
Catégorie : Pédiatrie
Dépistage de la surdité chez les nouveau-nés à risque par otoémissions acoustiques provoquées et potentiels évoqués auditifs précoces du tronc cérébral Étude de prévalence, à propos de 231 cas
La carence en fer chez l’enfant avec ou sans anémie Quoi de nouveau ?
Les photodermatoses : démarche diagnostique
Les photodermatoses sont nombreuses. On distingue : les photodermatoses par déficience d’un des moyens de photoprotection naturelle
Hammadi, Service de Dermatologie, Hôpital Central de l’Armée, Aïn Naâdja, Alger.
Date de soumission : 23 Juin 2019.
Résumé : Les photodermatoses sont nombreuses. On distingue : les photodermatoses par déficience d’un des moyens de photoprotection naturelle ; les photosensibilisations endogènes ou exogènes de contact ; les lucites idiopathiques ; les dermatoses révélées ou aggravées par la lumière. Le diagnostic d’une photodermatose repose essentiellement sur l’interrogatoire et l’examen clinique. Accessoirement, l’exploration photobiologique et éventuellement d’autres examens paracliniques permettront de mieux classer l’affection. L’âge du patient est un bon élément d’orientation.
Mots clés : photodermatose, lucite polymorphe, phototoxique, photoallergique, UV, exploration photobiologique
Abstract: Photodermatosis are numerous. We distinguish: photo dermatoses by deficiency of one of the means of natural photoprotection; endogenous or exogenous photosensitization of contact, polymorphous light eruption; skin diseases revealed or aggravated by light.
The diagnosis of a photodermatosis is essentially based on the patient interview and the clinical examination. Then, photobiological testing and possibly other paraclinical examinations will better classify the condition.The age of the patient is a good starting point guiding element.Key words: photodermatosis, polymorphous light eruption, phototoxic, photoallergic, UV, photobiological testing
Introduction. Généralités
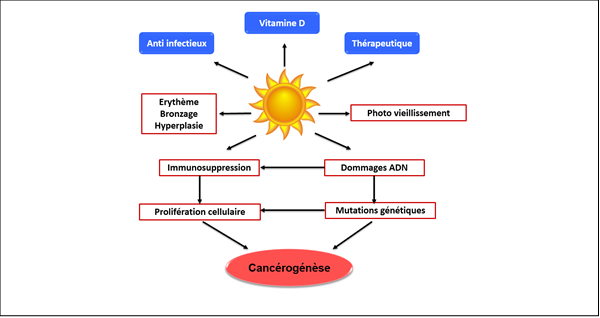
Les effets biologiques du soleil sur la peau sont pour une part, bénéfiques et associent la synthèse de la vitamine D, des effets anti-infectieux vis-à-vis de germes pathogènes et des effets thérapeutiques dans certaines maladies comme le psoriasis. D’autre part, ils peuvent avoir des effets néfastes impliqués dans la cancérogénèse (Fig. 01). Les effets des UV visibles à court terme incluent les coups de soleil et le bronzage, une hyperplasie épidermique, des réponses pro-inflammatoires et une immunodépression.
Les effets biologiques du rayonnement solaire au niveau de la peau sont liés aux plus grandes longueurs d’onde UVB (290-320 nm), et UVA (320-400 nm). La profondeur de pénétration dans la peau est proportionnelle à la longueur d’onde. Ce qui explique que 85% des UVB sont arrêtés par l’épiderme alors que 50% des UVA atteignent le derme [1].
Coup de soleil : Il est surtout induit par les UVB plutôt que les UVA, il apparaît dans les 30 minutes à 8 heures d’une exposition suffisante, le pic est à 12/24 heures, avec diminution progressive de la rougeur et de l’inflammation.
La DEM (dose érythèmale minimale) : la plus faible dose d’UV capable d’induire un coup de soleil qui se traduit cliniquement par un érythème.
Figure 01 : Effets biologiques du soleil [2].
Les photodermatoses regroupent toutes les maladies dans la genèse desquelles le soleil intervient [3].
Le diagnostic d’une photodermatose repose d’abord sur l’interrogatoire et sur l’examen clinique. L’interrogatoire confirme le rôle de la lumière dans le déclenchement de l’éruption.
Il faut préciser les caractéristiques de l’exposition (type et intensité de l’exposition lumineuse), ainsi que le délai d’apparition par rapport à l’exposition, les antécédents personnels et familiaux, les médicaments utilisés et l’application de topiques, l’évolution selon la saison et au cours des années. Les signes fonctionnels tels que le prurit et la sensation de brûlure sont à préciser [4].
La topographie des lésions a une grande valeur d’orientation ; les zones photo-exposées atteintes sont souvent nettement délimitées par les vêtements. Le visage, nuque, décolleté, avant-bras et dos des mains sont les zones les plus atteintes avec respect relatif des régions sous-arénaires et sous-mentonnières, du sillon rétro-auriculaire et de la lisière du cuir chevelu. Le visage peut être respecté dans la lucite estivale bénigne [4].
À noter qu’une extension secondaire aux parties couvertes est possible (mécanisme photo-allergique) mais toujours avec une prédominance lésionnelle aux parties découvertes.
Certains patients peuvent ne pas avoir de manifestations cutanées au moment de la visite et le diagnostic repose sur l’historique, en particulier ceux qui ont une lucite polymorphe ou une urticaire solaire.
L’exploration photobiologique complète éventuellement l’interrogatoire et peut avoir plusieurs buts : confirmer le diagnostic, établir le degré de photosensibilité, déterminer le spectre d’action, évaluer l’efficacité du traitement, suivre l’évolution de la photodermatose dans le temps.
Nous aborderons ici les principaux signes cliniques des photodermatoses les plus courantes.
|
Dermatoses photo-aggravées |
· Lupus érythémateux · Dermatomyosite · Rosacée · Dermatite atopique · Dermite séborrhéique |
|
Dermatoses photo-induites : Photosensibilité induite chimiquement ou par des médicaments |
Exogène : · Médicament par voie générale · Par application de topique · Cosmétique · Végétaux Endogène : · Porphyrie · Érythème pellagroide |
|
Photodermatoses idiopathiques |
· Lucite polymorphe · Prurigo actinique · Hydroa vacciniforme · Dermatite actinique chronique · Urticaire solaire |
|
Affections héréditaires caractérisées par un défaut de réparation de l’ADN ou une instabilité chromosomique |
· Xerodermapigmentosum · Syndrome de Cockayne · Syndrome de Bloom · Syndrome de Rothmund-Thomson |
Tableau 01 : Classification des photodermatoses. Classification des photodermatoses (tableau 01) [4].
Dermatoses photo-aggravées
Certains lupus sont très photosensibles (lupus subaigu) et peuvent prendre l’aspect d’une lucite polymorphe. La recherche d’anticorps antinucléaires solubles (anti-Ro) et la biopsie avec immunofluorescence permettront de préciser le diagnostic en tenant compte du contexte clinique et biologique [5]. Certaines affections sont volontairement caractérisées par l’aggravation de leurs lésions des zones exposées au soleil comme la dermatomyosite, la rosacée, la dermatite atopique et la dermite séborrhéique.
Dermatoses photo-induites
Exogène : Selon le mécanisme de la photosensibilité, les lésions diffèrent : aspect de fort coup de soleil et sensation de brulure dans les réactions phototoxiques, réaction eczémateuse prurigineuse dans les réactions photo allergique [4].
La photo toxicité résulte des blessures tissulaires et cellulaires directes consécutives à l’activation d’un agent phototoxique par les UV ; elle ressemble à un coup de soleil (Fig. 2).
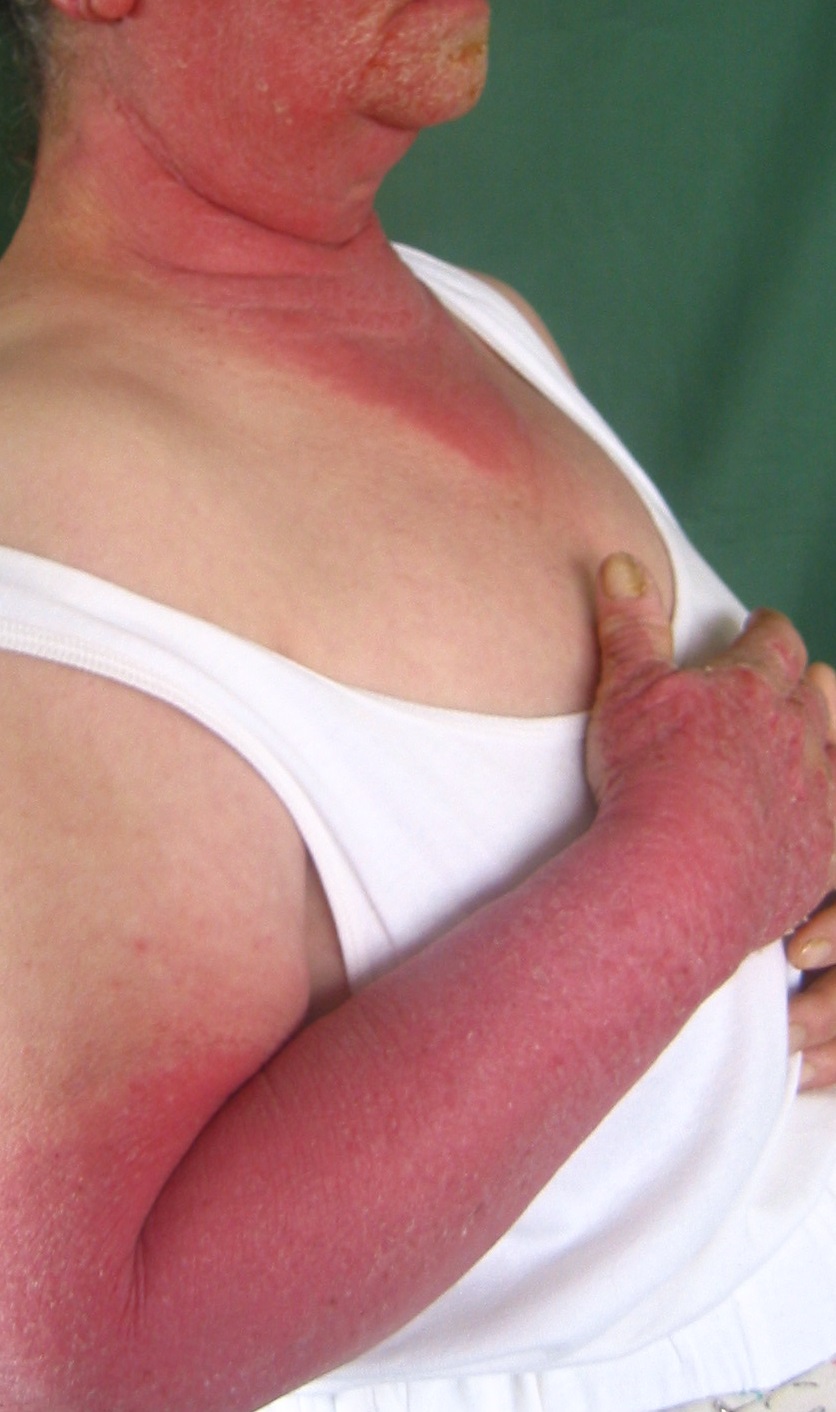
Figure 2 : réaction phototoxique, notant la limite nette de l’érythème
La photoallergie est une réponse par hypersensibilité de type retardé qui nécessite à la fois un photo-allergène et une exposition aux UV ; elle ressemble à un eczéma (Tableau 02).
De nombreuses substances peuvent être responsables, les produits les plus classiques sont : (phénothiazines, cyclines, AINS, quinolones, sulfamides).
Les topiques médicamenteux les plus incriminés restent les AINS (ketoprofene en particulier) et certains filtres solaires (octocrylène) [6]. La phyto-photodermatose est une dermatose qui résulte du contact avec la peau d’une plante (phyton = végétal en grec ancien) et du soleil. C’est une réaction phototoxique, due aux actions associées d’un agent chimique photosensibilisant contenu dans la plante et de la longueur d’onde d’UV (Fig 3).
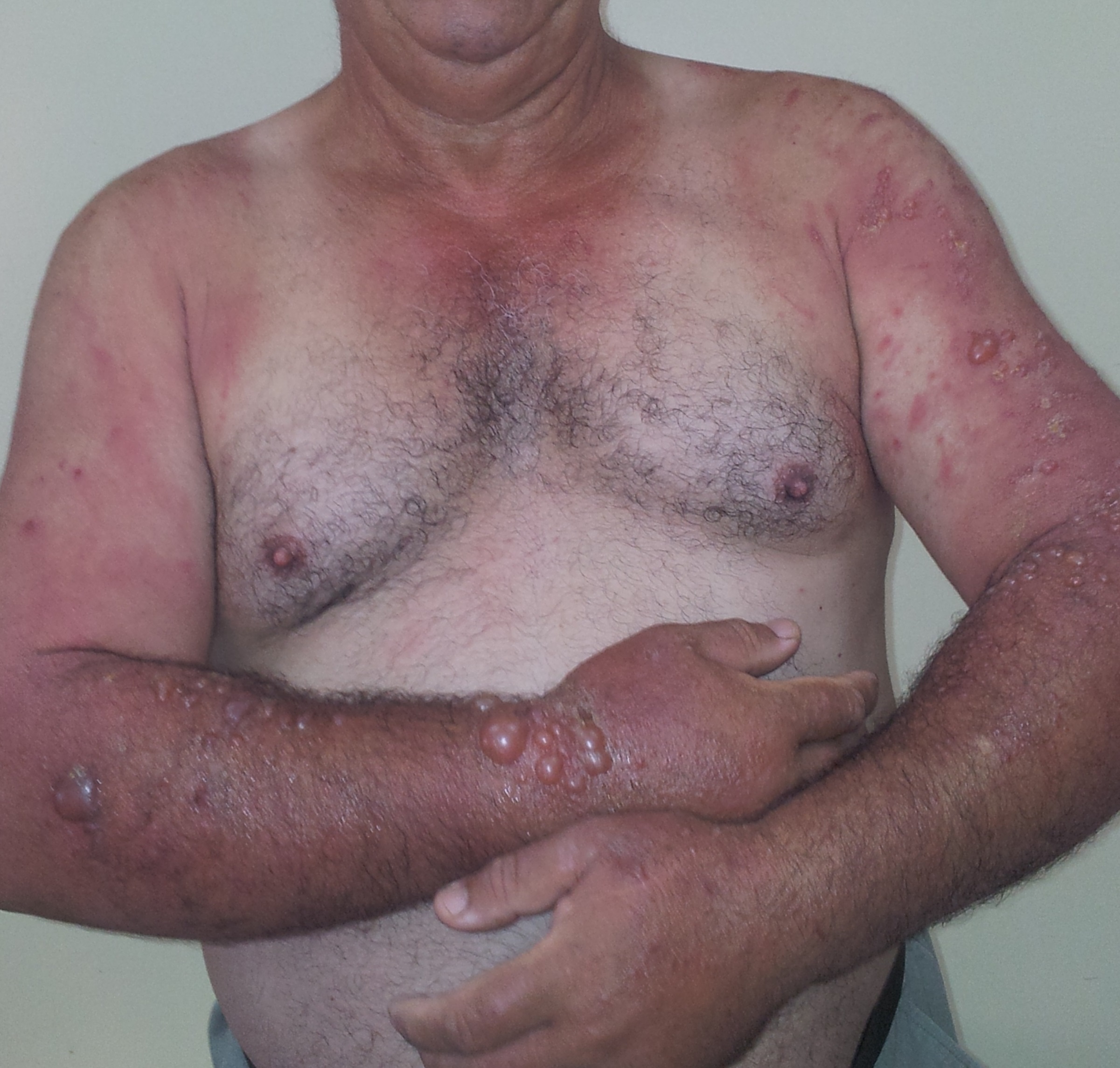
Figure 3 : phytophotodermatose, rougeur initiale et parfois décollements bulleux
Le diagnostic différentiel peut se poser avec une dermite de contact aéroportée.
|
Phototoxicité |
Photo-allergie |
|
|
Troubles pigmentaires |
++, plusieurs semaines ou mois |
+/- |
|
Signes fonctionnels |
Brûlures, sensation de cuisson |
Prurit par fois intense |
|
Exploration |
Photo-patch-tests : – |
Photo-patch-tests : + |
|
Histologie |
Altération épidermique Œdème dermique et vasodilatation |
Spongiose, exocytose. Infiltrat lympho-histiocytaire périvasculaire |
|
Mécanisme |
Toxique |
Immuno-allergique |
|
Évolution |
Disparition après élimination de la drogue photosensibilisante |
Possibilité de photosensibilité persistante ou l’évolution vers une dermatite actinique chronique |
Tableau 02 : comparaison entre la réaction phototoxique et la réaction photoallergique
Endogène : Ce sont troubles métaboliques caractérisés par l’accumulation dans la peau de produits photoactifs à l’origine de réactions phototoxiques.
La liste des médicaments photosensibilisants est longue. Les produits classiques sont : phénothiazines, cyclines, AINS, quinolones, sulfamides, fibrates.
Porphyries : altération du métabolisme des porphyrines (substances qui interviennent dans la synthèse de l’hème).
La photodistribution des lésions de la porphyrie cutanée tardive (fragilité cutanée, vésicules, bulles, cicatrices, grains de milium) peut évoquer une photodermatose ; le dosage des porphyrines urinaires affirmera le diagnostic.
Au cours de la proto-porphyrie érythropoïétique, la photosensibilité débute dans la petite enfance et se traduit par des sensations de brulure des zones découvertes. Les lésions cutanées peuvent être absentes et l’on retrouve parfois de petites cicatrices péribuccales [7].
Le diagnostic se confirme par la recherche de proto-porphyrines dans les hématies.
Troubles du métabolisme du tryptophane (érythème pellagroïde) : L’érythème pellagroïde, se traduisant par des poussées de photosensibilité, aboutissant à un érythème sombre avec atrophie de la peau qui devient hyperpigmentée et couverte de squames grises. Il se voit chez les dénutris, les éthyliques, au cours des syndromes de malabsorption et après prise de médicaments qui entre en compétition avec la vitamine B6 (isoniazide, 6-mercaptopurine, 5-fluorouracile), et plus rarement, au cours des troubles congénitaux du métabolisme du tryptophane (la maladie d’Hartnup). Ils aboutissent à une carence en acide nicotinique (vitamine PP) avec accumulation de chromophores anormaux encore mal identifiés.
L’association à ces signes cutanés de troubles neurologiques et digestifs constitue la pellagre qui est liée à une carence polyvitaminique [4].
Photodermatoses idiopathiques
Groupe d’éruptions des parties découvertes directement en rapport avec l’ensoleillement. Le mécanisme est de type photoallergique mais l’agent photosensibilisant reste inconnu. En dehors de l’urticaire solaire de mécanisme immédiat, les lucites idiopathiques correspondent à une hypersensibilité retardée de type IV dirigée contre divers néoantigènes induits par les UV. L’individualisation des différentes variétés cliniques repose sur des critères cliniques (aspect des lésions, topographie, conditions d’ensoleillement, délai d’apparition, évolution), photobiologiques (Tableau 03) [4].
Affections héréditaires caractérisées par un défaut de réparation de l’ADN ou une instabilité chromosomique
Défaut de l’excision-réparation des nucléotides d’ADN [4] : Xerodermapigmentosum : sur le plan clinique, l’affection est caractérisée par une forte photosensibilité, la survenue précoce de des troubles pigmentaires (lentigines vers l’âge de 2 ans). La gravité de la maladie est liée à l’apparition des carcinomes et des mélanomes cutanés sur les zones exposées au soleil à l’âge de 6-8 ans. Les troubles visuels aggravent le tableau clinique (photophobie, kératite, opacification cornéenne) avec possibilité des tumeurs oculaires.
