentes, une prédisposition aux saignements et une détérioration neurologique tardive.
Khelafi, MS. Ladj, S. Benali Khodja, R. Belbouab, M. Noumi, S. Sokhal, R. Boukari, Service Pédiatrie CHU Mustapha, Alger
Date de soumission : 03 Avril 2019.
Résumé : Le syndrome de Chediak-Higashi (SCH) est une maladie génétique autosomique récessive rare, caractérisée par un albinisme oculo-cutané, une immunodéficience de l’activité cytotoxique des lymphocytes T et des cellules NK, responsables d’infections récurrentes, une prédisposition aux saignements et une détérioration neurologique tardive. Selon International Union of Immunological Societies, le SCH est une immunodéficience primaire par dysrégulation immunitaire appartenant aux syndromes d’hématophagocytose lymphohistiocytaires familiale (HLF) avec hypopigmentation [1]. Le gène LYST-CHS1 (Lysosomal Trafficking Regulator Gene) a été identifié sur le bras long du chromosome 1 en 1q42-q43 [2,3]. Ce gène code pour la protéine CHS dont la fonction exacte reste imprécise. Environ 500 cas ont été signalés [4,5]. Le diagnostic est orienté par les signes cliniques et facilité par l’étude de l’aspect microscopique du cheveu qui met en évidence la présence d’agrégats pigmentaires. Mais le signe pathognomonique de la maladie est la présence de granulations intracytoplasmiques géantes dans la plupart des cellules de l’organisme [6], notamment dans le sang périphérique ou la moelle osseuse. Environ 85% des patients développent une phase d’accélération caractérisée par un syndrome d’hémophagocytose lymphohistiocytaire (HLH), qui survient au cours de la première décennie, rarement présent au début de la maladie [7]. Elle est fatale en l’absence de traitement [8]. Actuellement, la seule option thérapeutique efficace est la greffe de moelle osseuse, qui améliore les anomalies hématologiques et immunitaires, mais n’empêche pas la détérioration neurologique ultérieure. Le pronostic reste mauvais en l’absence de greffe de moelle osseuse, le décès survenant souvent avant l’âge de dix ans.
Mots clés : syndrome de Chediak-Higashi, dysrégulation immunitaire, LYST-CHS1, albinisme oculo-cutané, immunodéficience, détérioration neurologique, HLH, greffe de moelle osseuse.
Abstract: Chediak-Higashi syndrome (SCH) is a rare autosomal recessive genetic disorder characterized by oculo-cutaneous albinism, immunodeficiency by cytotoxic activity of T lymphocytes and natural killer cells responsible for recurrent infections, predisposition to bleeding, and late neurological deterioration. According to the International Union of Immunological Societies, the SCH is a primary immunodeficiency by immune dysregulation belonging to familial lymphohistiocytic haematophagocytosis syndromes (HFH) with hypopigmentation [1]. The LYST-CHS1 (Lysosomal Trafficking Regulator Gene) gene was identified on the long arm of chromosome 1 in 1q42-q43 [2,3]. This gene encodes the CHS protein whose exact function remains imprecise. About 500 cases have been reported [4,5]. The diagnosis is oriented by the clinical signs and facilitated by the study of the microscopic aspect of the hair which highlights the presence of pigment aggregates. But the pathognomonic sign of the disease is the presence of giant intracytoplasmic granulations in most cells of the organism [6], especially in peripheral blood or bone marrow. Approximately 85% of patients develop an acceleration phase characterized by a syndrome of lymphohocytic hemophagocytosis (HLH), which occurs during the first decade, rarely present at the onset of the disease [7]. It is fatal in the absence of treatment [8]. Currently, the only effective therapeutic option is bone marrow transplantation, which improves haematological and immune abnormalities, but does not prevent subsequent neurological deterioration. The prognosis remains poor in the absence of a bone marrow transplant, the death often occurring before the age of ten years.
keywords: Chediak-Higashi syndrome, immune dysregulation, LYST-CHS1, oculo-cutaneous albinism, immunodeficiency, neurological deterioration, HLH, bone marrow transplantation.
Introduction
Le syndrome de Chediak-Higashi (SCH) est une maladie génétique autosomique récessive rare caractérisée par un albinisme oculo-cutané, une immunodéficience par défaut d’activité cytotoxique des lymphocytes T et des cellules natural killer, responsable d’infections récurrentes, une prédisposition au saignement, et une détérioration neurologique tardive. Selon l’International Union of Immunological Societies, le SCH est un déficit immunitaire primaire par dysrégulation immunes faisant partie des syndromes d’hematophagocytoses lymphohistiocytaires familiaux (FHL) avec hypopigmentation (1).
Le gène LYST-CHS1 (Lysosomal Trafficking regulator gene) de la maladie a été identifié 1996 sur le bras long du chromosome 1 en 1q42-q43 (2 ,3). Ce gène code pour la protéine CHS dont la fonction exacte reste imprécise.
Environ 500 cas ont été rapportés (4,5). Le diagnostic est orienté par les signes cliniques et facilité par l’étude de l’aspect microscopique des cheveux qui met en évidence la présence d’agrégats de pigment ; mais le signe pathognomonique de la maladie est la présence de granulations intracytoplasmiques géantes dans la plupart des cellules de l’organisme (6), surtout au niveau du sang périphérique ou de la moelle osseuse. Environ 85% des patients développent une phase d’accélération caractérisée par un syndrome d’hémophagocytose lymphohistiocytaire (HLH), qui survient pendant la première décennie, rarement présente au début de la maladie (7) ; elle est mortelle en l’absence de traitement (8).
Actuellement, la seule option thérapeutique efficace est une greffe de moelle osseuse, qui améliore les anomalies hématologiques et immunitaires, mais n’empêche pas la détérioration neurologique ultérieure. Le pronostic reste mauvais en l’absence de greffe de moelle, le décès survenant souvent avant l’âge de dix ans.
Patients et méthodes
Il s’agit de quatre enfants suivis dans le service de pédiatrie du CHU Mustapha d’Alger pour syndrome de Chediak-Higashi entre 2014 et 2017. Le diagnostic a été porté sur les manifestations cliniques, la présence de granulations géantes intra-leucocytaires. Une étude génétique faite chez trois enfants a confirmé le diagnostic.
Résultats
Il s’agit de trois garçons et une fille dont l’âge au diagnostic est en moyenne de 3,2 ans (7 mois – 6 ans). Une consanguinité est retrouvée dans tous les cas ainsi qu’une forme familiale (2 frères). Trois patients ont des antécédents d’infections répétées, de fièvre inexpliquée avec un décès par septicémie dans la forme familiale. Les enfants qui ont été vaccinés n’ont pas présenté d’incidents particuliers. Au plan clinique, l’albinisme oculo-cutané est présent chez 3 enfants et une mélanodermie avec un iris très pigmenté dans 1 cas (Tableau 1).
Le frottis sanguin périphérique a permis de faire le diagnostic en montrant les granulations géantes intra-cytoplasmiques chez tous les patients. L’étude microscopique des cheveux a retrouvé des dépôts de mélanine en mottes irrégulières au niveau de la tige pilaire en faveur du syndrome de Chediak-Higashi. Un syndrome d’hémophagocytose lymphohistiocytaire (HLH) (Tableau 2) est présent dans 3 cas puis dans un cas 8 mois après le diagnostic.
Deux enfants ont présenté des signes neurologiques à type de convulsions et de nystagmus horizontal associé à une dyslexie dans un cas. Le diagnostic d’activation lymphohistiocytaire (HLH) est porté selon les critères de la Hystiocyte Society 2004, confirmant la phase accélérée de la maladie. L’étude de l’activité des lymphocytes NK est abaissée dans un cas. La confirmation génétique a été faite chez 3 patients (haplotype homozygote dans 2 cas et partiellement homozygote dans un cas). Le LCR est normal chez tous les patients et un patient a eu une sérologie EBV positive a IgM (VCA). Tous les patients avec des signes d’activation lymphohistiocytaire ont été mis protocole HLH 2004. L’évolution est marquée par une bonne tolérance du traitement. Un enfant a bénéficié d’une greffe de moelle allogénique, mais est décédé quelques mois après, les trois autres patients sont toujours vivants et stables sur le plan clinique.
Tableau 1 : Données cliniques, cytologique et génétiques
|
|
Cas 1
|
Cas 2
|
Cas 3
|
Cas 4
|
|
Âge
|
7 mois
|
6 ans
|
2 ans
|
3,5 ans
|
|
Sexe
|
♂
|
♂
|
♀
|
♂
|
|
Consanguinité
|
+
|
+
|
+
|
+
|
|
Infections répétées
|
+
|
+
|
+
|
+
|
|
Cas familial
|
+
|
+
|
–
|
–
|
|
Albinisme cutané
|
Présent
|
Présent
|
Présent
|
Mélanodermie
|
|
Cheveux gris argentés
|
+
|
+
|
+
|
+
|
|
Albinisme oculaire
|
Absent
|
Hétérochromie
|
Absent
|
Iris très pigmenté
|
|
Photophobie
|
–
|
+
|
–
|
–
|
|
Nystagmus horizontal
|
–
|
–
|
–
|
Présent
|
|
Signes neurologiques
|
–
|
–
|
–
|
+
|
|
Granulations géantes
(Frottis sanguin)
|
+
|
+
|
+
|
+
|
|
Génétique
|
Haplotype homozygote
|
Haplotype homozygote
|
Haplotype partiellement homozygote
|
–
|
|
|
Cas 1
(a 15 mois)
|
Cas 2
|
Cas 3
|
Cas 4
|
|
Fièvre
|
+
|
+
|
+
|
+
|
|
Splénomégalie
|
+
|
+
|
+
|
+
|
|
Signes neurologiques
|
–
|
–
|
–
|
+
|
|
Neutrophiles (/mm3)
|
570
|
700
|
120
|
450
|
|
Hémoglobine (g/dl)
|
5.8
|
9.7
|
9.5
|
8.7
|
|
Plaquettes (/mm3)
|
56 000
|
104 000
|
28 000
|
66 000
|
|
Hémophagocytose (PMO)
|
+
|
+
|
+
|
+
|
|
Fibrinogène (g/l)
|
1.9
|
2.76
|
1.34
|
?