Syndrome de Cockayne : il existe une photosensibilité sans troubles pigmentaires. Les signes progressifs de vieillissement prématuré sont associés à un nanisme cachectique. L’incidence des tumeurs malignes n’est pas augmentée.
Instabilité chromosomique
Syndrome de Bloom : Il se caractérise par un retard de croissance intra-utérin, un érythème télangiectasique du visage apparaissant dans les premières semaines de la vie, exacerbées par l’exposition solaire. Des taches café au lait sont retrouvées dans la moitié des cas environ. La photosensibilité s’atténue avec l’âge mais l’incidence des tumeurs malignes est augmentée (notamment les leucémies).
Syndrome de Rothmund-Thomson : Apparaît dans les tout premiers mois de vie avec un érythème télangiectasique photodistribué, un œdème et des vésicules sur les joues et la face qui évoluent vers une atrophie. Secondairement, l’éruption s’étend à l’ensemble du visage, au cou, aux membres supérieurs, aux cuisses et aux fesses et prend un caractère poïkilodermique. Le risque accru d’ostéosarcome pendant l’enfance fait la gravité de ce syndrome.
Diagnostic et démarche étiologique d’une photodermatose
Un examen général permet de faire un diagnostic positif facilement en mettant en évidence le rôle de la lumière dans le déclenchement de l’éruption et devant sa distribution qui atteint les parties découvertes.
Le bilan métabolique est demandé en fonction des éléments d’orientation à fin confirmer le trouble métabolique à l’origine d’une photosensibilisation endogène. Un bilan hépatique, martial, une sérologie VIH et du VHC, le dosage de porphyrines dans les urines sont demandés en cas de suspicion de porphyrie cutanée tardive.
En cas de suspicion de lupus, des facteurs antinucléaires, une NFS et plaquettes, une VS, une urée, une créatinine sanguine, un dosage du complément et de la protéinurie des 24 heures peuvent être demandés.
Les explorations photobiologiques sont nécessaires devant toute éruption cutanée évoquant une photodermatose. Elle comprend deux étapes :
Dose érythémale minimal : La détermination de la sensibilité actinique du sujet aux UVB repose sur le calcul de la dose érythémale minimale (DEM), c’est-à-dire de la plus petite dose capable d’induire un érythème couvrant toute la surface d’irradiation à la 24ème heure.
Reproduction expérimentale des lésions : Elle repose sur l’utilisation de la lumière seule ou en présence d’un photosensibilisant : La lecture s’effectue un quart d’heure, 72 heures plus tard, et de manière retardée 15 à 20 jours après le test. Le choix du lieu de testage dépend de la photodermatose suspectée, par exemple la zone habituellement concernée dans une lucite polymorphe et plutôt une zone ordinairement couverte dans l’urticaire solaire. La dose lumineuse administrée dépend aussi de l’affection à reproduire. La réalisation de photo-patch-tests est l’indication de choix pour les réactions photoallergiques.
La réalisation d’une exploration photobiologique demande une heure par jour trois jours de suite, puis une lecture tardive. Ces tests nécessitent d’avoir à disposition un matériel délivrant une lumière artificielle du spectre d’émission proche du spectre lumineux naturel [8]
La biopsie cutanée est utile pour confirmer la positivité d’un phototest ou d’un photo-épidermo-test dans les cas cliniquement douteux.
L’âge du début de la maladie est un élément de diagnostic intéressant et on peut schématiquement distinguer trois circonstances : enfance, âge adulte, sujet âgé [5] :
- Chez l’enfant on évoquera plutôt une dermite printanière juvénile, une lucite polymorphe, un prurigo actinique, une proto-porphyrie érythropoiétique, hydroa vacciniforme (Fig 4).
- Chez l’adulte une lucite polymorphe, une photosensibilisation médicamenteuse ou de contact, une urticaire solaire.
- Chez le sujet âgé une photosensibilisation médicamenteuse, une dermatite actinique chronique (Fig 5).
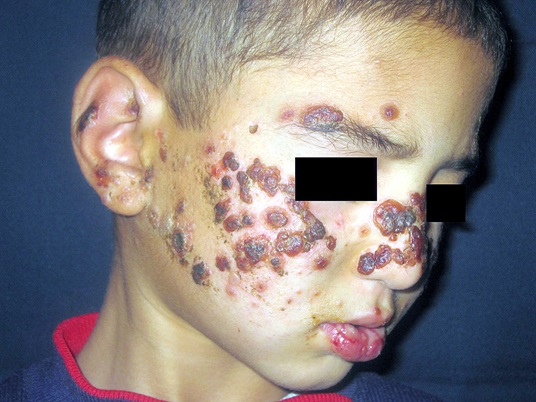
Figure 4 : hydroa vacciniforme chez un enfant
Diagnostic différentiel des photodermatoses [4]
Certains diagnostics sont facilement éliminés :
- Les dermatoses liées à la chaleur et les dermatoses aggravées ou révélées par le soleil à cause du cortège de signes qui les accompagne ;
- Les dermites de contact du visage ;
- Les dermites infectieuses du visage (érysipèle, staphylococcie maligne) ;
En fait, le diagnostic différentiel le plus délicat est l’eczéma aéroporté, car les circonstances de survenue sont identiques et l’éruption peut concerner toutes les parties découvertes. L’analyse sémiologique (en particulier l’atteinte du fond des plis est évocatrice d’une dermatose aéroportée) ; et dans l’interrogatoire permettent de se sortir de ce piège.
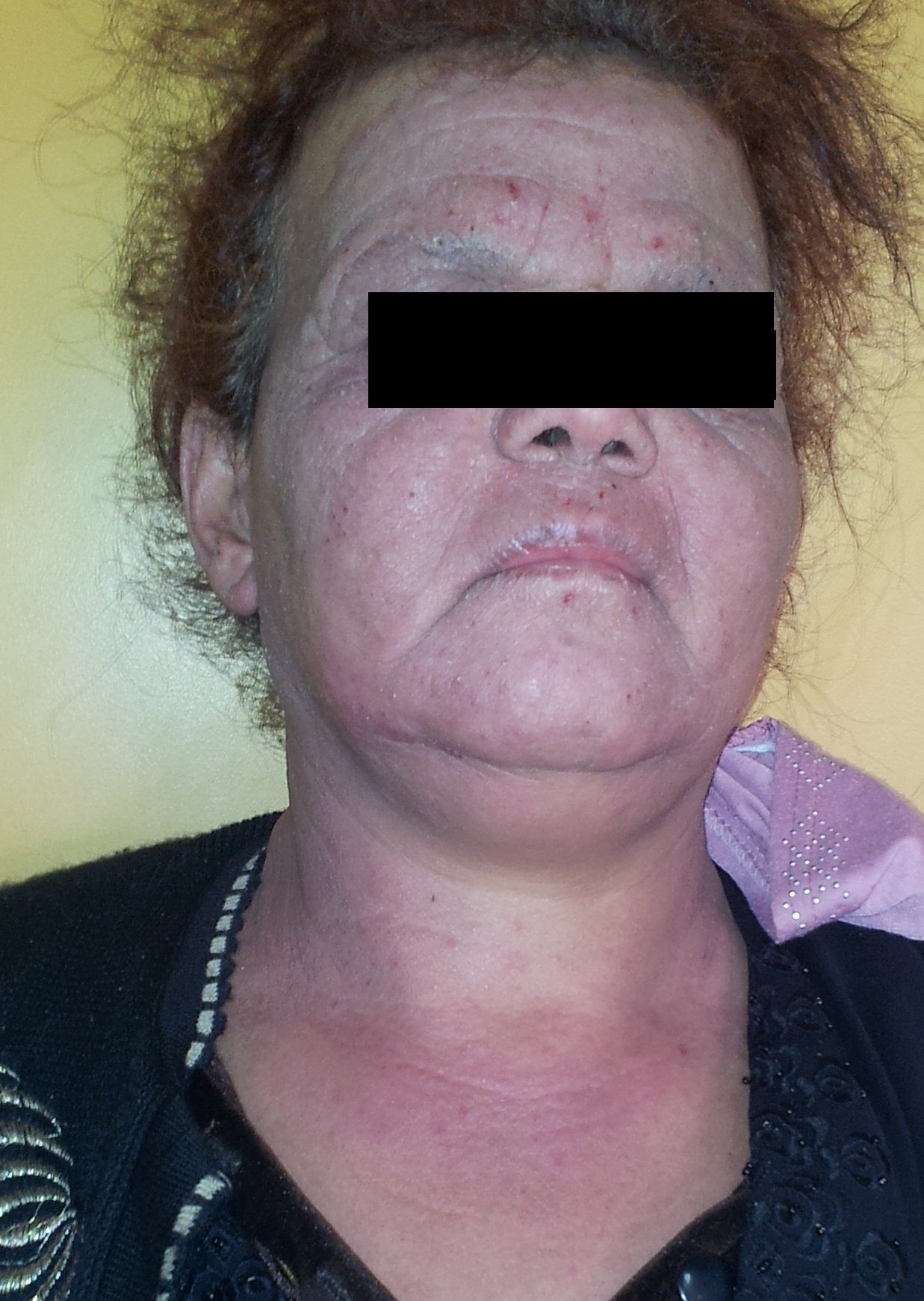
Figure 5 : dermatite actinique chronique
Conclusion
Les photodermatoses regroupent des affections déclenchées par les ultraviolets et aux cours desquelles la peau réagit de manière anormale. Différentes entités sont classées sur des caractéristiques cliniques et d’exploration photobiologique. Les plus fréquentes sont la lucite estivale bénigne et la lucite polymorphe. Le diagnostic d’une photodermatose repose sur l’histoire clinique qui incrimine les UV dans le déclenchement de la maladie, les données de l’examen clinique et de l’exploration photobiologique.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références :
- Diffey BL. Human exposure to UV radiation. In Photodermatology JM Hawk Ed London Arnold 1999
- Bédane, R. Roelandts; Rayonnement ultraviolet : effets biologiques ; Ann Dermatol Venereol ; 2007;134:4S9-4S11.
- Beani JC.Photodermatoses. EMC-Dermatologie 2015;10(1):1-26 [Article98-785-A-10].
- Charles, S. Mouret, J.-C. Beani, M.-T. Leccia ; Peau et soleil Dermatologie et infections sexuellement transmissibles. chapitre 4 © 2017.
- Peyron, J.-L. conduite à tenir devant une photodermatose. Revue Française d’Allergologie. Publié November 1, 2011. Volume 51, Supplement 1. Pages S7-S12. © 2011.
- Avenel-Audran M, Dutartre H, Goossens A, Jeanmougin M, ComteC, Bernier C, et al. Octocrylene: an emerging photo-allergen. Arch Dermatol 2010;146:753-7.
- Bolognia, Jean L., MD; Schaffer, Julie V., MD; Duncan, Karynne O., MD; Ko, Christine J., MD. Dermatologie: l’essentiel. Pages 718-734. © 2018.
- Peyron JL. Diagnostic d’une photodermatose. In: Société française de photo-dermatologie, editor. Photo-dermatologie. 3ème Montrouge: Doin John Libbey Eurotext;2018. p.61-8.
Dermatite atopique : Intérêt de l’éducation thérapeutique

L’éducation thérapeutique s’applique à de nombreuses maladies chroniques, son intérêt est capital dans la dermatite atopique permettant d’améliorer la qualité de vie des patients et de réduire la sévérité de l’eczéma.
S.ZOBIRI, Service de Dermatologie, CHU Mustapha Bacha, Alger
Date de soumission : 23 Juin 2019.
Résumé : L’éducation thérapeutique s’applique à de nombreuses maladies chroniques, son intérêt est capital dans la dermatite atopique permettant d’améliorer la qualité de vie des patients et de réduire la sévérité de l’eczéma.
Mots clés : Dermatite atopique, prurit, chronique, qualité de vie, éducation thérapeutique.
Abstract: Therapeutic education applies to many chronic diseases; its interest is crucial in atopic dermatitis to improve the quality of life of patients and reduce the severity of eczema
Key words: atopic dermatitis, chronic, pruritus, quality of life, therapeutic education.
La dermatite atopique (DA) est une dermatose inflammatoire chronique prurigineuse touchant le plus souvent l’enfant. Sa prévalence connait une augmentation notamment dans les pays industrialisés ou elle atteint 20 à 25% des enfants [1]. En Algérie, une étude faite dans la région de Constantine en 2012 a retrouvé une prévalence de 5,3 % chez les nourrissons [2].
L’âge moyen de survenue se situe entre 7 et 9 mois, les formes de l’adulte sont observées dans 10 % des cas. La physiopathogénie de la DA fait intervenir des anomalies de la barrière cutanée, des anomalies de l’immunité innée et des facteurs de l’environnement sur un terrain génétique particulier [1].
La DA se caractérise cliniquement par des poussées d’eczéma sur un fond de xérose cutanée permanente. Sa prise en charge est difficile du fait de son évolution chronique.
Rappel sur les manifestations cliniques de la dermatite atopique
L’aspect clinique de la DA et la localisation des lésions varient avec l’âge. Chez le nourrisson, avant l’âge de 2 ans, la DA se manifeste par des lésions d’eczéma à type de plaques érythémateuses, œdémateuses à surface vésiculeuse, les vésicules se rompent donnant des lésions suintantes dont l’assèchement aboutit à des croûtes. Ces lésions siègent de façon symétrique sur les zones convexes du visage (front, joues, menton, cuir chevelu) avec un respect des zones médio-faciales.
Après l’âge de 2 ans, l’aspect clinique et la topographie des lésions changent, il s’agit de lésions lichénifiées du fait de leur évolution chronique. La lichenification étant un épaississement de la peau qui prend un aspect pigmenté et fissuraire. Les lésions touchent les zones concaves : plis des coudes, creux poplités, poignets, plis du cou et plis rétro-auriculaires, l’atteinte du visage à cet âge est moins fréquente.
Chez l’adolescent et adulte jeune, en plus des placards de lichénification, on peut retrouver des lésions de prurigo (séropapules prurigineuses), notamment aux membres, une atteinte prédominante au visage et au cou, et même une érythrodermie.
Le diagnostic d’une dermatite atopique est clinique, aucun examen complémentaire n’est utile pour confirmer le diagnostic. Il est aisé devant des antécédents familiaux d’atopie, des lésions d’eczéma sur une topographie évocatrice en fonction de l’âge, un prurit et une xérose cutanée.
L’évolution de la DA est chronique par poussées/rémissions. Les poussées d’eczématisation aiguë avec vésicules et suintement peuvent être déclenchées par divers facteurs : les infections ORL, pulmonaires ou cutanées, la vaccination, les poussées dentaires et les changements d’environnement [3].
Retentissement de la dermatite atopique
Le prurit est un signe fonctionnel constant au cours de la DA, il est insomniant rendant l’enfant irritable, fatigué au cours de sa journée avec un retentissement sur le rendement scolaire. De plus, les lésions cutanées sont souvent visibles sur le visage, les mains ou lorsque l’enfant se déshabille rendant la pratique de sport en collectivité pénible du fait du retentissement psychologique de ces lésions et de la détérioration de l’image du soi et des relations sociales pouvant aller jusqu’à des troubles du comportement et des conduites d’évitement. Le retentissement familial est également majeur au cours de la DA. Il devient alors évident que la DA altère la qualité de vie de l’enfant et son entourage (qualité de vie plus altérée que lors du diabète) [4].
Prise en charge de la DA
Le traitement médical de la DA a pour but de traiter les poussées d’eczéma et d’éviter les récidives, il fait appel aux traitements locaux principalement les dermocorticoïdes appliqués sur les lésions et les émollients étalés sur le reste du tégument pour lutter contre la xérose cutanée [4].
Ceci nécessite une participation active de l’enfant et de son entourage, il doit savoir quand, comment et où appliquer son traitement, il doit comprendre son traitement pour l’appliquer au long cours d’où l’intérêt d’une éducation thérapeutique. Les échecs thérapeutiques sont souvent liés à une mauvaise observance du traitement. La prise en charge doit également aborder les facteurs de l’environnement, la toilette, le sport, l’alimentation et l’habillement. Tous ces points ne peuvent être abordés lors d’une consultation de ville mais nécessitent un temps plus long [5,6].
Éducation thérapeutique
Elle consiste à éduquer le patient et ses parents sur sa maladie et sa prise en charge. Il s’agit d’un transfert de compétences au patient afin de l’aider à mieux gérer sa maladie chronique. Sa place est capitale dans la prise en charge de la DA.
Ce processus d’apprentissage a conduit au concept de l’école de l’atopie qui accueille l’enfant et ses parents au cours d’ateliers éducatifs. Ces ateliers sont animés par un dermatologue, un psychologue et une infirmière et durent en moyenne une heure et demie. Les ateliers peuvent être individuels ou collectifs regroupant en moyenne cinq enfants et leurs parents permettant ainsi des échanges d’expérience entre les différents patients [6].
Des objectifs éducatifs à atteindre sont définis au préalable à savoir, être capable d’adapter les soins de sa peau à son état cutané, être capable de gérer sa douleur, être capable d’effectuer son traitement sans l’aide d’autrui.
Le programme éducatif visant à atteindre ses objectifs comprend 3 étapes (Tableau 1) : le savoir, le savoir-faire et le savoir-être. Comme à l’école, des moyens d’apprentissage sont utilisés pour chaque étape en fonction de l’âge.
Le savoir : cette étape consiste à expliquer les connaissances théoriques sur la maladie, pour commencer l’éducateur peut recueillir les interrogations des parents et des enfants qui sont toujours les mêmes « quelle est la cause de la DA ? », « est-ce que c’est contagieux », « j’ai lu sur internet tous les effets des dermocorticoïdes, est-ce dangereux pour mon enfant ? », « est-ce que ça va guérir ? ». Répondre à ses questions permettra d’animer un débat et d’expliquer plusieurs notions à savoir la notion d’eczéma atopique, la notion de barrière cutanée, la chronicité de l’eczéma, les facteurs aggravants, le terrain génétique, la sécheresse cutanée et les différents traitements.
On expliquera l’importance du maintien de l’intégrité de la barrière cutanée. Des photographies sont utilisées pour permettre à l’enfant de reconnaitre une lésion inflammatoire, une peau sèche, une lésion suintante ou une lésion inhabituelle (surinfection bactérienne ou herpétique) (photo 1).
Photo 1 & 2 : Outils illustrant les différents aspects cliniques de la DA
Un dépliant classeur (photo 2) est utilisé pour expliquer la physiopathologie de la DA avec des notions ludiques, par exemple, l’inflammation est représentée par le feu et le pompier pour l’éteindre n’est autre que le dermocorticoïde qui doit être appliqué dès que l’incendie se déclare, et non pas quelques jours après du fait de la corticophobie des parents ou de leur méconnaissance de la maladie [6] (Photo 3). La corticophobie est abordée afin d’atténuer l’appréhension des parents quant aux effets secondaires des dermocorticoïdes qui sont le plus souvent bien tolérés.

Photo 3 : L’inflammation cutanée illustrée par le feu [10]
La deuxième étape de l’apprentissage est le savoir-faire : l’enfant doit être capable de réaliser ses soins lui-même.
Au cours de cette étape, l’importance d’appliquer une crème hydratante sur l’ensemble du corps est expliquée avec des démonstrations sur l’enfant lui-même ou sur des poupées. L’enfant doit savoir quelle quantité mettre, à quel moment de la journée, et il doit comprendre que c’est capital de le faire tous les jours, même en dehors des poussées car une bonne hydratation cutanée chez l’atopique prévient les poussées d’eczéma. Des coloriages représentant des enfants sont utilisés, on demandera à l’enfant de colorier en rouge les zones où il a de l’eczéma.
Le savoir-faire doit aborder les moyens de lutte contre le prurit qui est souvent invalidant, on proposera des alternatives au grattage comme des massages avec des émollients froids ou avec le dos d’une cuillère, les antihistaminiques plus pour leur effet sédatif, la vaporisation de spray d’eau thermale gardée au froid ou l’application de galets froids sur la peau. Une trousse anti grattage est proposée ainsi au patient [6,7].
La lutte contre le prurit est capitale, car le prurit entretient l’inflammation et favorise les infections cutanées, créant ainsi un cercle vicieux que l’on doit rompre impérativement.
Le savoir être : il s’agit de savoir vivre avec sa maladie, l’expliquer à son entourage, à l’école. L’enfant devra exprimer ses émotions et ses difficultés, ses croyances et son vécu familial. L’aide d’un psychologue est capitale dans cette étape du diagnostic éducatif [8].
L’évaluation des compétences acquises fait partie intégrante de l’éducation thérapeutique, elle sera faite sur l’état clinique du patient, sa qualité de vie ainsi que celle de son entourage, l’intensité du prurit et les troubles du sommeil.
Résultats de l’école de l’atopie : les études ont montré une amélioration du score de sévérité de la DA appelé SCORAD chez 97% des patients bénéficiant d’une éducation thérapeutique ainsi qu’une amélioration de la qualité de vie. Une diminution globale de l’hospitalisation et du coût sont également notés [9].
Éducation thérapeutique en Algérie :
En Algérie, une école de l’atopie a été inaugurée en Avril 2018 par la fondation dermatite atopique (photo 4), le coordinateur national étant le Pr A. Ammar-Khodja. Quatre centres sont actifs à Alger, Oran, Tlemcen et Constantine. Les ateliers se déroulent dans les services de dermatologie de ces régions avec un retour excellent, les enfants et leurs parents sont très satisfaits à la fin de la séance, ils se sentent écoutés, pris en considération car l’éducation thérapeutique n’est pas simplement une information donnée (enseignant → élève), mais un partage, un échange entre le personnel soignant et l’enfant. A l’issue des ateliers collectifs, un suivi individuel est proposé par le psychologue aux enfants en difficulté.
Le bénéfice apporté par ces écoles de l’atopie est évident, les confrères pourront adresser leurs patients atopiques pour bénéficier de ce programme d’éducation thérapeutique aux services de dermatologie des régions cités plus haut[1].

Photo 4 : Déroulement d’une séance d’éducation thérapeutique au service de dermatologie du CHU Mustapha
Conclusion : Tout comme un diabétique doit savoir gérer sa maladie tout seul, un atopique doit bénéficier d’une éducation thérapeutique lui permettant de mieux soigner son eczéma car la prise en charge de la DA est bien plus qu’une simple ordonnance.
|
Savoir |
· Expliquer l’eczéma atopique · Expliquer la non contagiosité · Reconnaitre la sécheresse cutanée · Reconnaitre l’eczéma · Connaitre les facteurs aggravants · Reconnaitre quelque chose d’inhabituel (herpes, impétigo) · Reconnaitre les différents traitements et expliquer leurs actions |
|
Savoir faire |
· Appliquer les émollients seul · Se laver et se sécher seul · Savoir que faire en cas de prurit · Débuter les dermocorticoïdes si poussée, · Savoir adapter son traitement à la plage, à la piscine, au sport |
|
Savoir être |
· Expliquer la maladie à la famille, aux amis · Exprimer ses émotions · Donner son avis sur la prise en charge médicale · Évoquer les difficultés |
Tableau 1 : Étapes du programme éducatif
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références :
- Dammak, G. Guillet, Dermatite atopique de l’enfant Journal de Pédiatrie et de Puériculture (2011),24, 84-102.
- Baghou, Prévalence et profil clinique de la dermatite atopique en Algérie. Ann. Dermatol. Vénéréol décembre 2012, vol 139, n 12S.
- Barbarot, Dermatite atopique, Encycl Med Chir Dermatolo; 2016, 98-150 A-10
- Misery les ateliers de l’atopie, Journal de Pédiatrie et de Puériculture, (2011),24,273-275
- Peyron JL. Prise en charge thérapeutique de l’eczéma atopique. Rev Fr Allergol. Immunol. Clin. 2006;46(Suppl. 1):S18-21.
- Barbarot, dermatite atopique : Un référentiel d’éducation thérapeutique. Ann Dermatol. Venerol. 2007 ;134 :121-7
- François LAUNAY. Actualités pharmaceutiques. 16. Supplément formation au n° 534. 1er trimestre 2014.
- F. Stalder Ecole de l’atopie : Éducation thérapeutique de l’atopique. Rev. Prat. 2006;56:273-6
- Chavigny J.M., Adiceom F., Bernier C., Debons M., Stalder J.F. « École de l’atopie », évaluation d’une expérience d’éducation thérapeutique chez 40 malades. Ann Dermatol. Venereol. 2002;129:1003-7.
- GET, www.edudermatologie.com, Groupe Éducation Thérapeutique, CHU Nantes.
Photo 1 & 2 : Outils illustrant les différents aspects cliniques de la DA
Photo 3 : L’inflammation cutanée illustrée par le feu [10]
Photo 4 : Déroulement d’une séance d’éducation thérapeutique au service de dermatologie du CHU Mustapha
[1] Numéro de téléphone de l’école de l’atopie : 05.56.84.00.59
Cholestéatome de l’oreille moyenne : nos résultats fonctionnels
Nous avons mené une étude descriptive prospective longitudinale monocentrique ayant durée 3 ans et portés sur 87 patients, tous présentaient une indication de chirurgie de l’oreille moyenne dans le cadre de la prise en charge d’un cholestéatome ; soit 100 oreilles
Medkour (1), Tabouche (2) Y. Laid (3)
(1) Service ORL CHU Mohamed Lamine Debaghine, Bab El Oued,
(2) Service de Radiologie CHU Mohamed Lamine Debaghine, Bab El Oued,
(3) Département de Prévention, Ministère de la Santé, Alger.
Date de soumission : 18 Novembre 2019.
Résumé : Objectifs : comparer les résultats obtenus dans la chirurgie du cholestéatome en technique fermée (TF), par l’utilisation de cartilage ou de prothèses partielles (PORP) ou totales (TORP) positionnés sur la platine ou l’étrier. Patients et méthodes : Nous avons mené une étude descriptive prospective longitudinale monocentrique ayant durée 3 ans et portés sur 87 patients, tous présentaient une indication de chirurgie de l’oreille moyenne dans le cadre de la prise en charge d’un cholestéatome ; soit 100 oreilles, qui ont bénéficié d’une tympanoplastie en technique fermée sous anesthésie générale dont 13 cholestéatomes bilatéraux. Par un seul otologiste et en première main. En pré opératoire, la fonction auditive a été évaluée chez tous les patients à l’aide d’un audiogramme étalonné. Résultats : On a noté une amélioration du rinne moyen dans 71,29% par rapport au rinne moyen préopératoire, une aggravation dans 1 ,99%. Le rinne était inférieur ou égal à 20 dB dans 36 cas ; entre 20 et 30 dB dans 26 cas. On a noté une chute qui varie entre 20 et 40 dB de la conduction osseuse sur les fréquences 2.000 et ou 4.000 Hz dans 4 cas. On a comparé dans la même population, les résultats audiométriques avec PORP et TORP versus cartilage et il n’y avait pas de différence significative. Conclusion : l’utilisation de cartilage sous forme de lamelles positionnées sur la tête de l’étrier ou la platine pour rétablir l’effet columellaire donne des résultats fonctionnels similaires à ceux des PORP et TORP, n’engendrant aucun surcoût, et doit à notre avis être privilégiée ; le cartilage, étant facilement disponible, totalement biocompatible, et simple à manipuler.
Mots clés : ossiculoplastie, prothèse partielle, prothèse totale, cartilage, technique fermée.
Abstract: objectives: The objective of this study is to compare the results obtained with the canal wall up technique in cholesteatoma surgery, using cartilage or partial dentures (PORPs) or total dentures (TORP) positioned on platinum or stirrup. Patients and methods: We conducted a 3-year, single-centre longitudinal prospective descriptive study involving 87 patients, all of whom presented an indication for middle ear surgery in the management of cholesteatoma; one hundred (100) ears underwent closed tympanoplasty under general anaesthesia, including 13 bilateral cholesteatomas. By a single otologist and in first-hand. The auditory function was evaluated preoperatively for all patients using a calibrated audiogram. Results: An improvement of the average Rinne was observed in 71.29% of the patients compared to the preoperative mean Rinne and an aggravation in 1.99% of the cases. Rinne was inferior or equal to 20 dB in 36 cases and between 20 and 30 dB in 26 cases. A fall which varies between 20 and 40 dB of bone conduction on the 2,000 and 4,000 Hz frequencies has been noted in 4 cases. The audiometric results with PORP and TORP versus cartilage were compared in the same population and there was no significant difference. Conclusion: the use of cartilage in the form of lamellae positioned on the stirrup or platinum head to restore the columellar effect gives functional results similar to those of the PORP and TORP. It does not generate any extra cost and must in our opinion be privileged because the cartilage is easily available, totally biocompatible and easy to handle.
Keyword: oculoplastic, partial prothesis, total prothesis, cartilage, canal wall up technique.
Introduction
Le cholestéatome de l’oreille moyenne est défini comme une otite chronique caractérisée par la présence généralement dans les cavités de l’oreille moyenne, d’un épithélium malpighien kératinisé, doué d’un potentiel de desquamation, de migration et d’érosion. C’est pourquoi l’otite est dite dangereuse car elle met en jeu le pronostic fonctionnel auditif et expose à des complications redoutables, justifiant pleinement le recours exclusif à un traitement chirurgical qui consiste souvent en une tympanoplastie en technique fermée (TTF).
La TTF permet de rétablir la fonction auditive assez satisfaisante en un seul temps grâce aux matériaux synthétiques ou l’utilisation de cartilage, car la restauration auditive est essentielle et ne devrait pas être considéré comme secondaire. S’il semble désormais que la biocompatibilité des prothèses ossiculaires ait été améliorée, reste le problème de leur coût.
Matériels et méthodes
Nous avons mené une étude descriptive prospective longitudinale monocentrique ayant duré 3 ans et ayant porté sur 87 patients, tous présentaient une indication de chirurgie de l’oreille moyenne dans le cadre de la prise en charge d’un cholestéatome ; soit 100 oreilles, qui ont bénéficié d’une tympanoplastie en technique fermée sous anesthésie générale dont 13 cholestéatomes bilatéraux, par un seul otologiste et en première main. En pré-opératoire, la fonction auditive a été évaluée chez tous les patients à l’aide d’un audiogramme étalonné.
Ce dernier est demandé en post-opératoire à partir de trois mois. L’audiomètre a été utilisé pour déterminer les seuils de la conduction osseuse (CO) et de la conduction aérienne. Le rinne moyen a été calculé en additionnant la moyenne des différences de la conduction aérienne (CA) et la conduction osseuse sur les fréquences 500, 1.000, 2.000, et 4.000. Conformément aux critères du bureau international d’audiophonologie (BIAP), la CO préopératoire a servi de référence pour le calcul du rinne postopératoire. La labyrinthisation a été définie comme une chute plus de 20 dB sur les fréquences 2.000 et 4.000 Hz, et qui ne remonte pas au-delà. Nous avons réalisé un rétablissement columellaire par différents procédés lors du premier temps chirurgical (photo 1,2).
Photo 1 : Mise en place d’une prothèse
Photo 2 : Position de la prothèse à la TDM
Résultats
Le déficit fonctionnel est variable, la chaine a été respectée ou bien le cholestéatome a joué un effet columellaire dans 11 cas. Une surdité de transmission dans 64 cas avec des extrêmes de la conduction aérienne moyenne allant jusqu’à plus de 40 dB (Graphe 2a, 2b). Une surdité de perception, une cophose, 23 surdités mixtes avec une conduction osseuse moyenne qui varie entre 10,5 et 48,5 en dehors de la cophose. Il y a eu labyrinthisation jusqu’à cophose.
Le déficit en conduction osseuse sur les fréquences 2.000 Hz atteint 20dB pour se creuser davantage sur les fréquences aiguës du spectre auditif ; c’est l’effondrement de la réserve cochléaire.
De ce bilan, résultait le déficit de l’oreille atteinte de cholestéatome traduisant une surdité de transmission dans la plupart des cas (Graphe 2).
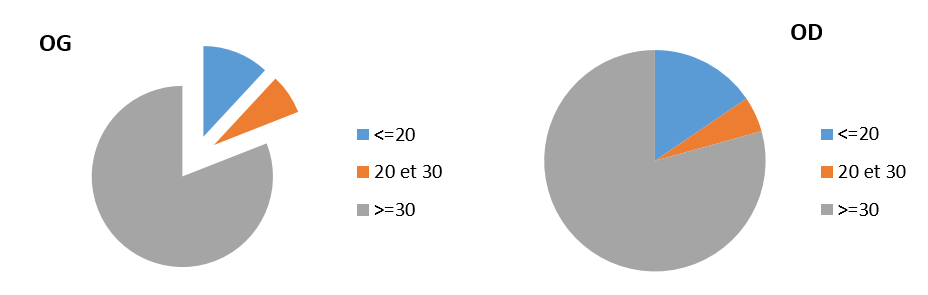
Graphe 2 : Répartition du Rinne audiométrique préopératoire
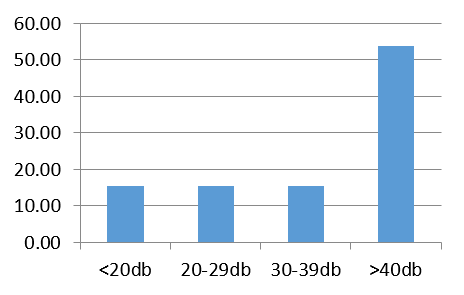
Graphe 2 a : Conduction osseuse de l’oreille droite
 Graphe 2 b : Conduction osseuse de l’oreille gauche
Graphe 2 b : Conduction osseuse de l’oreille gauche
En dehors, de la cophose et de la surdité de perception, le rétablissement columellaire a été réalisé par différents procédés lors du premier temps chirurgical. La chaine a été conservée dans près de 8% des cas (n=8) ; et on a dû déposer la chaine ossiculaire dans 13% des cas (n=13), par nécessité chirurgicale. Les osselets n’ont pas été utilisés pour rétablir l’effet columellaire, vu le risque d’inclusion microscopique par le cholestéatome et le risque d’ankylose lors des reprises chirurgicales.
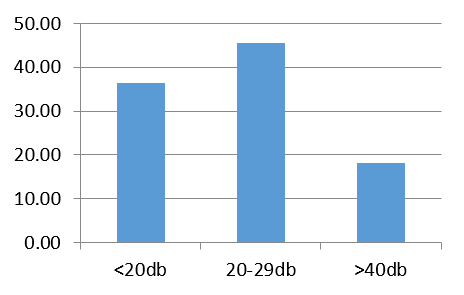
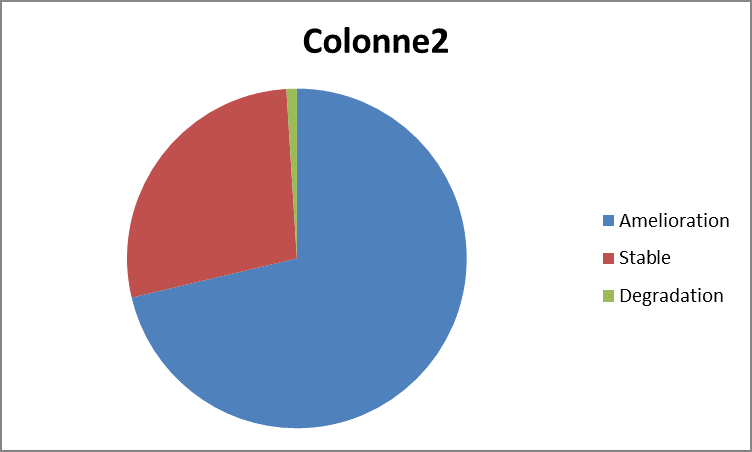
On s’assure à la fin que le montage, notamment la prothèse, est bien placé sous lamelle cartilagineuse et vient en contact du greffon afin d’éviter l’extrusion ; soit par voie du conduit auditif externe, et surtout par tympanotomie postérieure lorsque celle-ci est réalisée. On a noté une amélioration du rinne moyen dans 71,29% par rapport au rinne moyen préopératoire, et une aggravation dans 1,99% des cas (Graphe 3).
Le rinne était inférieur ou égal à 20 dB dans 36 cas ; entre 20 et 30 dB dans 26 cas (Graphe 4) ; et on a noté une chute qui varie entre 20 et 40 dB de la conduction osseuse sur les fréquences 2.000 et/ou 4.000 Hz dans 4 cas (Graphe 4).
 Graphe 3 : Résultats audiométriques
Graphe 3 : Résultats audiométriques
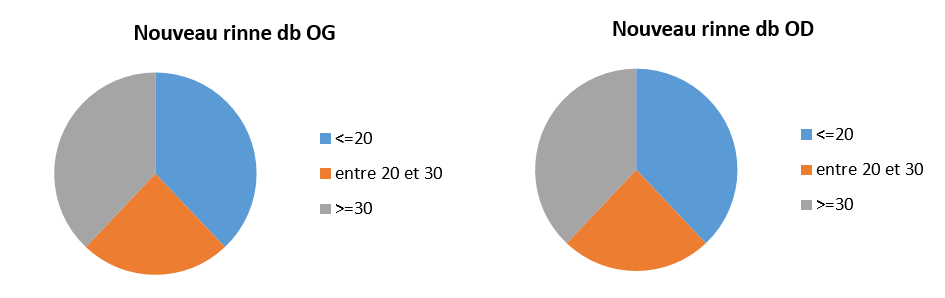
Graphe 4 : Nouveau rinne dB OG-OD
Discussion
L’audiométrie tonale liminaire préopératoire permet une quantification et le suivi du devenir fonctionnel du patient, les examens réalisés ont donné des résultats proches de la littérature : dans près de 65% de surdité de transmission ; on note 1 cophose ; 14 patients avec rinne inférieur ou égal à 20 dB, dont 11 présentent une bonne audition.
On a comparé dans la même population les résultats audiométriques avec PORP et TORP versus cartilage et il n’y avait pas de différence significative (Tableau 1a, 1b) ; pour nos résultats PORP versus cartilage le p était de 0,735 et pour le TORP versus cartilage le p est de 0,0942 sans différence significative.
Tableau 1 a : Gain Oreille droite
|
|
Effectif |
Moyenne |
Minimum |
Maximum |
Écart Type (SD) |
P |
|
Porp |
9 |
23,33 |
17 |
32 |
4,80 |
0,7353 |
|
Rehaussement étrier |
10 |
22,05 |
13 |
46,5 |
10,18 |
|
|
Torp |
6 |
25,5 |
10 |
47 |
12,28 |
0,0942 |
|
Rehaussement Platinaire |
13 |
17,69 |
12 |
35 |
7,08 |
|
|
Total |
38 |
21,41 |
10 |
47 |
8,68 |
Tableau 1b : Gain Oreille gauche
|
Ossiculoplastie |
Effectif |
Moyenne |
Minimum |
Maximum |
Écart Type (SD) |
P |
|
Porp |
7 |
19,43 |
16 |
24 |
2,76 |
0,3557 |
|
Rehaussement étrier |
9 |
17,56 |
12 |
24 |
4,56 |
|
|
Torp |
2 |
20,5 |
19 |
22 |
2,12 |
0,7542 |
|
Rehaussement Platinaire |
13 |
19,23 |
12 |
30 |
5,40 |
|
|
Total |
31 |
18,87 |
12 |
30 |
4,44 |
Dans la chirurgie de l’otite chronique, la qualité des résultats fonctionnels obtenus par la mise en place de PORP et leur biocompatibilité ont déjà été démontrés, de même d’assez nombreuses publications ont montré l’intérêt de l’utilisation du cartilage pour renforcer la membrane tympanique et rétablir la transmission des sons. Bien que la technique de mise en place du cartilage varie selon les auteurs, celle que nous décrivons, est proche de celle de Nevoux et al. C. Querat, a comparé dans la même population les résultats fonctionnels avec juste PORP et rehaussement de cartilage par étrier, et il a trouvé qu’il n’y avait pas de différence significative. Nos résultats sont similaires ; PORP versus cartilage : le p est de 0,735 ; pour le TORP versus cartilage, le p est de 0,0942 (Tableau 1a, 1b).