|
|
Triglycérides (g/l)
|
9.5
|
2.28
|
5
|
2.9
|
|
Ferritine (ng/ml)
|
1388
|
512
|
1150
|
977
|
|
Activité NK
|
?
|
normale
|
Abaissée (1%)
|
?
|
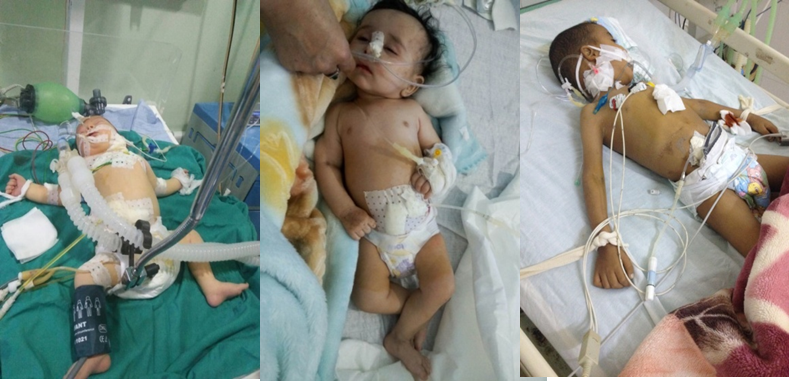
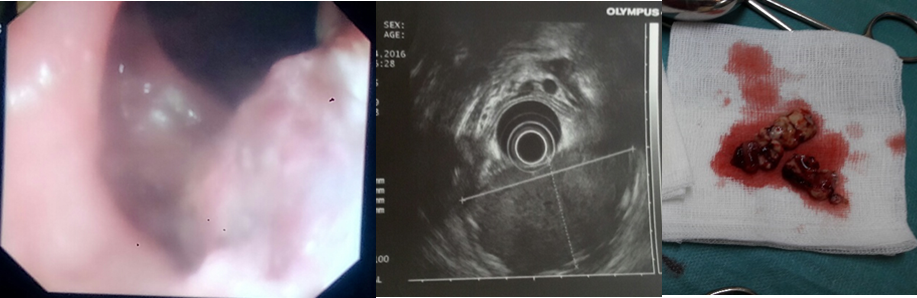
Fig 1 . Un de nos patient avec une hypopigmentation cutanée
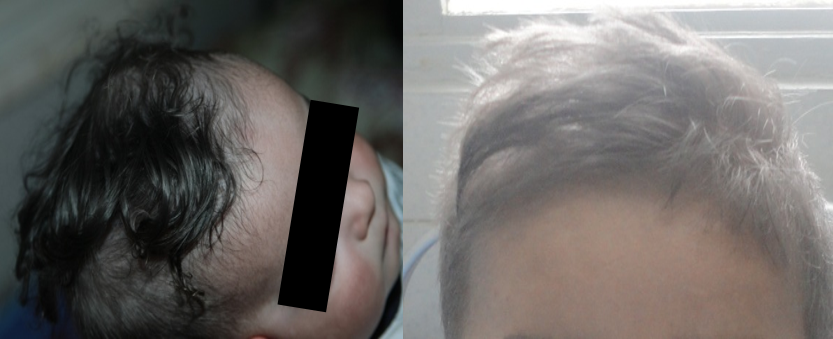
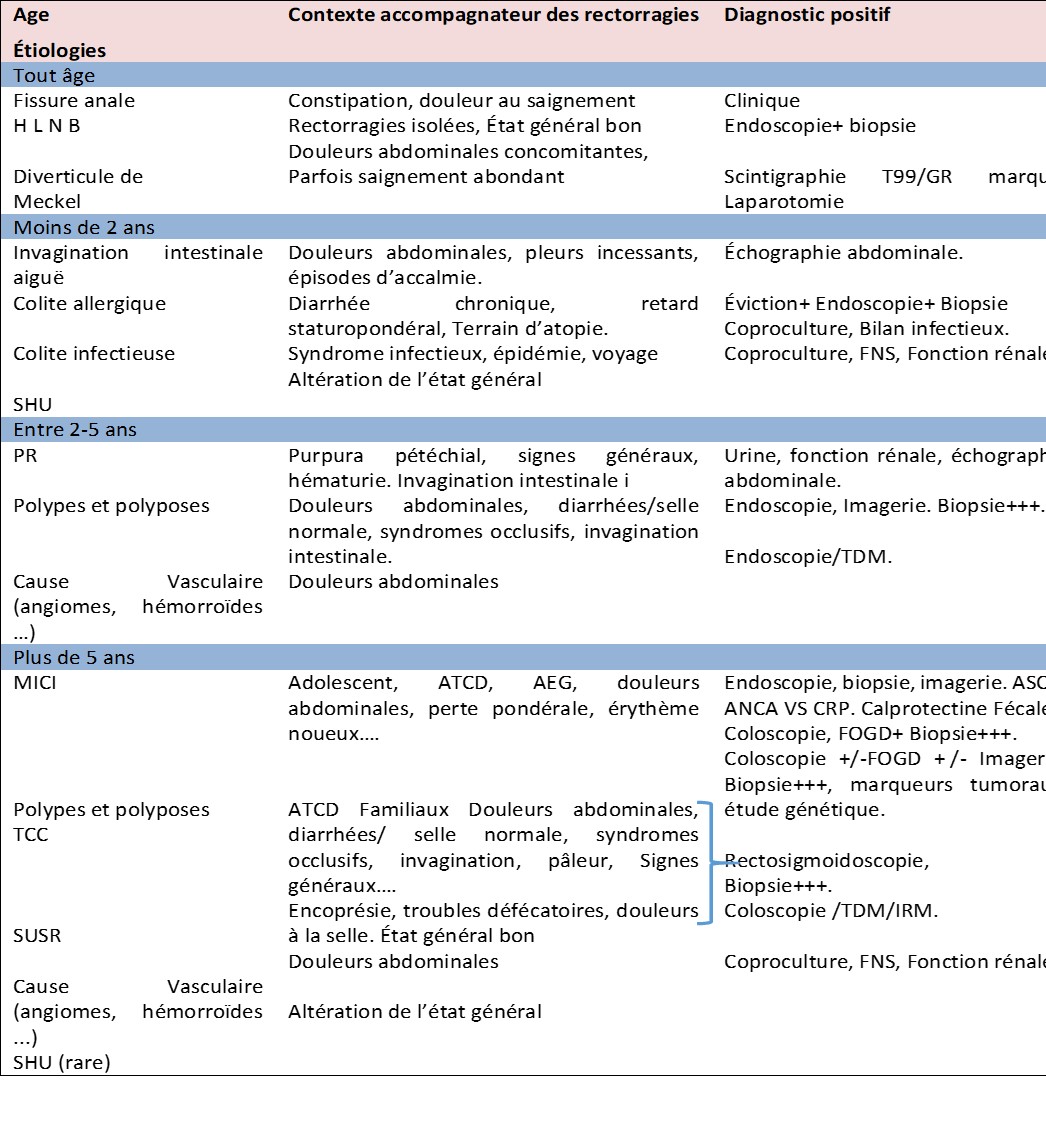
Fig. 2. Deux de nos patients avec un aspect « gris argenté » *des cheveux

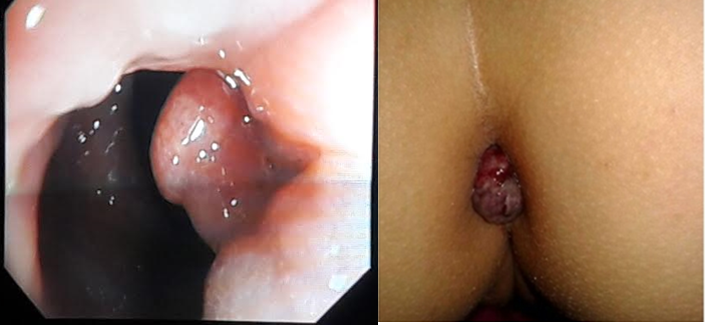
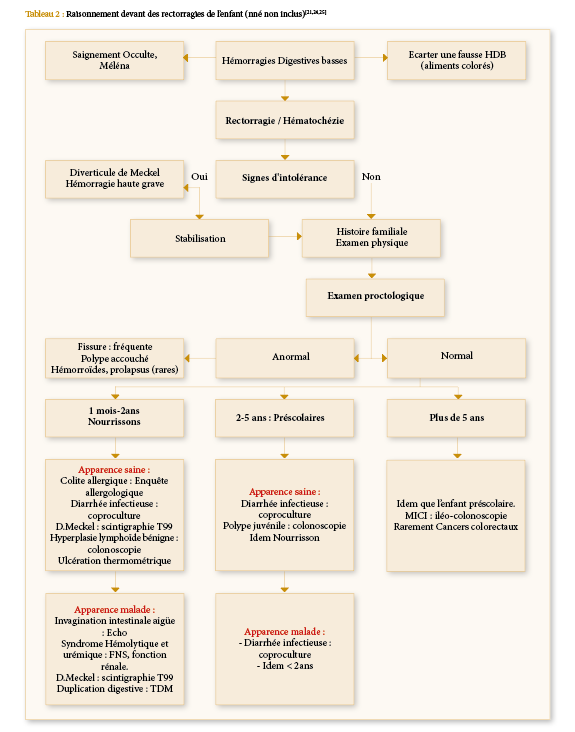
Fig 3. Tige pilaire de l’un de nos patients avec dépôt de mélanine
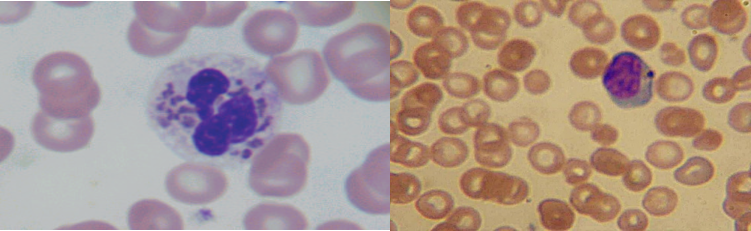
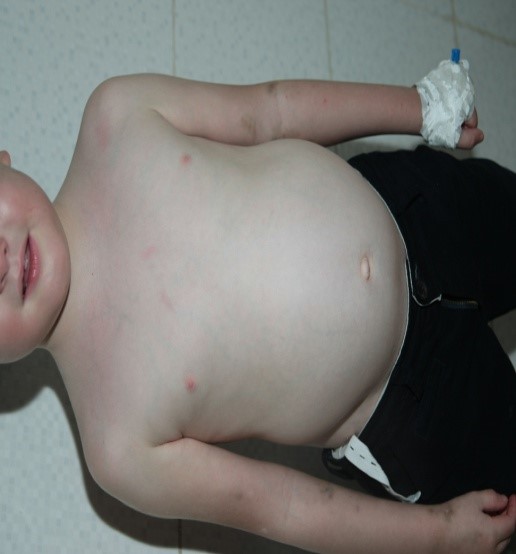
Fig.4 Frottis sanguin : granulations géantes au niveau des leucocytes et lymphocytes (In Indian J Hematol Blood Transfus (Sept 2014) 30(Suppl 1):S223–S226)
Discussion
Maladie génétique autosomique récessive très rare, le syndrome de Chediak-Higashi atteint toutes les races et tous les groupes d’âge. Moins de 500 cas ont été rapportés (4,5). Le plus souvent il s’agit de cas rapportés ou de petites séries publiées. La prévalence est difficile à déterminer en raison des cas rapportés plus d’une fois, et d’autres non déclarés. La plus grande série publiée (15 cas) a été rapportée au Japon sur une période de dix ans (9). Une consanguinité, présente chez tous nos patients, est rapportée dans 50 à 85% des cas (5).