Conclusion
Dans la chirurgie du cholestéatome opéré en TF, l’utilisation de cartilage sous forme de lamelles positionnées sur la tête de l’étrier ou la platine pour rétablir l’effet columellaire donne des résultats fonctionnels similaires à ceux des PORP et TORP, n’engendrant aucun surcoût, et doit à notre avis être privilégiée, le cartilage étant facilement disponible, totalement biocompatible, et simple à manipuler
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
- Palva A, Karma P, Karja J. Cholesteatoma in children. Arch Otolaryngol. Feb 1977 ;103(2) :74-77.
- Dornelles C de C, Da Costa SS, Meurer L, Rosito LP, da Silva AR, Alves SL. Comparison of acquired cholesteatoma between pediatric and adult patients. Eur Arch Otorhinolaryngol. Oct.2009;266(10) :1553-1561.
- Albino AP, Kimmelman CP, Parisier SC. Cholesteatoma: A molecular and cellular puzzle. Am J Otol. Jan 1998 ;19(1) :7-19.
- Fisch U. Tympanoplasty and stapedectomy. 1980; Georg Thieme Verlag.
- Chirurgie du cholesteatome opéré en technique fermée avec étrier conservé, comparaison des résultats auditifs entre cartilage et PORP sur étrier et influence de l’ablation du marteau et du renforcement total du tympan par du cartilage C. Querat, C Martin article original http://dx.doi.org /10.1016/janorl 2013.03.008
- Kabayashi T, Gyok, Shinohara T et al., Ossicular reconstruction using hydroxyapatite protheses with interposed cartilage am j otolaryngol 2002,23 :222-7
- Kyrodimos E, Sismanis A, Santos D, Type III cartilage schield tympanoplasty: an effective procedure for hearing improvement otolaryngol head neck surg 2007,136 :982-5.
- Nevoux J, Roger G, Chauvin P et al., Cartilage shield tympanoplasty in children review of 268 consecutives cases Arch Otolaryngol Head Neck Surg 2011;137:24-9
- Samari H, otite moyenne chronique cholestéatomateuse, thèse de la faculté de médecine de Casablanca, 2001. N°278
- Saida roida, le cholestéatome bilateral à propos de 24 cas, Thèse de médecine de Casablanca, N°255, 2005
Le reflux gastro-œsophagien de l’enfant
Le reflux gastro-œsophagien est très fréquent au cours des premiers mois de la vie, et est souvent physiologique. Le plus souvent, il n’affecte pas la croissance, ne provoque pas de symptômes et disparaît généralement de lui-même à l’âge de 12 mois.
Bekkat Berkani, N. Cherif, Service de Pédiatrie B, CHU Issaad Hassani, Beni Messous, Alger.
Date de soumission : 28/12/2019
Résumé : 6:32 Le reflux gastro-œsophagien est très fréquent au cours des premiers mois de la vie, et est souvent physiologique. Le plus souvent, il n’affecte pas la croissance, ne provoque pas de symptômes et disparaît généralement de lui-même à l’âge de 12 mois. Une alimentation épaissie, un traitement postural et des changements de style de vie doivent être envisagés si les symptômes sont fréquents et gênants. La présence de signes d’alerte doit faire rechercher des complications du reflux ou d’autres pathologies pouvant mimer le reflux. La pH-métrie / impédancemétrie pour les manifestations extra-œsophagiennes et l’endoscopie pour l’œsophagite sont les investigations de choix. Le traitement empirique par les inhibiteurs de la pompe à protons pendant 4 à 8 semaines est justifié chez les enfants plus âgés et les adolescents présentant des symptômes classiques, mais pas chez les nourrissons. La chirurgie anti-reflux doit être envisagée pour les patients présentant une sténose œsophagienne, des complications potentiellement mortelles et des vomissements persistants ne répondant pas à un traitement médical.
Mots-clés: reflux gastro œsophagien, régurgitation, alimentation épaissie, traitement posturale, antagonistes des récepteurs H 2, inhibiteurs de la pompe à protons, impédancemétrie, chirurgie anti-reflux
Abstract: Gastroesophageal reflux is very common in the first months of life and is often physiological. It most often does not affect growth, does not cause symptoms, and usually goes away on its own at 12 months of age. A thickened diet, postural treatment and lifestyle changes should be considered if symptoms are frequent and bothersome. The presence of warning signs should lead to the search for complications of reflux or other pathologies that may mimic reflux. The pH-metrics / impedance for extra-oesophageal manifestations and endoscopy for esophagitis are the investigations of choice. Empirical treatment with proton pump inhibitors for 4 to 8 weeks is warranted in older children and adolescents with classic symptoms, but not in infants. Anti-reflux surgery should be considered for patients with oesophageal stenosis, life-threatening complications, and persistent vomiting that does not respond to medical treatment.
Keywords: gastroesophageal reflux, regurgitation, thickened diet, postural therapy, H 2 antagonists, proton pump inhibitors, impedance, anti-reflux surgery.
Introduction
Le reflux gastro-œsophagien (RGO) est l’une des principales causes de symptômes gastro-intestinaux chez l’enfant. Défini comme le passage du contenu gastrique dans l’œsophage, il est un processus physiologique normal se produisant plusieurs fois par jour chez des nourrissons, des enfants et des adultes en bonne santé.
Il est dit pathologique lorsque le reflux du contenu gastrique dans l’œsophage provoque des symptômes gênants et/ou des complications 1- 2-3.
Cette définition du RGO a été adoptée dans les lignes directrices publiées par la Société Nord-Américaine de Gastroentérologie, d’Hépatologie et de Nutrition Pédiatriques (NASPGHAN : North American Society for Paediatric Gastroenterology, Hepatology, and Nutrition) et la Société Européenne de Gastroentérologie, d’Hépatologie et de Nutrition Pédiatriques (ESPGHAN : European Society for Paediatric Gastroenterology, Hepatology, and Nutrition) 1.
À ce jour, aucune autre définition du RGO pédiatrique n’a été proposée et aucune étude de validation de cette définition n’a été réalisée 4.
On sait que les symptômes du RGO varient considérablement selon l’âge et ne sont pas spécifiques. Il peut souvent être difficile de distinguer le RGO physiologique du RGO maladie, en particulier chez les nourrissons.
Un diagnostic correct de RGO est crucial afin de cibler le traitement, en évitant la surutilisation de médicaments antiacides, qui représente actuellement une source de préoccupation majeure.
Epidémiologie
Des études épidémiologiques ont montré que le reflux gastro-œsophagien se produit chez environ 50% des nourrissons de moins de 2 mois, 60 à 70% des nourrissons de 3 à 4 mois et 5% des nourrissons de 12 mois 5. L’évolution spontanée du RGO est favorable chez pratiquement tous ces enfants. Les nourrissons allaités au sein sont moins susceptibles d’avoir un reflux gastro-œsophagien que les nourrissons nourris au lait artificiel 6.
Les données sur l’incidence du reflux gastro-œsophagien dans le groupe d’âge pédiatrique au-delà de la petite enfance sont rares.
Une maladie de reflux se manifeste plus fréquemment chez des enfants présentant un retard psychomoteur, des pneumopathies récidivantes (asthme ou la mucoviscidose), des malformations (la hernie hiatale ou une atrésie œsophagienne opérée), une cardiopathie congénitale ou une obésité 2.
Physiopathologie
Le sphincter œsophagien inférieur est le composant majeur de la barrière anti-reflux. Le diaphragme, qui se contracte lors d’une augmentation de la pression intra-abdominale, l’angle de His (incisura cardialis ; angle aigu entre l’œsophage et le fundus gastrique) ; et une motricité intacte de l’œsophage constituent d’autres mécanismes anti-reflux. Le tonus du sphincter s’élève à environ 25 mm de Hg à tout âge 2.
Pendant le péristaltisme, le sphincter se détend brièvement. Une relaxation transitoire du sphincter œsophagien inférieur à celui de la pression gastrique peut entraîner un passage rétrograde du contenu gastrique dans l’œsophage.
En fait, la plupart des épisodes de reflux gastro-œsophagien sont causés par cette relaxation transitoire du sphincter œsophagien inférieur, déclenchée par une distension gastrique postprandiale. Cependant, le reflux gastro-œsophagien peut également se produire avec une pression du sphincter inférieur de l’œsophage normale s’il y a une pression importante intra-abdominale ou s’il y a un retard de la vidange gastrique, plus rarement lors de troubles de la motilité d’origine neurogène ou myogène de l’œsophage2. Sans traitement, le RGO évolue vers une inflammation chronique de la muqueuse œsophagienne avec des œsophagites érosives ou ulcéreuses avec un risque sténose œsophagienne. Une transformation de l’épithélium œsophagien stratifié en épithélium cylindrique spécialisé peut survenir constituant la muqueuse dite de Barrett 7.
Manifestations cliniques
Les présentations cliniques varient avec l’âge
- Tableau clinique du RGO chez les nourrissons
Les régurgitations et les vomissements sont très fréquents chez les nourrissons en bonne santé, principalement pendant les premiers mois de la vie. Environ 70% des nourrissons en bonne santé régurgitent physiologiquement plusieurs fois par jour, et dans environ 95% d’entre eux, les symptômes disparaissent spontanément à l’âge de 12 à 14 mois 8.
La grande majorité de ces nourrissons se développent bien et n’ont aucun autre symptôme ; ils ne nécessitent aucune intervention.
Certains nourrissons et de jeunes enfants atteints de RGO peuvent présenter une irritabilité, des pleurs excessifs, un manque d’appétit, un refus d’alimentation, un retard de croissance, des troubles du sommeil, une toux chronique, une respiration sifflante ou un stridor 1-4.
Le syndrome de Sandifer, caractérisé par une dystonie spasmodique en torsion avec une inclinaison latérale intermittente de la tête et du cou avec un aspect de torticolis et une courbure importante de la colonne vertébrale, est très spécifique de la maladie du RGO.
Cependant, ni les régurgitations et les vomissements, ni l’irritabilité et les pleurs excessifs, quelle que soit leur ampleur, ne sont suffisants pour diagnostiquer le RGO.4
- Tableau clinique du RGO chez les enfants
Les enfants plus âgés et les adolescents atteints de RGO peuvent présenter des régurgitations chroniques, des nausées, une dysphagie, des brûlures d’estomac, des douleurs rétro sternales ou épigastriques, une toux chronique, un enrouement et des érosions dentaires 2.
Les nourrissons et les enfants présentent un large éventail de symptômes non spécifiques qui peuvent être interprétés comme des symptômes de RGO, mais la relation de ces manifestations cliniques avec le RGO n’est pas toujours claire. Étant donné que cela peut conduire à la fois à des sur- et sous-diagnostics et à un traitement, pour aider à faire la différence, les dernières directives internationales sur le RGO ont établi une liste de signes d’alarme (tableau 1) nécessitant des investigations pour rechercher des pathologies autres, ou des complications du reflux gastroœsophagien.4
Le rôle majeur de l’anamnèse et de l’examen physique avec une attention aux signes d’alerte suggérant d’autres diagnostics 1-4 (Tableaux 1,2) ; est généralement suffisant pour établir un diagnostic clinique de RGO infantile non compliqué.
L’anamnèse doit inclure l’âge d’apparition des symptômes, la durée de la période d’alimentation, le volume de chaque repas, l’intervalle de temps entre les repas , le type et les méthodes de mélange des préparations, les additifs aux aliments, la restriction des allergènes, les caractéristiques des régurgitations ou vomissement (par exemple nocturne, immédiatement post-prandial, longtemps après les repas, digérés ou non digérés), des antécédents médicaux familiaux, des facteurs déclenchants environnementaux possibles, y compris les antécédents psychosociaux de la famille et des facteurs tels que le tabagisme et l’exposition à la fumée de tabac, la courbe de croissance du patient. Le RGO physiologique commence rarement avant l’âge d’une semaine ou après l’âge de six mois.
Tableau 1 : Symptômes, des signes d’alarme ou de « Red Flags» qui suggèrent des troubles autres que le RGO4
Tableau 2 : Diagnostics différentiels du reflux gastro-œsophagien (RGO) 1
|
Obstruction gastro-intestinale Sténose pylorique Malrotation avec volvulus Maladie de Hirschsprung Corps étranger Hernie étranglée Syndrome de l’artère mésentérique supérieure Autres troubles gastro-intestinaux Achalasie Gastro parésie Gastro-entérite Ulcère peptique Œsophagite éosinophile Allergie / intolérance alimentaire Maladie inflammatoire de l’intestin Pancréatite Appendicite Neurologique Hydrocéphalie Hématome sous-dural Hémorragie intracrânienne Tumeur intracrânienne Infectieux Septicémie / méningite Infection urinaire Infection des voies aériennes supérieures / inférieures Otite moyenne Hépatite |
Métabolique / endocrinien Galactosémie Intolérance héréditaire au fructose Défauts du cycle de l’urée Amino-acidopathies Troubles de l’oxydation des acides gras Acidose métabolique Hyperplasie surrénale congénitale / crise surrénale Autres Négligence ou maltraitance envers les enfants Vomissements cycliques Mérycisme Toxique Intoxication au plomb Autres toxiques Rénal Uropathie obstructive Insuffisance rénale Cardiaque Insuffisance cardiaque Arcs vasculaires Dysfonctionnement autonome |
Examens complémentaires
Une anamnèse approfondie et un examen physique complet restent la pierre angulaire du diagnostic 4-9. Lorsque le diagnostic est incertain ou lorsque des complications sont suspectées, d’autres investigations peuvent être justifiées 9.
- Transit œsophago-gastro-duodénal (TOGD) :
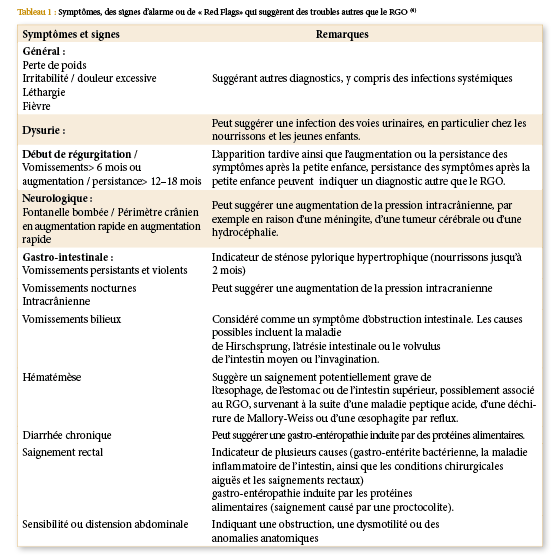
Le TOGD n’est pas recommandé pour diagnostiquer le reflux gastro-œsophagien chez les nourrissons et les enfants. Il n’est ni sensible ni spécifique. Il donne une image instantanée. Les épisodes isolés de reflux que l’on peut observer ne sont pas obligatoirement l’expression d’une maladie des reflux.
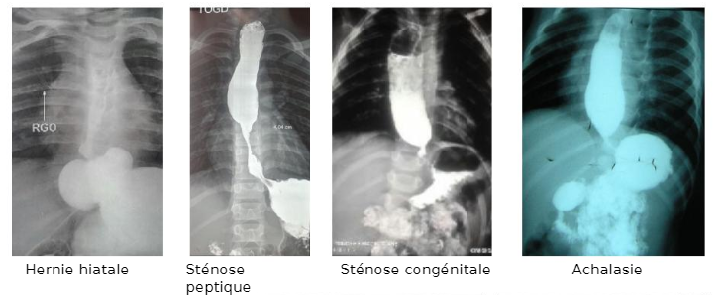
Figure 1 : Anomalies anatomiques de l’œsophage
Cette technique d’imagerie peut être utilisée pour détecter des anomalies anatomiques (Figure 1), telles qu’une sténose œsophagienne, une compression extrinsèque œsophagienne, une achalasie, une sténose pylorique, une sténose duodénale, une hernie hiatale, une malrotation et des pancréas annulaires qui peuvent provoquer des vomissements2-4.
- L’échographie
Il n’y a aucune preuve que l’échographie soit un outil de diagnostic de la maladie de RGO chez les nourrissons et les enfants 4. L’échographie Doppler couleur a une sensibilité de 95,5% et une spécificité de 11%, avec une valeur prédictive positive de 84,3% et une valeur prédictive négative de 33,3% 2. L’échographie peut être utilisée pour détecter une sténose du pylore, qui peut imiter le RGO.
- Manométrie œsophagienne
La manométrique œsophagienne de la fonction du sphincter œsophagien inférieur est utilisée pour exclure les troubles de la motilité œsophagienne tels que l’achalasie œsophagienne, dont les symptômes peuvent imiter le RGO. Les preuves sont insuffisantes pour soutenir l’utilisation de la manométrie œsophagienne pour le diagnostic du RGO 1-4.
- pH-métrie œsophagienne de longue durée (24h) (Figure 2)
La surveillance du pH œsophagien s’est révélée à la fois sensible et spécifique pour détecter le RGO. L’examen est utile pour diagnostiquer le RGO, déterminer sa gravité, évaluer s’il contribue à une pathologie extra-œsophagienne et évaluer l’efficacité du traitement de suppression acide 10. La principale limite de la surveillance du pH œsophagien est qu’elle ne détecte pas les épisodes de reflux alcalin.
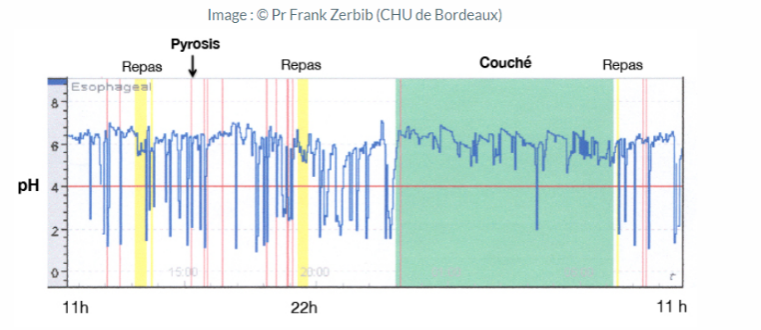
Figure 2 : Tracé de pH-métrie œsophagienne
- Impédance électrique œsophagienne intraluminale multicanaux MII
Figure 3 : pH-impédancemétrie11.
L’impédance est la résistance au passage du courant électrique entre deux électrodes reliées à un générateur de courant. Dans un organe creux, lorsqu’il y a présence de gaz entre deux électrodes d’enregistrement, la résistance au passage du courant augmente et par conséquent l’impédance augmente. Lorsqu’il y a du liquide entre les électrodes, le passage du courant est facilité et l’impédance diminue 11 (figure 3). Les mesures d’impédance intra-œsophagienne sont couplées à une mesure du pH œsophagien. La sonde d’impédancemétrie comporte une électrode de pH placée 5 cm au-dessus du sphincter inférieur de l’œsophage. D’autres électrodes de pH peuvent être rajoutées et placées dans le pharynx, l’œsophage ou l’estomac.
Elle détecte à la fois le reflux acide et non-acide en capturant les changements d’impédance électrique lors du mouvement d’un bolus liquide, solide et/ou gazeux entre les électrodes de mesure à différents niveaux œsophagiens, quelles que soient les caractéristiques physiques ou chimiques du bolus7-12-13. C’est l’outil le plus sensible pour évaluer le RGO chez les patients présentant à la fois des symptômes atypiques et typiques. Ce dispositif combiné est la pierre angulaire de l’évaluation moderne du reflux gastro-œsophagien 7-13.
- Endoscopie oeso-gastro-duodénale avec ou sans biopsie
NASPGHAN et ESPGHAN ne recommandent pas l’utilisation de l’endoscopie digestive haute pour diagnostiquer le reflux gastro-œsophagien chez les nourrissons et les enfants1-4. La valeur prédictive négative d’une endoscopie normale est faible. Le RGO peut être présent malgré l’apparence endoscopique normale de la muqueuse œsophagienne ainsi qu’en l’absence d’anomalies histologiques 2.
Elle doit être envisagée dans certains cas avec biopsie œsophagienne pour exclure d’autres affections (par exemple, œsophagite éosinophile, ulcère gastroduodénal) ; qui imitent le RGO et pour évaluer les complications du reflux gastro-œsophagien : par exemple, œsophagite, sténose œsophagienne, œsophagite de Barrett (Figure 4).
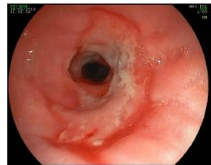
Figure 4 : Œsophagite peptique ulcérée avec sténose (grade 4 de Savary).
Prise en charge
- Mesures hygiéno-diététiques
Dans la majorité des cas, aucun traitement n’est nécessaire pour le reflux gastro-œsophagien, sauf pour rassurer les parents sur la nature bénigne de la maladie.4 Des repas épaissis, un traitement postural et des changements de style de vie doivent être envisagés si les régurgitations sont fréquentes et gênantes 4-14.
- Repas épaissis
Chez les nourrissons, l’épaississement des formules de lait avec des céréales est recommandé 1-4. Les épaississants incorporés dans le lait augmentent la viscosité dans le but de limiter les régurgitations. L’épaississant est soit constitué de caroube, qui va s’épaissir au niveau de l’estomac en absorbant l’eau au contact de l’acidité ; soit d’amidon, dont la structure se modifie sous l’effet de la chaleur ou de l’hydratation. Des formules épaissies commerciales sont disponibles à cet effet 5. La suralimentation doit être évitée, car cela peut aggraver le reflux 2.
- Traitement posturale
Le maintien d’un nourrisson en position verticale pendant 20 à 30 minutes après la tétée aide à réduire les épisodes de régurgitation 8. Le traitement postural (par exemple, élévation de la tête, position latérale et couchée) n’est pas recommandé pour traiter les symptômes du RGO chez les nourrissons endormis, en raison du risque de syndrome de mort subite, mais peut être envisagée pour le traitement du RGO chez les enfants 1-4.
- Changements de style de vie
Étant donné que les nourrissons allaités sont moins susceptibles d’avoir un RGO que les nourrissons nourris au lait artificiel, l’allaitement maternel doit être encouragé 8. Certains nourrissons allergiques au lait de vache (APLV), présentent des régurgitations et des vomissements, qui peuvent imiter le reflux gastro-œsophagien. Les deux situations APLV et RGO peuvent coexister 8. Le NASPGHAN et l’ESPGHAN suggèrent un essai de 2 à 4 semaines d’une formule hypoallergénique largement hydrolysée ou d’une formule à base d’acides aminés chez les nourrissons nourris au biberon, soupçonnés d’avoir un RGO ne répondant pas aux formules épaissies et au traitement postural 1-4. Chez les nourrissons allaités, les mères devraient envisager d’éliminer le lait de vache et les substances potentiellement allergènes de leur alimentation (par exemple les noix, les œufs, le chocolat).
D’autres mesures dans l’hygiène de vie consistent en la réduction de poids pour les personnes en surpoids, l’éviction de la suralimentation, du tabagisme actif/passif, de l’alcool, de la prise de repas avant le coucher et certains aliments 8. Les aliments épicés et gras peuvent ralentir la vidange gastrique et doivent donc être évités. Les boissons caféinées, la menthe poivrée et le chocolat peuvent abaisser la pression du sphincter œsophagien et doivent également être évitées 2.
- Traitement médical
En général, le traitement médical n’est pas indiqué dans le traitement du RGO non compliqué chez les nourrissons, car les symptômes ont tendance à disparaître avec le temps.
Il doit être envisagé dans le traitement du RGO chez les patients qui ne répondent pas aux mesures hygiéno-diététiques. Les antagonistes des récepteurs H2 et les inhibiteurs de la pompe à protons se sont révélés sûrs et efficaces pour les nourrissons et les enfants dans la réduction de leur production d’acide gastrique 15. La durée du traitement varie de quelques semaines à quelques mois. Une évaluation régulière est nécessaire pour déterminer si un traitement continu est nécessaire 6.
- Antagonistes des récepteurs H2
Les antagonistes des récepteurs H2 qui sont utilisés pour le traitement du reflux gastro-œsophagien comprennent la cimétidine (enfants : 30 à 40 mg/kg/jour en 4 doses), la ranitidine (enfants : 5-10 mg/kg/jour en 2 à 3 doses) 2. Ces médicaments réduisent la sécrétion d’acide gastrique par inhibition compétitive de l’interaction entre l’histamine et le récepteur H2 situé sur les cellules pariétales gastriques. Ils réduisent également la production de pepsine et le volume d’acide gastrique. Ils ne réduisent cependant pas la fréquence du RGO 2-4. Ils sont moins efficaces que les inhibiteurs de la pompe à protons. Ils ont un début d’action relativement rapide 2. Leur utilisation à long terme est limitée par la tachyphylaxie et l’hypochlorhydrie. Cette dernière peut entraîner une colonisation bactérienne gastrique. Il existe également un risque accru de pneumonie d’origine communautaire et d’infection entérique, en particulier, Clostridium difficile.
- Les inhibiteurs de la pompe à protons
Ceux qui sont utilisés pour le traitement du RGO sont l’oméprazole, l’esoméprazole et le lansoprazole ; Généralement, ils sont administrés une fois par jour, idéalement 30 minutes avant le repas. Ces médicaments inhibent sélectivement la sécrétion d’acide en bloquant les pompes hydrogène-potassium-adénosine triphosphatase (H + –K + –ATPase) ; qui se situent sur la membrane des cellules pariétales gastriques. Comme les antagonistes des récepteurs H2, ils ne réduisent pas la fréquence du RGO 10.
Ils aident à améliorer la dyspepsie, à prévenir les lésions œsophagiennes induites par l’acide et à accélérer la guérison de l’œsophagite 4-10.
Chez les enfants, les inhibiteurs de la pompe à protons sont préférés aux antagonistes des récepteurs H2 en raison de leur meilleure efficacité. Ces médicaments sont sûrs et bien tolérés. Ils sont les médicaments de choix pour le traitement du reflux gastro-œsophagien, surtout s’il existe des signes d’œsophagite 2-4.
Les événements indésirables des inhibiteurs de la pompe à protons sont la somnolence, les étourdissements, les maux de tête, des éruptions cutanées, des nausées, des douleurs abdominales, la diarrhée et la constipation.
- Les antiacides
Les antiacides tels que l’hydroxyde d’aluminium, le carbonate de calcium et l’hydroxyde de magnésium ne sont pas utiles dans le traitement du reflux gastro-œsophagien chez les nourrissons, mais peuvent être utilisés pour une courte période chez les enfants plus âgés pour le soulagement des brûlures d’estomac 1-4. Les antiacides agissent en neutralisant l’acide gastrique, diminuant ainsi l’exposition de la muqueuse œsophagienne à l’acidité gastrique lors d’épisodes de reflux gastro-œsophagien. Leur utilisation au long cours peut entraîner un syndrome lait et alcalins, un rachitisme hypophosphatémique ou une toxicité à l’aluminium (p. ex. ostéopénie, neurotoxicité, anémie microcytaire) 2.
- Les agents prokinétiques
Les agents prokinétiques tels que le métoclopramide (0,1-0,3 mg/kg/dose 3 à 4 fois par jour), la dompéridone (0,3–0,6 mg/kg/dose 3 fois par jour) ; favorisent la vidange gastrique et pourraient théoriquement être utiles pour le traitement du reflux gastro-œsophagien. Cependant, l’utilisation d’agents prokinétiques ne s’est pas avérée utile 1-4-15. Les effets secondaires potentiels de chaque prokinétique actuellement disponible l’emportent sur les avantages potentiels. Les preuves sont insuffisantes pour justifier l’utilisation systématique du métoclopramide ou de la dompéridone 6.
- Les agents de barrière de surface
Les agents de barrière de surface agissent comme une barrière physique pour empêcher les dommages à la muqueuse œsophagienne par l’acide gastrique au reflux. L’alginate de sodium (exemple le Gaviscon®), un polysaccharide dérivé des algues brunes, a été utilisé dans le traitement du reflux gastro-œsophagien. En présence d’acide gastrique dans l’estomac, l’alginate précipite et forme un gel visqueux, augmentant ainsi la viscosité des aliments et réduisant le reflux gastro-œsophagien 16. Le NASPGHAN et ESPGHAN ne recommandent pas l’utilisation d’alginates pour le traitement du RGO 1-4-15.
- Le traitement chirurgical
Les indications absolues pour la chirurgie anti-reflux, généralement une fundo-plicature laparoscopique, sont les événements potentiellement mortels tels qu’une insuffisance cardio-pulmonaire, une apnée attribuable à un reflux gastro-œsophagien 1-4. Les enfants présentant un retard de croissance, une œsophagite, des sténoses œsophagiennes, des vomissements récurrents ou des pathologies chroniques (par exemple, maladies neurologiques, fibrose kystique) présentant un risque important de complications qui ne répondent pas au traitement médical doivent également faire discuter la chirurgie anti-reflux.
Le taux de réussite de la fundo-plicature varie de 60 à 90% 4-15. Les complications du traitement chirurgical comprennent une infection au niveau du site chirurgical, une hémorragie, une perforation digestive, un pneumothorax, une hernie hiatale, une sténose œsophagienne, une lésion du nerf vague, des brûlures d’estomac et un dumping syndrome 17.
Approche diagnostique
L’approche diagnostique du RGO chez les nourrissons souffrant fréquemment de régurgitations ou de vomissements est présentée dans l’algorithme 1. En l’absence de signes d’alerte, des examens complémentaires et/ou des traitements médicamenteux ne sont pas nécessaires s’il n’y a pas d’impact des symptômes sur l’alimentation, la croissance et le développement. En présence de signes d’alertes « Red Flags » (tableau 1), des pathologies autres que le RGO peuvent être en causes (tableau 2).
Les recommandations actuelles ne soutiennent pas un essai empirique par des inhibiteurs de la pompe à protons en tant que test de diagnostic du RGO chez les nourrissons et les jeunes enfants, car les symptômes évocateurs de la maladie de reflux gastro-œsophagien ne sont pas, ou moins spécifiques chez les enfants de ce groupe d’âge 1-4-17.
L’approche diagnostique des enfants souffrant de symptômes évoquant un RGO est présentée dans l’algorithme 2. Le NASPGHAN et ESPGHAN suggèrent un essai de 4 à 8 semaines avec les inhibiteurs de la pompe à protons chez les enfants plus âgés présentant des symptômes typiques de RGO (tels que des brûlures d’estomac rétro-sternales ou des douleurs épigastriques) comme test de diagnostic pour le reflux gastro-œsophagien 1-4.
L’impédancemétrie couplée à la pH-métrie est la référence pour le diagnostic du RGO en cas de doute. Lorsque cet examen n’est pas disponible, le NASPGHAN et ESPGHAN suggèrent d’envisager une surveillance du pH œsophagien pour corréler les symptômes gênants persistants avec les événements de reflux gastro-œsophagien acide 4.
Pronostic
Le reflux gastro-œsophagien disparait à l’âge de 12 mois chez environ 95% des nourrissons en général lorsque la station assise est acquise. Les enfants dont les symptômes de reflux persistent au-delà de 18 mois sont plus susceptibles de souffrir de reflux gastro-œsophagien chronique 14.
Les enfants ayant une déficience neuro-développementale et des antécédents familiaux de reflux gastro-œsophagien ont un pronostic moins favorable que les nourrissons atteints de reflux gastro œsophagien physiologique 4-14.
Conclusion
En l’absence de signes cliniques d’alerte tels que le retard de croissance, l’hématémèse, une posture anormale, la suffocation ou la toux pendant l’alimentation ; la régurgitation chez le nourrisson ne nécessite aucune investigation ni traitement. Chez les enfants plus âgés et les adolescents présentant des symptômes de reflux typiques, un traitement empirique par inhibiteurs de la pompe à protons est justifié. Pour les RGO à manifestations atypiques ou ne répondant pas au traitement médical, une endoscopie et un examen de pH/impédance sont les investigations de choix. Le traitement chirurgical n’est pas toujours la solution car il entraine une morbidité importante et échoue souvent chez ceux qui en ont le plus besoin.
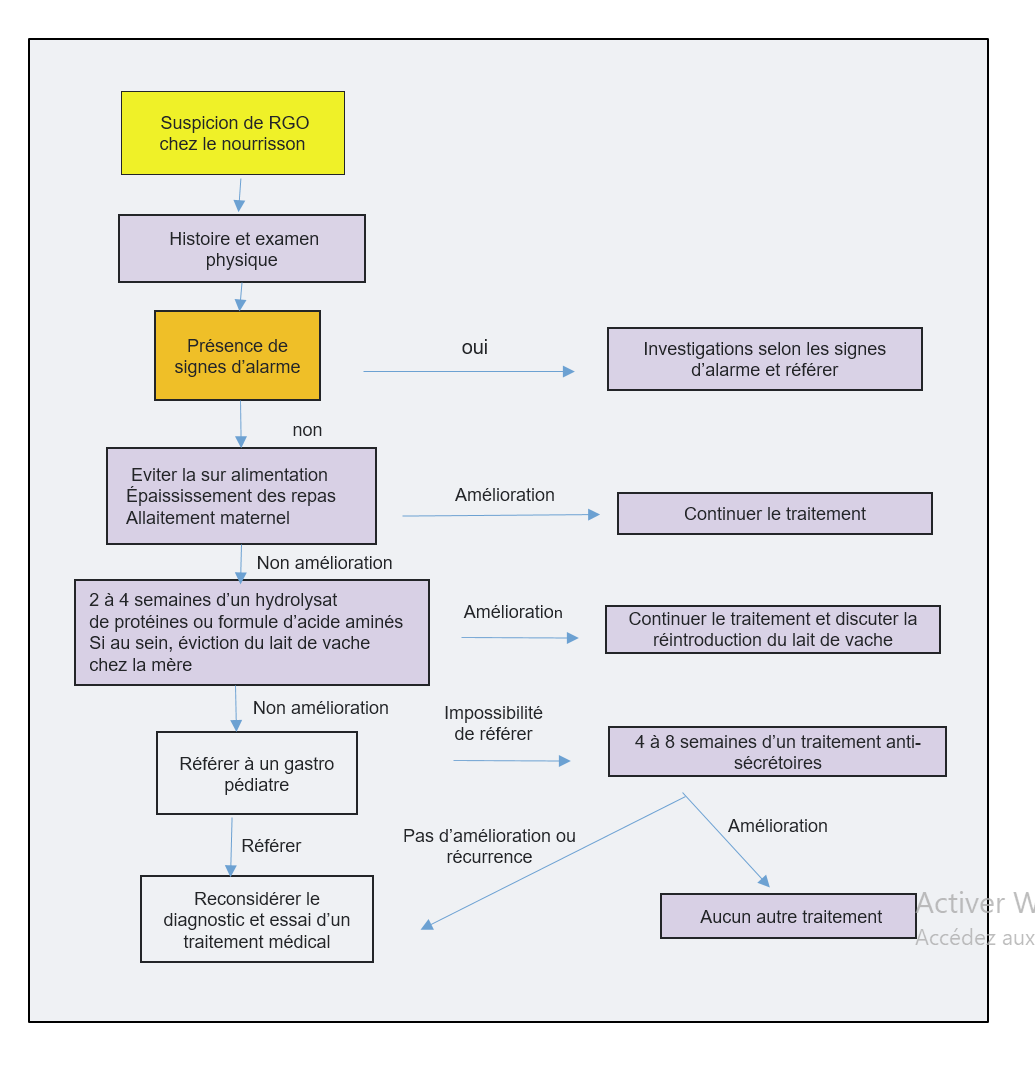
Algorithme 1 : Approche de la prise en charge du RGO chez les nourrissons2
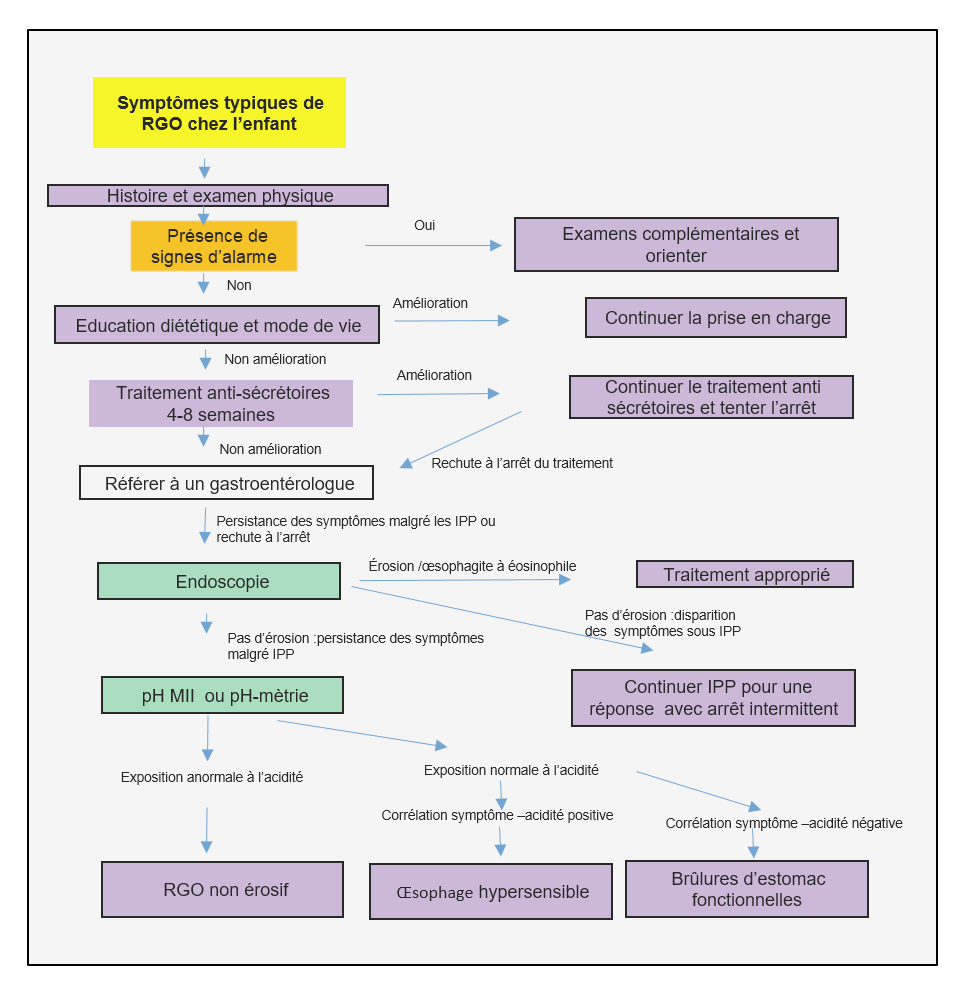
Algorithme 2 : Approche de la prise en charge du RGO chez l’enfant2
Conflit d’intérêt : aucun
Références
- Vandenplas Y, Rudolph CD, Di Lorenzo C, et al. Pediatric gastroesophageal reflux clinical practice guidelines: joint recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition (NASPGHAN) and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN). J Pediatr Gastroenterol Nutr 2009;49 :498-547.