Le gène LYST/CHS1 de la maladie code pour la protéine CHS cytosolique dont la fonction reste imprécise. Elle aurait un rôle dans l’exocytose de protéines à partir des endosomes multivésiculaires tardifs. Plus d’une soixantaine de mutations ont été rapportées dans la littérature (faux-sens, non-sens, délétion, insertions) (10). Les mutations du gène LYST/CHS1 entrainent une fonction anormale de la protéine CHS avec une altération du transport intra cytoplasmique, une séquestration de protéines dans les structures intra cytoplasmiques géantes et le blocage de la fonction sécrétoire notamment celle des leucocytes et des mélanocytes. Des corrélations phénotypes/génotypes ont étés rapportées, ainsi une mutation de type délétion est corrélée avec la survenue précoce et fulminante de la phase d’accélération, alors qu’une mutation de type faux-sens est corrélée avec un meilleur pronostic avec absence de phase d’accélération et de détérioration neurologique (11).
Les signes cliniques débutent après la naissance, ou avant l’âge de 5 ans. L’albinisme oculo-cutané (AOC) est un signe important d’orientation diagnostique, présent chez trois de nos patients (Fig.1), il se caractérise par une hypopigmentation qui affecte généralement la peau, les cheveux et les yeux. Elle est en rapport avec l’agrégation pathologique et une répartition inégale des mélanosomes.