- Winter HS. Clinical manifestations and diagnosis of gastro-esophageal reflux in children and adolescents. In: Post TW, ed. UpToDate. Waltham 2019
- Mousa H., Hassan M. Gastroesophageal reflux disease. Pediatr Clin North Am. 2017;64(3):487–505.
- Rosen R, Vandenplas Y, Singendonk M, et al. Pediatric gastroesophageal reflux clinical practice guidelines: joint recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN) and the European Society for Pediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN). J Pediatr Gastroenterol Nutr. 2018;66:516–554.
- Nelson SP, Chen EH, Syniar GM, Christoffel KK. Prevalence of symptoms of gastro-esophageal reflux during infancy. A pediatric practice-based survey. Pediatric Practice Research Group. Arch Pediatr Adolesc Med. 1997;151(6):569–572.
- Randel A. AAP releases guideline for the management of gastro-esophageal reflux in children. Am Fam Physician. 2014;89 :395–397.
- Onyeador N, Paul SP, Sandhu BK Paediatric gastro-esophageal reflux clinical practice guidelines. Dis Child Educ Pract Ed. 2014 ; 99:190-3.
- Winter HS. Gastro-esophageal reflux in infants. In: Post TW, ed. UpToDate. Waltham 2019.
- Sullivan JS, Sundaram SS. Gastro-esophageal reflux. Pediatr Rev. 2012;33 :243–253.
- Van Der Pol RJ, Smits MJ, Venmans L, Boluyt N, Benninga MA, Tabbers MM. Diagnostic accuracy of tests in pediatric gastro-esophageal reflux disease. J Pediatr. 2013;162 :983–987.
- David Bertolini. pH-impédancemétrie : quelle place en clinique ? Rev Med Suisse 2008;4:1873-1878.
- Tong S, Mallitt KA, Krishnan U. Evaluation of gastro-esophageal reflux by combined multi channel intra-luminal impedance and Ph monitoring and oesophageal motility patterns in children with oesophageal atresia. Eur J Pediatr Surg. 2016;26 :322–331.
- Safe M, Cho J, Krishnan U. Combined multi channel intra-luminal impedance and pH measurement in detecting gastro-esophageal reflux disease in children. J Pediatr Gastro-enterol Nutr. 2016;63(5):e98–e106.
- Alexander KC Leung, Kam Lun Hon. Gastro-esophageal reflux in children : an updated review Drugs Context 2019; 8: 212591.
- Mattos ÂZ, Marchese GM, Fonseca BB, Kupski C, Machado MB. Anti-secretory-treatment for pediatric gastro-esophageal reflux disease – a systematic review. Arq Gastro-enterol. 2017;54(4):271–280.
- Salvatore S, Ripepi A, Huysentruyt K, et al. The effect of alginate in gastro-oesophageal reflux in infants. Paediatr Drugs 2018;20(6):575–583.
- Wakeman DS, Wilson NA, Warner BW. Current status of surgical management of gastroesophageal reflux in children. Curr Opin Pediatr. 2016;28:356–362.
Rôle du médecin généraliste dans la prise en charge du diabète de l’enfant
Le diabète de type 1 chez l’enfant et l’adolescent est une maladie chronique qui est devenue une préoccupation majeure de santé publique dans le monde et en Algérie. Sa prise en charge est complexe faisant intervenir une équipe multidisciplinaire.
Bekkat Berkani, N. Cherif, Service de Pédiatrie B, CHU Issaad Hassani, Beni Messous, Alger.
Date de soumission : 13 Décembre 2019.
Résumé : Le diabète de type 1 chez l’enfant et l’adolescent est une maladie chronique qui est devenue une préoccupation majeure de santé publique dans le monde et en Algérie. Sa prise en charge est complexe faisant intervenir une équipe multidisciplinaire. Le médecin généraliste, médecin de premier recours, associé à cette prise en charge, doit avoir accès aux services et aux conseils d’une équipe spécialisée en diabétologie pédiatrique. Il jouerait un rôle à plusieurs niveaux de la prise en charge. La connaissance des signes du diabète et un diagnostic précoce pourraient éviter l’évolution vers l’acidocétose diabétique, mode de révélation grave de la maladie, et permettre une annonce du diagnostic dans un climat plus favorable pour le patient et ses parents. Sa participation au parcours de soins lors des consultations et aux urgences réduit les visites aux urgences liées au diabète, et les taux d’hospitalisation. Il pourra ainsi surveiller l’observance du traitement insulinique de l’adolescent en réduisant le risque de comorbidités à long terme du diabète. Il devrait également veiller à ce que le dépistage correct des complications dégénératives et des maladies associées ait été entrepris. Il informera les parents que leur enfant doit être vacciné normalement et que la vaccination contre la grippe est recommandée. Il doit savoir gérer les jours de maladies de l’enfant diabétique pour éviter le déséquilibre du diabète et l’absentéisme scolaire. De par sa proximité et sa disponibilité, il donne des conseils et oriente le patient et sa famille sur les évènements et activités de la vie courante pour faciliter l’insertion scolaire et sociale. Conclusion : Le médecin généraliste fait partie intégrante de l’équipe multidisciplinaire qui s’occupe de l’enfant et de l’adolescent diabétique. Il devrait être associé aux formations d’éducation thérapeutique et de transition des soins pédiatriques vers les soins d’adultes.
Mots clés : diabète type 1, suivi ambulatoire, parcours de soins, médecin généraliste.
Abstract: Type 1 diabetes in children and adolescents is a chronic disease that has become a major public health concern worldwide and in Algeria. Its management is complex involving a multidisciplinary team. The general practitioner, primary care physician, associated with this care, must have access to the services and advice of a specialized team in paediatric diabetology. He would play a role at several levels of care. Knowledge of the signs of diabetes and early diagnosis could prevent progression to diabetic ketoacidosis, a mode of serious revelation of the disease, and allow the diagnosis to be announced in a more favourable climate for the patient and his parents. Its participation in the care pathway during consultations and emergencies reduces visits to diabetes-related emergencies and hospitalization rates. He will thus be able to monitor adherence to adolescent insulin treatment by reducing the risk of long-term comorbidities of diabetes. He should also ensure that proper screening for degenerative complications and associated diseases has been undertaken. He will inform parents that their child should be vaccinated normally and that vaccination against influenza is recommended. He must know how to manage the days of illness of the diabetic child to avoid the imbalance of diabetes and school absenteeism. Due to its proximity and availability, he gives advice and guides the patient and his family on events and activities of daily life to facilitate academic and social integration. Conclusion: The general practitioner is an integral part of the multidisciplinary team that deals with children and adolescents with diabetes. He should be associated with therapeutic education and transition training from paediatric to adult care.
Key words: type 1 diabetes, delivery of ambulatory care, care pathway, general practitioner.
- Introduction
Le diabète de type 1 est l’endocrinopathie la plus fréquente chez l’enfant. C’est une maladie chronique qui est devenue actuellement une préoccupation majeure de santé publique.
La prévalence du diabète type 1 de l’enfant dans le monde est de plus d’un demi-million (586.000) d’enfants âgés de moins de 15 ans. L’incidence mondiale augmente d’environ 3 à 5% par an (tableau 1)1. En Algérie, l’incidence augmente comme partout dans le monde. Elle compte aujourd’hui parmi les pays qui sont au Top 10 en nombre de nouveaux cas de DT1 âgés moins de 15 ans par an.1 Selon le registre du DT1 de l’enfant d’Oran, l’incidence a doublé (12/100.000 en 1993 à 30/100.000 enfants moins de 15 ans en 2015). À Alger, elle est également à 30/100.000 nouveaux cas par an2.
Tableau N°1 : Top 10 des pays/territoires en nombre de nouveaux cas de diabète de type 1 (enfants et adolescents < 15 ans) par an1
Les soins aux enfants atteints de diabète de type 1 (T1D) sont complexes. Les objectifs de la prise en charge sont d’organiser le parcours de soins de tous ces enfants et d’améliorer leur qualité de vie.
Le médecin généraliste est le médecin de premier recours de par sa proximité et sa disponibilité. Pour s’occuper d’enfants et d’adolescents diabétiques, il doit avoir accès aux services et aux conseils de l’équipe de diabétologie du centre régional de référence le plus proche3. Il pourrait jouer un rôle à plusieurs niveaux de la prise en charge3.
- Le diagnostic
Le diagnostic de diabète chez l’enfant est encore trop souvent tardif, établi devant une acidocétose pouvant mettre en jeu le pronostic vital.
Dans la wilaya d’Alger ; 17,6 % de nouveaux cas d’enfants présentant un DT1 sont diagnostiqués au stade d’ACD[1] (Fig 1).
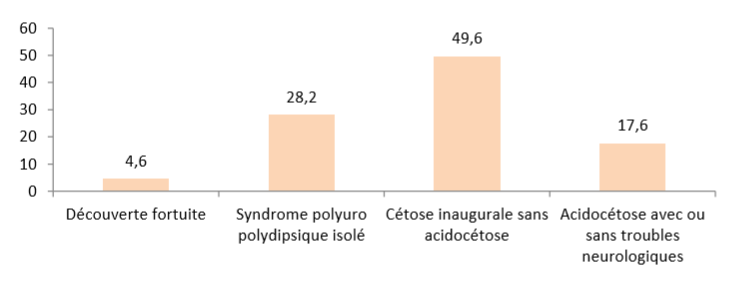
Fig. 1 : Répartition des nouveaux cas de diabète de type 1 selon les circonstances de découverte de la maladie dans la wilaya d’Alger en 20162
Un diagnostic précoce pourrait éviter l’ACD et permet une annonce dans un contexte plus confortable, point de départ de l’éducation dans laquelle l’enfant et sa famille seront plus à même de s’impliquer.
Le médecin généraliste est souvent le premier à faire le diagnostic. Son rôle est donc essentiel, dans le but de réduire l’incidence du mode de révélation grave du diabète de l’enfant qu’est l’acidocétose diabétique. Il doit être informé et sensibilisé, sur les signes et moyens simples et rapides de poser le diagnostic afin d’orienter immédiatement l’enfant vers l’hôpital : c’est une urgence vitale4.
- Le diagnostic est principalement clinique4,5. Il est extrêmement facile chez les enfants car le début est stéréotypé :
- Une polyurie (ce qui se remarque plus facilement la nuit)
- Une polydipsie, un amaigrissement, une fatigue importante
Ceci se déroule en quelques jours.
Sans une attention particulière, la présentation peut facilement être confondue avec une gastro-entérite ou une infection des voies urinaires. Un enfant qui urine la nuit plusieurs nuits de suite et qui perd du poids a presque toujours un diabète.
Si le diagnostic de diabète n’est pas évoqué à ce stade, l’évolution va se faire vers l’apparition de nausées, de vomissements, de douleurs abdominales et de troubles de la respiration pour arriver finalement au coma en quelques semaines constituant ainsi un tableau d’acidocétose diabétique.
- La confirmation du diagnostic est très simple.
Elle repose sur la simple réalisation de :
- Une bandelette urinaire (recherche de sucre et de corps cétoniques dans les urines ++) (Fig 2)
- Une glycémie capillaire

Fig. 2 : Bandelette urinaire
En général, le diagnostic est confirmé rapidement par la mesure d’une élévation marquée de la glycémie.
Dans cette situation, s’il y a des corps cétoniques dans le sang ou l’urine, le traitement est urgent. Attendre un autre jour pour confirmer l’hyperglycémie peut être dangereux car l’acidocétose peut évoluer rapidement.
Aucun passage en laboratoire d’analyses médicales n’est nécessaire.
Le diagnostic de diabète, en l’absence d’hyperglycémie évidente, doit être confirmé par une des trois méthodes présentées dans les critères diagnostics suivants (Tableau N° 2) :
Tableau N° 2 : Critères pour le diagnostic de diabète sucré 5
|
1. Symptômes de diabète plus glycémie au hasard > 11,1 mmol/l (2 g/l) au hasard est défini comme à n’importe quelle heure de la journée et sans considération de temps après un repas. Ou |
|
2. Glycémie à jeun > 7,0 mmol/l (> 1,26 g/l) à jeun signifie aucun apport calorique depuis au moins 8h. Ou |
|
3. Glycémie 2 h après la prise de glucose > 11,1 mmol/l (> 2 g/l) au cours d’une HGPO. Le test devrait être réalisé selon les critères de l’OMS, avec l’équivalent de 75 g de glucose anhydre dissous dans l’eau ou 1,75 g/kg de poids avec un maximum de 75 g |
L’annonce du diagnostic est une mission difficile pour le médecin, douloureuse pour les parents. Elle est souvent faite dans le cadre de l’urgence.
Des erreurs peuvent être commises, du fait que le médecin traitant diagnostique rarement un diabète chez un jeune : de faux espoirs sont donnés, des idées fausses sont véhiculées, sur le lien entre le diabète et un évènement récent.
Le médecin généraliste doit expliquer et orienter l’enfant et sa famille sans retard vers un pédiatre diabétologue qui sait très exactement ce qu’il doit dire pour mieux les préparer à gérer la prise en charge de la maladie de leur enfant en tenant compte de leur contexte familial et socioprofessionnel.
- Le parcours de soins et du suivi 6,7 :
Le médecin généraliste est en première ligne pour la surveillance et le suivi du bon développement du jeune patient. Il est le plus souvent le médecin traitant, qui connait l’enfant au sein de sa famille dans la réalité de leur vie.
Il peut participer au parcours de soins de l’enfant diabétique et avoir une place importante dans la continuité des soins lors des consultations et aux urgences réduisant ainsi des visites aux urgences liées au diabète et des taux d’hospitalisation.
Lors des visites de soins primaires, le médecin généraliste devrait surveiller l’observance du traitement insulinique de l’adolescent, notamment en évaluant la fréquence de la surveillance de la glycémie et de l’administration d’insuline. Il doit se familiariser avec les différents types d’insuline utilisés chez l’enfant (tableau N° 3). Les schémas d’insulinothérapie varient en fonction du mode de vie de l’enfant. Le schéma d’insulinothérapie le plus recommandé est le schéma d’insulinothérapie basal bolus à 3 ou 4 injections par jour (ISPAD 2018)3. En Algérie, le schéma thérapeutique le plus fréquemment utilisé est le basal-bolus tout analogue (63,0 %) selon l’étude DiabCare Pédiatrique Algérie 20148.
Les objectifs glycémiques recommandés (tableau N°4) permettent d’ajuster le schéma posologique d’insuline du patient dans le but d’améliorer le contrôle glycémique, en réduisant le risque de comorbidités à long terme du diabète6.
Le médecin généraliste devrait également veiller à ce que le dépistage correct du maintien de la santé avec le diabète ait été entrepris (tableau N°5).
Le médecin généraliste et le pédiatre diabétologue doivent communiquer régulièrement, au moins une fois par an, pour discuter de tout problème lié à la gestion du diabète de l’enfant.
En plus de la surveillance de la glycémie et de l’observance du traitement, les patients doivent faire l’objet d’une surveillance fréquente pour détecter les complications du diabète. Cette surveillance, guidée par les recommandations de l’ISPAD 2018, est décrite dans le tableau N° 3.
Tableau N° 3 : Les insulines les plus fréquemment utilisées en pédiatrie (disponibles en Algérie)
|
Type d’insuline début d’action pic d’action (h) Durée d’action (h) |
|
Analogues rapides 10 à 1-3 3-5 (Aspart, Llispro, Glulisine) 20 minutes |
|
Insuline humaine rapide 30 à 2-4 5-8 60 minutes |
|
Insuline intermédiaire 2 à 4 h 4-8 12-16 (NPH) |
|
Insuline de longue durée d’action |
|
Glargine 2 à 4 heures Pas de pic 20-24 |
|
Detemir 1-2 heures 6 12-20 |
Tableau N° 4 : Objectifs glycémiques selon l’ISPAD 2018
|
Avant un repas = 4-7 mmol/l (70-130 mg/dl) Après chaque repas = 5 -10 mmol/l (90-180 mg/dl) A l’heure du coucher = 4,4 -7,8 mmol/l (80 -140 mg/dl) HbA1c < 7 %quel que soit l’âge |
Tableau N° 5 : Recommandations de surveillance de l’enfant diabétique (ISPAD 2018)3
|
|
Examens |
Rythme |
|
Auto-surveillance |
Glycémie Glycosurie, Acétonurie |
Pluriquotidiens Si glycémie > 2,5g/l
|
|
Examen clinique Contrôle glycémique |
Poids – Taille – TA – Puberté – recherche de lipodystrophies – état bucco-dentaire, complications aigus (hypo-hyper), problèmes psychologiques, tenue du carnet de surveillance Hémoglobine glyquée ≤ 7 %
|
Trimestrielle |
|
Dyslipidémie Rétinopathie Néphropathie Maladie thyroïdienne Maladie cœliaque |
HDL-LDL cholestérol FO ACR (Ration albuminurie/Créatinurie) sur un échantillon d’urine le matin TSH et ATPO TTG et AC anti IgA. |
Après le diagnostic puis tous les 2 ans après l’âge de 11 ans et 2 et 5 d’évolution Tous les ans à partir de 11ans et à 2et 5 ans d’évolution Tous les ans à partir de 11ans et 2 à 5ans d’évolution. Au diagnostic et tous les 2ans Au diagnostic et à 2 et 5 ans |
HDL-C : cholestérol à lipoprotéines de haute densité ; LDL-C : cholestérol à lipoprotéines de basse densité ; FO : Fond d’œil, ATPO : anticorps de la peroxydase thyroïdienne ; TTG : transglutaminase tissulaire ; TSH : hormone stimulant la thyroïde.
Mise en œuvre de la vaccination
Toutes les vaccinations obligatoires du calendrier national sont autorisées. Le diabète n’est pas une contre-indication aux vaccinations.
Le médecin généraliste informe les parents que leur enfant qui a un diabète doit être vacciné comme les autres et que la vaccination contre la grippe est recommandée : le risque de grippe n’est pas plus élevé chez l’enfant diabétique que chez les autres, mais, comme toute maladie fébrile, la grippe risque de déséquilibrer le diabète.
- La gestion des affections intercurrentes 6,7
Le médecin généraliste est le gestionnaire des événements intercurrents susceptibles d’occasionner un déséquilibre glycémique.
De nombreuses maladies, en particulier celles associées à la fièvre, élèvent la glycémie en raison de l’effet des hormones de stress. La résistance accrue à l’insuline peut augmenter la production de cétone.
Les maladies avec des symptômes gastro-intestinaux (par exemple la diarrhée ou encore les vomissements) peuvent conduire à une hypoglycémie due à une diminution de l’apport alimentaire, à une mauvaise absorption et à des modifications de la motilité intestinale.
Des vomissements, chez un enfant qui a un diabète, devraient être considérés comme un signe d’acidocétose jusqu’à preuve du contraire.
Le médecin généraliste peut se retrouver devant plusieurs situations qu’il doit gérer chez son patient diabétique :
- Si infections associées à des nausées, des vomissements ± de la diarrhée (cas de la gastroentérite)
- L’insuline ne doit pas être arrêtée.
- La dose totale quotidienne d’insuline est réduite de 20-50 % si nécessaire.
- Les repas sont remplacés par la prise fréquente de boissons sucrées, en petites quantités.
- Surveillance régulière de la glycémie.
- Si infections associées à une hyperglycémie, avec ou sans cétose
- Des injections supplémentaires d’insuline d’action rapide.
- Une surveillance stricte est recommandée pour faire baisser la glycémie, prévenir l’acidocétose, et éviter l’hospitalisation.
- La maladie sous-jacente devrait être traitée comme elle le serait chez un enfant qui n’a pas de diabète.