L’AOC peut être présent dès la naissance, et concerner les trois organes ou certains d’entre-deux, total ou partiel ou même être absent (12). Parfois une hyperpigmentation comme chez l’un de nos patients peut exceptionnellement être vue, faisant retarder le diagnostic (13,14). La plupart des patients présentent une photosensibilité. Certains patients ont un phénotype atypique avec une forme atténuée où l’AOC est subtile, voire absent, et qui sont probablement méconnues (11,15). Rarement d’autres lésions cutanées sont observées comme une hyperhydrose, un érythème polymorphe.
Au niveau des cheveux l’hypopigmentation leur donne une couleur blonde, grise (Fig. 2) ou blanche, souvent avec un éclat argenté ou métallique. Les yeux sont de couleur bleue et l’hypopigmentation de l’iris peut être associée à une diminution de la pigmentation de la rétine, et des manifestations oculaires comme la photophobie, diminution de l’acuité visuelle, nystagmus, et le strabisme.
Les infections fréquentes et récurrentes sont habituelles pendant l’enfance. Souvent graves, elles sont en rapport avec un défaut de cytotoxicité des lymphocytes T, de la fonction NK, et une diminution de l’activité chimiotactique et bactéricide des granulocytes (16,17). Les infections pyogènes sont les plus fréquentes, en particuliers au niveau de la peau, des voies aériennes supérieures et des muqueuses. Les germes les plus souvent isolés sont le staphylocoque aureus, streptocoques ß-hémolytiques et le pneumocoque.