- Indications d’une hospitalisation dans les circonstances suivantes
- Très jeunes enfants atteints de diabète, qui peuvent se déshydrater plus rapidement que les enfants plus âgés ou les adolescents.
- Nausée ou vomissements qui empêchent l’enfant de boire
- Incapacité des parents à vérifier la glycémie à la maison
- Si les soins de soutien ne peuvent pas être assurés à domicile
- Si la maladie aiguë est grave
- S’il y a une cétonurie persistante
- Conduite à tenir devant une hypoglycémie modérée
- Hypoglycémie < 0,70g/l
- L’hypoglycémie est légère lorsque le patient en reconnaît les signes et est capable de se traiter sans aide extérieure.
- L’hypoglycémie est sévère lorsque le patient perd conscience et il a besoin de l’aide d’une tierce personne
- Signes cliniques
- Malaise, tremblement, pâleur, sueur, fatigue, vertige, somnolence, vision floue ou double, sensation de froid, picotement des lèvres, trouble de la parole, faiblesse musculaire, faim impérieuse, irritabilité, comportement, bizarre, nervosité.
- La nuit : sommeil agité, cauchemar, réveil nocturne, mal de tête au réveil
- Chaque enfant diabétique a un seul et même signe d’hypoglycémie qu’il faut reconnaitre.
- Les principales causes de d’hypoglycémie sont les suivantes
- Repas retardés ou supprimés (examiner les raisons de cette situation)
- Activité physique (si possible la glycémie doit être vérifiée avant l’exercice, et des glucides supplémentaires devraient être consommés, basés sur d’une part la glycémie mesurée, d’autre part l’intensité et la durée attendues de l’exercice).
- Absorption insuffisante d’hydrates de carbone (évaluer le moment, la quantité et l’effet glycémique maximal des aliments consommés)
- Trop d’insuline injectée (évaluer le profil d’insuline, l’heure d’administration, le pic et l’intensité de l’action)
- La conduite devant une hypoglycémie
- Agir rapidement, cesser toute activité, s’assoir ++++.
- Faire une glycémie si possible.
- Prendre du sucre : 1 morceau de 5 g/20 kg de poids.
- Contrôle glycémie capillaire au bout de 10- 15 mn.
- Si pas de réponse, reprendre du sucre.
- Contrôle glycémie capillaire au bout de 15 mn.
- Prendre ensuite un repas ou collation (ex : fruit, pain, céréales, lait) pour prévenir la rechute de l’hypoglycémie.
- Tout enfant diabétique doit avoir de sucre sur lui.
- CAT devant une hypoglycémie sévère : urgence
- Ne rien donner par la bouche +++.
- Faire injection de Glucagon en IM /SC
Posologie : Poids < 12 ans (>25 kg) : 0,5 mg de Glucagon (1/2 amp.)
Poids > 12 ans (< 25 kg) : 1mg de Glucagon (1 amp.)
Quand l’enfant se réveille lui donner du sucre puis un sucre lent
- CAT devant une hyperglycémie (> 2,5 g/l) avec cétonurie
- Faire une injection supplémentaire d’insuline rapide.
- Combien : 1/10ème de la dose totale (rapide et lente) de 24 H.
- Répéter les analyses de sang et d’urine toutes les 4 heures (même la nuit).
- Manger normalement malgré hyperglycémie.
- Ne pas faire de sport jusqu’à ce que l’acétone ait disparu.
- Si pas de négativation au bout du 2ème appoint, faire le 1/5ème et partir à l’hôpital.
- L’erreur qui est faite le plus souvent est de conseiller de ne pas faire l’insuline parce que l’enfant est malade et ne mange pas, augmentant ainsi le risque d’acidocétose diabétique.
- Une surveillance plus fréquente de la glycémie et des corps cétoniques est nécessaire.
- La vie avec le diabète à la maison et à l’école3:
Le médecin généraliste donne des conseils et oriente le patient et sa famille sur les évènements et activités de la vie courante pour faciliter l’insertion scolaire et sociale.
- La diététique
Les messages à donner aux familles sont un repas équilibré, partagé en famille autour d’une table, pas de grignotage et de la discipline.
- Le sport
L’activité physique est indispensable à l’équilibre de l’enfant diabétique. Elle doit être encouragée.
Tous les sports sont autorisés sauf les sports en individuel ex. : plongée sous-marine, alpinisme). La dose d’insuline est diminuée de 10% ou plus, celle qui agit pendant, voire après un effort important ; éventuellement plus d’aliments sucrés à consommer avant, pendant et après le sport.
La glycémie avant la séance doit être >1,20g/l et < 2,5 g/l.
- L’école
Le médecin de santé scolaire est aussi un médecin généraliste.
Il y a une complémentarité entre le médecin de santé scolaire et le pédiatre hospitalier qui prend en charge l’enfant pour une continuité dans les soins.
Son rôle est de veiller à ce que l’enfant ait une scolarité normale
Les objectifs sur le plan pédiatrique, sont d’une part, de communiquer les signes évocateurs d’un diabète de type 1 et favoriser l’intégration de l’enfant diabétique au milieu scolaire et dans ses activités extrascolaires.
Il doit :
- Organiser une réunion avec les parents/tuteurs au début de l’année afin de convenir du plan de prise en charge du diabète.
- Entrer en contact avec les parents pour s’informer des habitudes de l’enfant.
- S’assurer qu’ils disposent des numéros de téléphone des parents/tuteurs.
- Convenir des situations où les parents/tuteurs doivent être contactés.
- Soutenir l’enfant atteint de diabète à l’école.
- Permettre à l’enfant de contrôler sa glycémie aux moments prévus.
- Veiller à ce que l’enfant dispose d’un endroit sûr pour ses injections d’insuline.
- S’assurer qu’un enfant en état d’hypoglycémie est pris en charge et n’est pas laissé seul.
- Permettre à l’enfant d’accéder de manière illimitée à l’eau et aux sanitaires.
- Diabète et Ramadhan
Les enfants diabétiques ne doivent pas faire le ramadhan. Le risque de survenue d’hypoglycémies sévères en cas de jeun ou d’hyperglycémie en cas d’arrêt de l’insuline est très important et à l’origine d’un déséquilibre total du diabète. Le médecin généraliste devrait pouvoir les en dissuader. Les adolescents vont quand même tenter l’expérience : il faut les accompagner tout en essayant de les convaincre.
- La transition
Un dernier axe repose sur l’amélioration de la prise en charge en pédiatrie au passage à l’âge adulte. En effet, la transition du suivi pédiatrique de jeunes patients diabétiques vers celui des adultes représente une période à risque pouvant aller jusqu’à l’interruption d’un suivi médical spécialisé, voire la perte de vue des patients.
Cela représente une piste potentielle : le médecin généraliste, même s’il est peu consulté pendant cette période, pourrait néanmoins avoir un rôle d’accompagnement et de liaison dans le suivi du jeune diabétique auprès du diabétologue adulte, moment généralement appréhendé de son parcours.
- Conclusion
Le médecin généraliste participe au parcours de soins en complémentarité avec tous les autres acteurs de santé dans la prise en charge au quotidien des besoins de l’enfant diabétique et l’amélioration de sa qualité de vie. Il devrait être associé aux formations d’éducation thérapeutique de l’enfant diabétique et de transition vers les soins d’adultes.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts
Références
- IDF Diabetes Atlas 8th Edition (2017)
- Incidence du diabète de l’enfant dans la wilaya d’Alger en 2016. Registre du diabète de l’enfant. INSP d’Alger 2018
- ISPAD Clinical Practice Consensus Guidelines 2018 https://www.ispad.org›page›ISPAD Guidelines 2018
- Arati Mokashi, Michael Young. When you suspect diabetes in a child. Canadian Family Physician 2018 ; 64:32-34
- Mayer-Davis, A. Kahkoska, C. Jefferies, D. Dabelea, N. Balde, C.X. Gong, P. Schner, E. Craig. ISPAD Clinical Practice Consensus Guidelines 2018: Definition, epidemiology, and classification of diabetes in children and adolescents Pediatric. Diabetes 2018; 19:7-19.
- Pihoker, G. Forsander, B. Fantahun, A. Virmani, S. Corathers, P. Benitez-Aguirre, J. Fu, D. Maahs. ISPAD Clinical Practice Consensus Guidelines 2018: The delivery of ambulatory diabetes care to children and adolescents with diabetes Pediatric. Diabetes 2018; 19:84–104.
- Nue and all. Diagnoses, Thérapie and Control of Diabetes Mellitus in Children and Adolescents Clin Endocrinol Diabetes 2014;122: 425–434
- Bensenouci, M. Achir, R. Boukari, Z. Bouderda, F. Lacete, C. Kaddache, B. Bioud, D. Bekkat-Berkani, Y. Aouabed, C. Nasri, S. Zinai. La prise en charge du diabète de type 1 chez l’enfant en Algérie (DiabCare Pédiatrique) Médecine des maladies Métaboliques 2014;8:646-651.
Interprétation d’un ECG normal chez l’enfant
L’électrocardiogramme (ECG) est l’une des investigations cardiaques les plus simples et les plus anciennes disponibles, mais il peut fournir une mine d’informations utiles et reste une partie essentielle de l’évaluation des patients cardiaques.
Haddad, Service de Pédiatrie, CHU Issaad Hassani, Beni Messous, Alger.
Date de soumission : 13 Décembre 2019.
Résumé : L’électrocardiogramme (ECG) est l’une des investigations cardiaques les plus simples et les plus anciennes disponibles, mais il peut fournir une mine d’informations utiles et reste une partie essentielle de l’évaluation des patients cardiaques. Un ECG est simplement une représentation de l’activité électrique du muscle cardiaque, imprimée sur du papier pour une analyse plus facile. Le muscle cardiaque se contracte en réponse à la dépolarisation électrique des cellules musculaires. Par convention, nous enregistrons l’ECG de surface standard en utilisant 12 directions. Six sont placées sur le thorax (les dérivations précordiales). Quatre sont placées au niveau des 4 membres (les dérivations périphériques). Il est essentiel que chacune des 10 électrodes d’enregistrement soit placée dans sa position correcte. L’amplitude du signal électrique enregistré est exprimée sur un ECG dans la dimension verticale et est mesurée en millivolts (mV). Sur du papier ECG standard, 1mV est représenté par une déviation de 10 mm. Le papier ECG standard se déplace à la vitesse de 25 mm par seconde pendant l’enregistrement. Cela signifie qu’une distance de 25 mm le long de l’axe horizontal représente 1 seconde. La première structure à dépolariser pendant le rythme sinusal normal est l’oreillette droite, suivie par l’oreillette gauche. Ainsi, le premier signal électrique sur un ECG normal provient des oreillettes et est représenté par l’onde « P ». L’enregistrement d’un ECG sur papier standard permet de mesurer le temps des différentes ondes et intervalles en millisecondes et de calculer la fréquence cardiaque. Il existe des tables de référence des normes en fonction de l’âge.
Mots clés : ECG, enfant, interprétation normale.
Abstract: The electrocardiogram (ECG) is one of the simplest and oldest cardiac investigations available, yet it can provide a wealth of useful information and remains an essential part of the assessment of cardiac patients. An ECG is simply a representation of the electrical activity of the heart muscle as it changes with time, usually printed on paper for easier analysis. Like other muscles, cardiac muscle contracts in response to electrical depolarization of the muscle cells. By convention, we record the standard surface ECG using 12 different recording lead directions. Six of these are recorded from the chest overlying the heart (precordial derivations). Four are recorded from the limbs (peripheral derivations). It is essential that each of the 10 recording electrodes is placed in its correct position. The amplitude, or voltage, of the recorded electrical signal is expressed on an ECG in the vertical dimension and is measured in millivolts (mV). On standard ECG paper 1 mV is represented by a deflection of 10 mm. Standard ECG paper moves at 25 mm per second during real-time recording. This means that when looking at the printed ECG a distance of 25 mm along the horizontal axis represents 1 second in time. The first structure to be depolarised during normal sinus rhythm is the right atrium, closely followed by the left atrium. So the first electrical signal on a normal ECG originates from the atria and is known as the P wave. The recording of an ECG on standard paper allows the time taken for the various phases of electrical depolarisation to be measured, usually in milliseconds and an approximate estimation of the heart rate. There is a recognised normal range for such ‘intervals’.
Keywords: ECG, children, normal interpretation.
Introduction
Le rythme cardiaque normal naît dans des cellules dites automatiques (spontanément dépolarisées) du nœud sinusal.
A partir du nœud sinusal, la conduction se fait de proche en proche et de façon radiaire dans les oreillettes ; elle atteint ainsi le nœud auriculo-ventriculaire où elle ralentit, le faisceau de His puis ses branches, le réseau de Purkinje et les cellules ventriculaires sont ensuite excités.
- Définition de l’électrocardiogramme (ECG)
L’électrocardiogramme, c’est l’enregistrement sur un support papier standard de l’activité électrique du cœur sur un plan frontal (par les dérivations des membres) et sur un plan horizontal (par les dérivations précordiales).
L’abréviation usuelle utilisée pour parler de l’électrocardiogramme est ECG, en anglais comme en français. (Dans certaines sources anglo-saxonnes, on trouvera également l’abréviation EKG).
- A quoi sert l’électrocardiogramme ?
C’est un formidable outil diagnostic de pathologies cardiaques rythmiques (flutter, etc.), musculaires (infarctus, etc.), de problèmes extra cardiaques métaboliques (hyperkaliémie), médicamenteux (imprégnation digitalique, etc.), hémodynamique (hypovolémie), et autres (HTAP, etc.)
- Technique d’enregistrement
L’enregistrement de l’ECG chez l’enfant pose fréquemment quelques problèmes techniques, liés à l’agitation des enfants en bas âge ; en pratique, il est toujours possible d’obtenir un tracé si l’on fait preuve de patience. Il faut utiliser les petits moyens : biberon, jouets, présence de la mère, etc.
L’ECG standard est enregistré sur 12 dérivations (6 précordiales, 6 dérivations des membres), avec une vitesse de déroulement du papier à 25 mm par seconde et une amplitude de 10 mm pour 1 mV. Ce standard a été adopté en colloque international en 1938 en Angleterre, confirmé dans les dernières recommandations internationales.
Les différentes dérivations
- 6 dérivations périphériques ou standard (I, II, III, aVR, aVL, aVF) explorant le plan frontal
- 6 dérivations précordiales (V1, V2, V3, V4, V5, V6) explorant le plan horizontal.
Mise en place des électrodes
4 électrodes sur les membres = Dérivations périphériques
- Bras D …………. à Rouge
- Jambe D ……… à Noir
- Jambe G………. à Vert
- Bras G…………. à Jaune
6 autres électrodes sont placées sur le thorax = dérivations précordiales
- V1-V2 : ……….. 4ème EIC G D
- V4-V5-V6 : …… 5ème EIC respectivement sur la ligne médio claviculaire, axillaire antérieure et axillaire moyenne
- V3 entre V2 et V4
Calibrage ECG
Étalonnage du temps : vitesse de déroulement du papier
Standard : 25mm/sec (1mm = 0,04s)
Étalonnage de l’amplitude : Déflection test standard = 10 mm = 1 millivolt
Enregistrer des tracés à 25 mm (0,04 s/petit carreau) et éventuellement 50 mm/sec (0,02 s/petit carreau).
L’enregistrement en demi-amplitude est fréquemment nécessaire pour les dérivations précordiales gauches en raison de la grande amplitude des voltages.
NB : dans les appareils actuels, l’étalonnage est automatique
Le tracé ECG obtenu
L’onde P représente la contraction des oreillettes. Elle est de forme arrondie, souvent positive et de faible amplitude.
Le complexe QRS correspond à la contraction des ventricules.
L’onde T est la période de repos du cœur.
Le segment ST correspond à la période d’excitation des ventricules jusqu’à la phase de repos. Un sus-décalage ou un sous-décalage de plus de 1 mm par rapport à la ligne isoélectrique est anormal.
Une méthode de lecture
Le tracé de l’enfant est différent de celui de l’adulte et il se modifie beaucoup de la naissance à l’adolescence : l’interprétation en est donc délicate et doit toujours tenir compte de l’âge du patient.
L’analyse de l’ECG montre autant d’ondes P que de QRS, chaque onde P entraînant un QRS fin. Le segment ST est isoélectrique.
Comment lire un électrocardiogramme
- Vérifiez que les 12 dérivations sont bien enregistrées et que l’ECG ne comprend pas trop d’artéfacts qui compliquent sa lecture.
- Il faut être rigoureux, systématique et méthodique dans la lecture :
- Lire l’E.C.G. dans l’ordre des dérivations de D1 à V6.
- Lire chaque segment de l’E.C.G. de gauche à droite (de l’onde P vers l’onde T)
- Ne pas s’attacher à une anomalie dans une dérivation unique souvent sans valeur.
- Toujours penser que l’E.C.G. correspond à l’activité électrique du myocarde et qu’il est à confronter à la clinique.
- Éviter les pièges liés à des inversions d’électrodes +++
- Ne pas hésiter à refaire un tracé s’il y a un doute ou s’il est parasité.
- Enfin, toujours avoir en tête ce que peut révéler un ECG +++
Toutes les dérivations de l’électrocardiogramme doivent être analysées dans leur ensemble.
- Calcul de la fréquence cardiaque
- Analyse du rythme cardiaque
- Évaluation de l’intervalle PR
- Évaluation de l’intervalle QT
- Détermination de l’axe électrique
- Analyse du segment ST
- Autres altérations électrocardiographiques
- Fréquence cardiaque
La première chose qu’il faut déterminer sur un électrocardiogramme, c’est la fréquence cardiaque. La fréquence de base au repos s’accélère de la naissance à la fin du premier mois ; elle est voisine de 120 à 150 bats/mn ; puis elle diminue régulièrement à partir du 3ème mois, et ce jusqu’à l’âge de 16 ans, passant progressivement de 120 à 85 bats/mn environ.
Une tachycardie sinusale est très fréquente, souvent ≥ à 180 bats/mn chez le nouveau-né, ≥ à 150 bats/mn chez le nourrisson, ≥ à 120 bats/mn chez l’enfant, dès que le patient s’agite, sans que cela ait de signification pathologique.
Une bradycardie sinusale est plus rare, habituellement rencontrée chez le grand enfant et chez l’adolescent.
L’arythmie sinusale respiratoire est très répandue ; elle peut être très marquée et donner l’impression d’un trouble du rythme cardiaque.
Tout professionnel doit connaître les différentes méthodes de calcul de fréquence cardiaque car l’analyse automatique n’est pas toujours juste ou parce l’électrocardiogramme n’affiche pas la valeur de la fréquence cardiaque.
- La méthode des 300 est la plus rapide et la plus utilisée: On localise sur l’ECG une onde R qui puisse coïncider avec une ligne épaisse, on compte le nombre de grands carreaux qu’il y a jusqu’à l’onde R suivante et on divise 300 par le nombre de grands carreaux (si rythme régulier).
Exemple : Si entre deux ondes R, il y a un carreau, cela fera 300 bpm ; deux carreaux, 150 bpm, trois, 100 bpm, quatre. Comment avez-vous deviné ? : 75 bpm.
2. Et si la deuxième onde R ne coïncide pas ?
Dans un électrocardiogramme, souvent, la deuxième onde R ne coïncide pas exactement avec autre ligne épaisse.
On divise de nouveau 300 par le nombre de grands carreaux mais cette fois, nous ajoutons 0,2 chaque petit carreau restant.
Exemple : La distance entre deux ondes R est de 4 grands carreaux et 3 petits carreaux, on divise donc 300 par 4,6. Le résultat sera 65 bpm.
- Comment calculer la fréquence cardiaque si le rythme est irrégulier ?
Habituellement, les électrocardiogrammes enregistrent 10 secondes, c’est pourquoi il ne reste plus qu’à compter tous les complexes QRS et à les multiplier par 6.