Les atteintes de la cavité buccale, à type de gingivite, hémorragie gingivale, chute précoce des dents, aphtes, et ulcérations buccales ont été décrites. La parodontite a été identifiée comme une manifestation de la dysfonction immunitaire.
La tendance au saignement chez ces patients est en rapport avec un déficit du pool de stockage des granules denses et un défaut d’agrégation des plaquettes. Les manifestations hémorragiques sont en général bénignes et ne nécessitent habituellement pas de traitement.
La phase accélérée de la maladie représente la complication la plus importante et dangereuse du SCH. Elle est responsable d’un taux élevé de moralité en quelques mois (18). Elle peut survenir à tout âge mais surtout durant la première décennie (85%) (19).
Nos patients ont développé cette phase accélérée avant l’âge de 6 ans. Rarement, elle en est la première manifestation (7,20). Sa survenue précoce est associée à l’existence d’une mutation génétique de type délétion (11) ; et a une activité effondrée ou absente des LT cytotoxiques (21).
Elle se manifeste par un syndrome d’hématophagocytose lymphohystiocytaire (HLH) dont les facteurs déclenchant cette accélération ne sont pas clairs. Le rôle de l’infection à EBV retrouvé chez l’un de nos patients a été soulevé sans que cette relation ne soit établie (22). Le diagnostic de la maladie de la phase accélérée repose sur les critères de la Histiocytic Society 2004 (23).
Les manifestations neurologiques surviennent dans environ 50% des cas et peuvent apparaître à tout moment dans l’enfance ou à l’âge adulte, ils sont variables : neuropathie périphérique, coma, convulsions, ataxie, troubles cognitifs, troubles de l’équilibre, mouvements anormaux et altérations mentales. La greffe de moelle osseuse n’empêche pas leur apparition ultérieure (24).
Le diagnostic de SCH doit être précoce, souvent fait aux environs de 6 ans mais dans environ 25% des cas le diagnostic est tardif après l’âge de 10 ans (12), pour nos patients. La moyenne d’âge au diagnostic était de 3,2 ans. Il est suspecté sur les éléments cliniques, facilité par l’étude microscopique des cheveux (Fig.3) qui montre des agrégats de pigments de mélanine retrouvés chez tous nos patients, cet aspect permet le diagnostic différentiel avec d’autres types d’hypopigmentation cutanée. Mais le signe pathognomonique de la maladie est la présence de granulations géantes intracytoplasmiques (Fig.4) dans la plupart des cellules de l’organisme (6) mais souvent elles sont identifiées au niveau du sang périphérique comme ce fut le cas de nos patients ; ou au niveau de la moelle osseuse. Le diagnostic est confirmé par un test génétique de recherche de la mutation LYST. Le diagnostic anténatal de la maladie est possible au niveau des cellules des villosités choriales, liquide amniotique, leucocytes du cordon fœtal (25).
Comme pour la maladie de Chediak-Higashi, d’autres déficits immunitaires génétiques s’accompagnent d’un albinisme oculo-cutané partiel, telles que la maladie de Griscelli et le syndrome Hermansky-Pudlak. La distinction peut se faire sans ambiguïté par l’aspect différent des amas de pigment dans la gaine du cheveu, beaucoup plus fins dans le cas du CHS, et surtout par la présence des granulations géantes intracytoplasmiques observées uniquement dans le CHS. Toutefois, dans certains cas de leucémies myéloïdes, on peut voir des granulations géantes dites anomalie pseudo-Chediak-Higashi (26).
Le traitement de la maladie de Chediak-Higashi est multidisciplinaire et repose sur la prise en charge des complications de la maladie, le traitement de la « phase accélérée » ou HLH et surtout la greffe de cellules souches hématopoïétiques. Le traitement symptomatique de la maladie de Chediak-Higashi repose sur une antibiothérapie efficace contre les infections et des transfusions de dérivés du sang pour lutter contre l’anémie et les complications hémorragiques. Les troubles oculaires doivent être corrigés. Les yeux et la peau doivent être protégés des rayons UV. Les vaccinations sont généralement bien tolérées comme ce fut le cas pour nos patients, et doivent être faites. L’hygiène et les soins buccodentaires sont primordiaux. La survenue de symptômes neurologiques et leur progression doivent être pris en charge assez tôt par un spécialiste en réadaptation. En cas de phase d’accélération (HLH, un traitement combinant corticoïdes, VP16, cyclosporine et des injections intrathécales de MTX (protocole HLH 2004) (23) est instauré pour obtenir une rémission. Celle-ci survient dans 75% des cas dans les huit semaines (27), mais les rechutes sont fréquentes et la réponse au traitement diminue au fil du temps. Une fois la rémission obtenue la greffe est recommandé. Chez les patients SCH avec HLH par EBV l’adjonction du Rituximab pourrait améliorer le traitement (28). En cas de HLH réfractaire, une autre option thérapeutique incluant un anticorps monoclonal l’anti CD52 (Alemtuzumab) (29) est possible comme traitement de seconde ligne avant la greffe de moelle osseuse.
La greffe allogénique de moelle osseuse (GMO) est le seul traitement efficace actuel qui guérit les anomalies hématologiques et immunologiques, mais sans résultats sur l’albinisme oculo-cutané ni sur la détérioration neurologique ultérieure (9,24,30) . Le régime de conditionnement pré-greffe comprend une combinaison d’étoposide, le busulfan, le cyclophosphamide (31). La réduction d’intensité du conditionnement pré-greffe avec fludarabine, melphalan, et alemtuzumab a entrainé une augmentation de la survie dans le HLH primaire ou forme familiale avec une plus faible toxicité (32,33). La greffe de moelle osseuse est d’autant plus efficace qu’elle est effectuée avant la survenue de la phase accélérée (31). Les patients ayant une diminution profonde de la fonction cytotoxique des lymphocytes T (CTL) ont un risque élevé de développer un syndrome d’activation lymphohistiocytaire (HLH), aussi leur dépistage peut constituer une indication à une greffe de moelle précoce (21). Le taux de survie globale après une greffe de moelle est 60-70% (30-32).
Le pronostic reste mauvais en l’absence de greffe de moelle, le décès survient fréquemment durant la première décennie par infections ou développement d’un HLH de la phase accélérée (34).
Environ 10% des patients survivent à la petite enfance, et développeront des troubles neurologiques graves à l’adolescence et au début de l’âge adulte.
Conclusion
Le syndrome de Chediak-Higashi est une maladie rare, dont le diagnostic est suspecté chez un enfant présentant un albinisme oculo-cutané avec des infections récurrentes. La majorité des formes cliniques sont précoce “infantiles” mortelles en l’absence de traitement. Une minorité de patients présentent une forme “atténuée” de la maladie survivent après l’enfance mais développent une maladie neuro-dégénérative associée. Dans tous les cas un diagnostic précoce doit être posé par un examen simple, le frottis de sang périphérique qui montre la présence de granulations géantes intracytoplasmiques pathognomonique de cette affection. Le seul traitement actuel efficace des anomalies hématologiques et immunologiques reste la greffe de moelle osseuse allogénique, mais sans impact sur les manifestations cutanées ou la détérioration neurologique ultérieure. Elle est d’autant plus efficace qu’elle est réalisée avant la survenue d’un syndrome HLH. En cas de d’apparition de la phase accélérée (HLH), un traitement selon le protocole HLH 2004 est instauré afin d’obtenir une rémission avant la greffe de moelle osseuse.
Le pronostic de la forme infantile est mauvais, le décès survenant fréquemment dans la première décennie de la vie par infections ou développement de HLH. La recherche de facteurs prédictifs de développement de HLH pourrait aider à poser l’indication d’une greffe de moelle osseuse précoce.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Bibliographie
- Picard C,Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME et al – Primary Immunodeficiency Diseases: an Update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol 2015 Nov;35(8):696-726).
- Barbosa MDFS et al – Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature 1996, 382:262–265
- Nagle DL et al – Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet 1996, 14:307–311
- Kaplan J, De Domenico I, Ward DM – Chediak-Higashi syndrome. Curr Opin Hematol 2008, 15:22–29
- Reddy RR, Babu BM, Venkateshwaramma B, Hymavathi Ch – Silvery hair syndrome in two cousins: Chediak-Higashi syndrome vs Griscelli syndrome, with rare associations. Int J Trichology 2011; 3(2):107-11
- C. Stinchcombe, L. J. Page, et G. M. Griffiths – Secretory lysosome biogenesis in cytotoxic T lymphocytes from normal and Chediak Higashi syndrome patients Traffic 2000; 1(5):435-444)
- Pooja Jaiswal, Yogesh Kumar Yadav, Nilam Bhasker, Rashmi Kushwaha – Accelerated Phase of Chediak-Higashi Syndrome at Initial Presentation: A Case Report of an Uncommon Occurrence in a Rare Disorder Journal of Clinical and Diagnostic Research. 2015 Dec, Vol-9(12): ED13-ED14)
- Introne WJ, Westbroek W, Golas GA, Adams D: Chediak-Higashi syndrome. In Gene Reviews™. Edited by Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K. Seattle (WA): University of Washington, Seattle; 2009 [updated 2012] [http://www.ncbi.nlm.nih.gov/books/NBK5188/]
- Nagai K, Ochi F, Terui K, Maeda M, Ohga S, Kanegane H, et al – Clinical characteristics and outcomes of Chediak–Higashi syndrome: a nationwide survey of Japan. Pediatr Blood Cancer 2013; 60: 1582–6)
- Maria L Lozano*, Jose Rivera, Isabel Sánchez-Guiu and Vicente Vicente Orphanet Journal of Rare Diseases 2014, 9:132
- Westbroek W, Adams D, Huizing M, Koshoffer A, Dorward H, Tinloy B, et al – The severity of cellular defects in Chediak–Higashi syndrome correlate with the molecular genotype and clinical phenotype. J Invest Dermatol 2007; 127: 2674–7
- Barak Y, Nir E. Chédiak-Higashi – Syndrome. Am J Pediatr Hematol Oncol 1987; 9(1):42-55
- Ho M-C, Hsieh Y-T – Mixed hyperpigmentation and hypopigmentation of iris and choroid in Chediak-Higashi syndrome. J AAPOS 2013, 17:558–560
- Pujani M, Agarwal K, Bansal S, Ahmad I, Puri V, Verma D, Pujani M – Chediak Higashi syndrome – a report of two cases with unusual hyperpigmentation of the face. Turkish J Pathol 2011, 27(3):246–248
- Karim MA, Suzuki K, Fukai K, Oh J, Nagle DL, Moore KJ et al – Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak Higashi syndrome.Am J Med Genet. 2002; 108: 16–22.
- Yenan T. Bryceson, Daniela Pende, Andrea Maul-Pavicic, Kimberly C. Gilmour , Heike Ufheil, Thomas Vraetz et al – A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes Blood 2012, 119:2754-2763;
- MC Dinauer -Disorders of Neutrophil Function: An Overview Methods Mol Biol. 2014, 1124, 501-515
- Pullarkat ST – Accelerated phase of Chediak-Higashi syndrome. Blood 2012; 119(1): 5.
- Lozano et al -Towards the targeted management of Chediak-Higashi syndrome Orphanet Journal of Rare Diseases 2014, 9:132.
- Gajendra S, Das RR, Chopra A, Singh A, Seth R – Accelerated Phase at Initial Presentation in Chédiak-Higashi Syndrome: Is It Really Uncommon? Pediatr Hematol Oncol 2014, 31:382–385.
- Birthe Jessen, Andrea Maul-Pavicic, Heike Ufheil, Thomas Vraetz, Anselm Enders, Kai Lehmberg, et al – Subtle differences in CTL cytotoxicity determine susceptibility to hemophagocytic lymphohistiocytosis in mice and humans with Chediak-Higashi syndrome Blood 2011, 118:4620–4629
- Wendy J Introne, Wendy Westbroek, Gretchen A Golas, et David Adams – Chediak HigashiSyndrome Gene Reviews® Last Update: January 15, 2015
- Henter JI, Home A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004 – Diagnostic and therapeutic guidelines for haemophagocytic lymphohistiocytosis. Paediatr Blood Cancer. 2007;48:124-31
- Tardieu M, Lacroix C, Neven B, Bordigoni P, de Saint Basile G, Blanche S, Fischer A – Progressive neurologic dysfunctions 20 years after allogeneic bone marrow transplantation for Chediak-Higashi syndrome Blood 2005;106:40–2.
- Diukman R, Tanigawa S, Cowan MJ, Golbus MS. Prenatal diagnosis of Chédiak-Higashi Syndrome. Prenat Diagn 1992; 12(11): 877-85
- Chang H, Yi QL – Acute Myeloid Leukemia With Pseudo–Chediak-Higashi Anomaly Exhibits a Specific Immunophenotype With CD2 Expression. Am J Clin Pathol 2006; 125: 791-4.
- Filipovich AH. Hemophagocytic lymphohistiocytosis and related disorders.Curr Opin Allergy Clin Immunol. 2006;6:410–5.
- Ogimi C, Tanaka R, Arai T, Kikuchi A, Hanada R, Oh-ishi T: Rituximab and cyclosporine therapy for accelerated phase Chediak-Higashi syndrome. Pediatr Blood Cancer 2011, 57:677–680
- Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, Lee ND, Khan SP, Lawrence J, Mo JQ, Bleesing JJ, Filipovich AH, Jordan MB: Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer 2013, 60:101–109
- Eapen M, DeLaat CA, Baker KS, Cairo MS, Cowan MJ, Kurtzberg J, Steward CG, Veys PA, Filipovich AH: Hematopoietic cell transplantation for Chediak-Higashi syndrome. Bone Marrow Transplant 2007, 39:411–415
- Haddad E, Le Deist F, Blanche S, Benkerrou M, Rohrlich P, Vilmer E, Griscelli C, Fischer A. Treatment ofChediak-Higashi syndrome by allogenic bone marrow transplantation: report of 10 cases Blood 1995, 85: 3328–33.
- Marsh RA, Vaughn G, Kim M-O, Li D, Jodele S, Joshi S, et al – Reduced-intensity conditioning (RIC) significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood 2010, 116:5824–5831.
- Marsh RA, Jordan MB, Filipovich AH: Reduced-intensity conditioning haematopoietic cell transplantation for haemophagocytic lymphohistiocytosis: an important step forward. Br J Haematol 2011, 154:556–563.
- Cooper N, Rao K, Gilmour K, et al. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic Blood 2006: 107: 1233–1236
- Laura Dotta, Silvia Parolini, Alberto Prandini, Giovanna Tabellini, Maddalena Antolini, Stephen F Kingsmore and Raffaele Badolato – Clinical, laboratory and molecular signs of immunodeficiency in patients with partial oculo-cutaneous albinism Orphanet Journal of Rare Diseases 2013, 8:168
Télécharger le PDF de cet article