Si l’ECG ne mesurait pas 10 secondes ou si vous ne savez pas combien il mesure, comptez 30 grands carreaux qui représentent 6 secondes – multipliez le nombre de complexes QRS par 10 et vous avez, approximativement, la valeur de la fréquence cardiaque.
Exemple : Comptez les complexes QRS sur 30 carreaux (6 secondes) et multipliez-les par 10 pour calculer la fréquence cardiaque : 11 complexes QRS x 10 = 110 bpm approximativement.
Rythme cardiaque
Le rythme cardiaque est-il sinusal ? Si oui :
- Chaque complexe QRS est précédé d’une onde P
- Et chaque P est suivie d’un QRS
- L’onde « P » est positive en DI – DII
Onde P est normalement négative en aVR et parfois en V1 et V2
- Si P négative en DI: Inversion électrodes, situs inversus et rythme ectopique (OG)
- Si P négative en DII: onde P rétrograde ou rythme du sinus coronaire
- Si P négative en V6 : rythme auriculaire gauche
Intervalle PR
On doit mesurer l’intervalle PR normal depuis le début de l’onde P jusqu’au début du complexe QRS.
Il s’allonge avec l’âge.
Il est normalement de 0,08 à 0,12 secondes de la naissance à 3 mois ; il s’allonge progressivement jusqu’à l’adolescence. Ainsi, un PR à 0,18s chez un nourrisson est un signe formellement pathologique.
Une prolongation de l’intervalle PR permet de diagnostiquer un bloc AV du premier degré. Un intervalle PR court permet de diagnostiquer un syndrome de Wolff-Parkinson-White.
Complexes QRS
Durée = 0,08s < 5 ans ; 0,09s entre 5 et 8 ans et < 0,10s de 8 – 16 ans
Le profil de QRS varie notablement avec l’âge, en raison des conditions de la circulation foeto-placentaire.
Les modifications hémodynamiques qui surviennent vont aboutir à une régression de la prédominance ventriculaire droite et au développement de la prépondérance VG normale chez l’adulte (car le VG est systémique).
- L’onde Q : habituelle en D2 D3 AVF à toutes les périodes de l’enfance ; elle peut être très profonde jusqu’à l’âge de 3 ans.
- L’onde R : ses variations en précordiales sont importantes à connaître et justifient l’usage de tables. Son amplitude diminue en V2 V3 alors qu’elle augmente progressivement en V5 V6 au cours de la croissance, en raison de l’installation de la prépondérance ventriculaire gauche.
- L’onde S : ses variations sont inverses à celle de l’onde R : elles augmentent d’amplitude en V1 et diminuent en V5 V6 (diminution de la prépondérance VD).
- Le rapport R/S : Il est intéressant de le calculer dans les dérivations précordiales, en V1 : 0-2 ans : > 1,2 à 4 ans : = 1 > 4 ans : < 1.
- L’indice de Sokolow : des valeurs importantes peuvent être atteintes, supérieures à celles de l’adulte : il a donc très peu de valeur chez l’enfant.
Intervalle QT
On doit mesurer l’intervalle QT depuis le début du complexe QRS jusqu’à la fin de l’onde T.
L’intervalle QT varie en fonction de la fréquence cardiaque, c’est pourquoi l’on doit corriger sa valeur selon la fréquence cardiaque. L’intervalle QT corrigé ou QTc est normal entre 350 ms et 440 ms.
Onde T
En V1 V2, l’onde T est positive à la naissance et se négative dans les 5 premiers jours.
L’existence d’ondes T positives en précordiales droites au-delà est pathologique et correspond habituellement à une HVD.
Plus tard, T est négative et souvent profonde en V1 V2 V3 parfois même en V4 (ceci est dû à la rotation horaire du cœur), pendant l’enfance.
Onde T : ischémie sous épicardique ou sous endocardique.
Axe électrique cardiaque
Chez le nouveau-né, l’axe de QRS est droit, voisin de + 135° (Il y a une prépondérance des cavités droites, qui diminue progressivement jusqu’à l’apparition d’un ECG de type adulte).
Dès le premier mois, AQRS se déplace vers la gauche ; de 6 mois à 16 ans, il est voisin de + 60° avec des variations individuelles importantes.
Une méthode sûre et rapide pour savoir s’il est normal consiste à vérifier que les dérivations D1 et aVF sont positives.
Analyse du segment ST
Le segment ST est la ligne entre la fin du complexe QRS et le début de l’onde T.
Il doit être isoélectrique +++.
Segment ST : lésion sous épicardique ou sous endocardique
En pratique pédiatrique, la seule façon d’éviter les erreurs, est de se reporter à des tables indiquant la valeur moyenne et les valeurs limites inférieures et supérieures des amplitudes de chaque onde pour l’âge considéré.
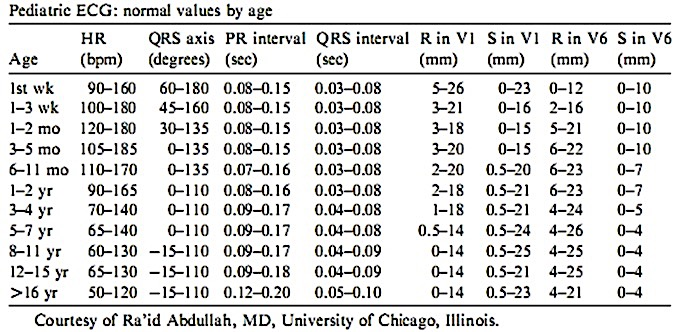
Conclusion
L’interprétation de l’ECG pédiatrique est parfois un exercice difficile. L’ECG est l’examen paraclinique primordial et irremplaçable dans l’analyse des troubles du rythme cardiaque.
Un ECG normal ne permet en aucun cas d’exclure la présence d’une cardiopathie. Il faut savoir qu’il peut être normal dans des cardiopathies sévères, surtout chez le nouveau-né et chez le nourrisson.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts
Références
- Dubin D. Rapid Interpretation of EKGs, 6th ed Tampa, FL: Cover Publishing, 2000.
- The Pediatric ECG Ghazala Q. and all Emerg Med Clin N Am 2006;24:195–208 (doi:10.1016/j.emc.2005.08.014)
- Kachaner. Cardiologie pédiatrique. Paris : Flammarion médecine-sciences ; 1987.
- Taboulet. Formation à « l’ ECG de A à Z » Disponible sur www.e-cardiogram.com
- Park MK, Gunteroth WG. How to Read Pediatric ECGs, 3rd Edn. Louis : Mosby Year Book, 1992.
Retard statural chez l’enfant
La croissance est le reflet de l’état de santé de l’enfant et sera modifiée par les maladies endocriniennes et la plupart des maladies chroniques.
Bouferoua, N. Bouterfas, K.N. Benhalla, Service de Pédiatrie A, CHU Issaad Hassani, Béni Messous, Alger.
Date de soumission : 13 Décembre 2019.
Résumé : La croissance est le reflet de l’état de santé de l’enfant et sera modifiée par les maladies endocriniennes et la plupart des maladies chroniques. Les attitudes thérapeutiques vis-à-vis d’un retard de croissance et/ou de puberté seront guidées par l’analyse physiopathologique. Les courbes d’évolution de la taille sont les documents clés qui permettent de savoir si un enfant donné a une croissance anormale. Les étiologies du retard statural sont multiples, pouvant être en rapport soit avec une maladie chronique, d’origine nutritionnelle, endocrinienne ou génétique. Le retard de croissance à début intra-utérin est la cause de 10 % des petites tailles. La petite taille peut être de type constitutionnel, comme elle peut être idiopathique lorsqu’aucune cause n’est identifiée.
Mots clés : croissance, déficit en hormone de croissance, facteurs de croissance, âge osseux, hormone de croissance, puberté, retard pubertaire, retard statural, syndrome de Turner.
Abstract: Growth is a reflection of the child’s health and will be modified by endocrine diseases and most chronic diseases. Therapeutic attitudes towards growth retardation and/or puberty will be guided by pathophysiological analysis. Size development charts are the key documents that can tell if a particular child is growing abnormally. The aetiologies of delay in stature are multiple, which can be related either to a chronic disease, of nutrional, endocrine or genetic origin. SGA is the cause of 10% of short stature. Small stature can be constitutional, as it can be idiopathic when no cause is identified.
Key words: growth, growth hormone deficiency, growth factors, bone age, puberty, pubertal delay, short stature, Turner’s syndrome.
Introduction
Le retard statural est défini par une taille inférieure à -2 SDS selon les courbes de référence de la population, il peut également s’agir d’un infléchissement de la courbe de croissance staturale, et une courbe staturale très inférieure à ce que l’on attend des tailles familiales, c’est à dire < -1,5 DS de la taille cible corrigée.
Facteurs de régulation de la croissance
Une croissance normale nécessite l’intervention de facteurs nutritionnels adéquats, un système endocrinien fonctionnel et un squelette normal, et tous ces facteurs sont sous la dépendance de facteurs génétiques dont l’influence débute à partir de l’âge de 2 ans.
Démarche diagnostique devant un retard de croissance staturale
Le retard statural est un motif de consultation fréquent, c’est un signe clinique qui peut être la conséquence de plusieurs pathologies, pour cela une enquête étiologique doit être menée afin de retenir un diagnostic.
Anamnèse
Dans une première étape, l’anamnèse doit préciser la taille des parents qui permet d’apprécier le rôle des facteurs génétiques en calculant la taille cible. Elle précise également la taille dans la fratrie et la famille, l’âge pubertaire dans la famille.
Les paramètres de naissance (poids, taille et périmètre crânien) doivent être précisés à la recherche d’un retard de croissance intra-utérin qui est défini par un poids et/ou une taille < -2 SDS par rapport à l’âge gestationnel selon la courbe de référence de population.
Rechercher l’existence d’une affection chronique, une prise médicamenteuse de longue durée (corticoïdes). Rechercher des signes fonctionnels d’orientation telle qu’une anorexie, troubles digestifs, syndrome polyuro-polydipsique, céphalées, troubles neurologiques.
Examen clinique
L’examen clinique doit être précis et minutieux à la recherche de signes d’orientation vers une étiologie : signes dysmorphiques pouvant faire évoquer un syndrome, une anomalie de la ligne médiane pouvant être associée à des anomalies hypothalamo-hypophysaires, des anomalies osseuses pouvant faire évoquer une maladie osseuse constitutionnelle, des signes cliniques pouvant faire évoquer une pathologie chronique, rénale, digestive, pulmonaire, cardiaque, métabolique.
Il permet également d’évaluer le stade pubertaire qui est essentiel pour interpréter le retard de croissance d’un enfant.
Courbe de croissance
C’est la pierre angulaire du diagnostic, elle apporte une notion dynamique au retard statural. Elle permet le calcul de la vitesse de croissance, cette dernière est calculée à partir de deux mesures précises de la taille espacées d’au moins 6 mois.
Elle permet de différencier les retards de croissance à vitesse de croissance normale, dans ce cas le retard de croissance est ancien et ne s’aggrave pas ; du retard de croissance à vitesse de croissance ralentie où le retard s’aggrave. La courbe de croissance peut exprimer soit une cassure ou un infléchissement.
Le retard statural avec vitesse de croissance régulière nous oriente vers une origine génétique et/ou constitutionnelle. Le retard statural avec ralentissement de la vitesse de croissance nous oriente vers une origine acquise.
L’évolution de la croissance staturale comparée à celle du poids nous permet également une orientation étiologique. En effet, lorsque le retard pondéral précède le retard statural, ceci nous oriente vers une origine énergétique (ex. maladie cœliaque), lorsque le retard statural est associé à une prise pondérale, ceci nous oriente vers une origine génétique (ex : syndrome de Prader-Willi) ou endocrinienne (ex : hypothyroïdie et hypercorticisme).
Maturation osseuse
Elle est évaluée par une radiographie de la main et du poignet gauche. La comparaison de cette radiographie à un atlas de référence (le plus utilisé est l’atlas de « Greulich et Pyle ») donne une valeur approximative, le plus souvent suffisante en pratique clinique courante. Des méthodes plus précises sont basées sur le calcul d’un score tenant compte de la taille et de l’aspect de chacun des os du carpe et de la main, telle la méthode de « Sempé ».
L’âge osseux (AO[1]) permet d’effectuer une prédiction de la taille finale, il a donc une valeur plus pronostique que diagnostique ; néanmoins, il nous permet une orientation étiologique très grossière qui ne doit en aucun cas être le seul juge dans l’orientation des investigations.
Examens complémentaires
Le plus souvent un interrogatoire et un examen clinique bien conduits, une courbe de croissance complète ainsi qu’un âge osseux suffisent pour orienter le diagnostic vers l’une des nombreuses causes de retard de croissance staturale.
Les examens complémentaires seront demandés en fonction de cette orientation initiale.
Dans le cas où les informations disponibles ne sont pas suffisantes pour avoir une orientation, des examens complémentaires de première intention seront demandés, une FNS, un bilan rénal, un bilan hépatique et phosphocalcique.
En deuxième intention la sérologie de la maladie cœliaque et, si nécessaire, une biopsie jéjunale et un bilan thyroïdien. Des radiographies du squelette, crâne, avant-bras, rachis dorso-lombaire, bassin et genoux. Un caryotype, un test de stimulation de l’hormone de croissance, et enfin l’étude génétique à la recherche de la mutation du gène SHOX.
Principales causes du retard de croissance statural
Retard de croissance secondaire à une maladie chronique. La courbe de croissance peut objectiver un infléchissement ou une cassure au début de la maladie chronique, l’âge osseux peut être retardé ou normal par rapport à l’âge chronologique.
Toutes les maladies chroniques peuvent être à l’origine d’un retard statural pouvant être soit en rapport avec leur sévérité, ou bien en rapport avec le traitement entrepris (ex : corticoïdes).
Maladie cœliaque. Son diagnostic est évident lorsque les manifestations digestives sont au premier plan, mais là encore, le retard de croissance peut être le premier signe d’appel, et seule la biopsie jéjunale pourrait affirmer le diagnostic en montrant une atrophie totale.
Les carences d’apport. Tous les syndromes de malabsorption peuvent retentir sur la croissance.
Insuffisance rénale chronique. Quelle que soit sa cause, elle s’accompagne d’un infléchissement de la courbe de croissance. Il en est de même des tubulopathies chroniques qui sont évoquées devant une polyurie et reconnues sur les explorations biologiques (glycosurie, protéinurie, calciurie, troubles ioniques, acidose, défaut de concentration des urines).
Le retard de croissance peut être le seul signe d’appel de ces maladies justifiant la pratique systématique d’un ionogramme, d’une créatininémie et bandelette urinaire.
Retards de croissance constitutionnels. Ils dépendent essentiellement de facteurs génétiques, ou sont secondaires à une affection anténatale. La courbe de croissance est habituellement assez régulière évoluant en dessous de -2 SDS sans cassure franche et un âge osseux égal ou légèrement inférieur à l’âge chronologique.
Maladies génétiques. Il peut s’agir d’un ensemble malformatif assez évocateur, tel que la trisomie 21. Chez la fille, le syndrome de Turner doit être soigneusement recherché car le syndrome dysmorphique peut être très discret dans les mosaïques. Devant tout retard statural isolé chez une fille, le caryotype doit être demandé systématiquement. Le syndrome de Prader-Willi qui associe un retard statural, une obésité et un retard mental. Le syndrome de Noonan qui est une maladie génétique non chromosomique touchant les deux sexes, sporadique ou de transmission autosomique dominante, due à des mutations situées en aval du récepteur de l’hormone de croissance associant un phénotype proche du syndrome de Turner et une cardiopathie spécifique (myocardiopathie ou sténose pulmonaire). Le traitement par GH est efficace mais n’a pas encore d’AMM. L’anomalie du gène SHOX est une chondrodysplasie qui se manifeste par un retard statural avec la présence à la radio de l’avant-bras d’une déformation au niveau du radius appelée déformation de « Madelung ».
Maladies osseuses constitutionnelles. Elles regroupent un ensemble de maladies osseuses constitutionnelles héréditaires, liées essentiellement à des anomalies du développement du cartilage de conjugaison des os longs et des vertèbres. Ce diagnostic peut être suspecté devant un nanisme dysharmonieux associé à des déformations osseuses variables, à préciser par des radiographies du squelette.
Si certaines maladies sont faciles à reconnaitre comme l’achondroplasie, hypochondroplasie, dyschondrostéose, ostéogénèse imparfaite, les muccopolysaccharidoses en raison des signes cliniques évocateurs, d’autres sont difficiles à reconnaitre du fait de leur caractère fruste.
Retard de croissance intra-utérin. Le diagnostic est facilement posé devant un nouveau-né prématuré ou né à terme, dont le poids et/ou la taille est inférieur à -2 SDS par rapport à l’âge gestationnel, selon les courbes de référence de la population.
Un rattrapage post-natal est la situation la plus fréquente et apparait avant l’âge de 2 ans, mais 10% de ces enfants ne rattrapent pas.
Il peut être isolé ou entrer dans le cadre de syndromes particuliers. L’évolution de la croissance est caractérisée par une vitesse régulière avec une courbe < à -2 SDS, et un âge osseux égal ou légèrement inférieur à l’âge chronologique.
Maladies endocriniennes
Hypothyroïdie congénitale. Le retard statural est constant du fait du diagnostic tardif à défaut du dépistage néonatal dans notre pays. L’hypothyroïdie est à l’origine d’un retard mental, qui fait toute la gravité de la maladie, ainsi qu’un retard statural et un retard de l’AO.
Le retard statural peut être le seul signe révélateur dans les cas d’ectopie thyroïdienne et de causes acquises.
Déficit en GH. Le retard statural se manifeste souvent entre 2 et 3 ans par un retard de croissance harmonieux avec une vitesse de croissance diminuée et retard de l’AO. La maladie peut se manifester dès la période néonatale par des hypoglycémies récurrentes.
A l’examen clinique, l’enfant présente un visage triangulaire, front bombant, petit menton, adiposité, l’examen des organes génitaux retrouve une ectopie testiculaire.
Le déficit en GH peut être congénital, acquis ou idiopathique. Le diagnostic est posé par 2 tests de stimulation de GH avec un pic de GH < 20 mUI/l et un taux d’IGF1 bas. L’IRM hypophysaire est réalisée à la recherche de l’étiologie, et l’exploration des autres axes hypophysaires est systématique.
Hypercorticisme. Le plus fréquent est l’hypercorticisme iatrogène secondaire à une corticothérapie prolongée. La maladie de Cushing est exceptionnelle chez l’enfant.
Retard simple de croissance et de puberté. C’est le diagnostic le plus fréquent devant un retard statural à l’adolescence. Il est plus fréquent chez le garçon que la fille, où le retard nécessitera le plus souvent une exploration plus poussée.
La notion de retard pubertaire est souvent retrouvée chez l’un des parents. La croissance de la petite enfance s’effectue normalement jusqu’à l’âge de 7-8 ans, puis la vitesse de croissance ralentit. Le ralentissement de la croissance staturale et de la maturation osseuse sont parallèles, l’AO est inférieur à l’AC, et concordant avec le stade pubertaire. A l’adolescence, la puberté tarde à se faire, entrainant par conséquent un retard à l’apparition du pic de croissance statural habituel, mais la courbe de croissance rejoindra son canal antérieur à la fin de la puberté.
Nanisme psycho- affectif. Il résulte des difficultés rationnelles entre l’enfant et son entourage ; il est évoqué sur le contexte familial et social ; les explorations biologiques sont variables. La séparation de l’enfant de son milieu permet de corriger la symptomatologie.
Petite taille familiale
C’est un diagnostic d’exclusion, la taille est < -2 DS sur la courbe de croissance, qui est normale pour la taille des parents, c’est-à-dire qui se situe dans la « taille cible », et la vitesse de croissance est normale. A partir de l’âge de 2-3 ans, la courbe suit en parallèle les lignes de percentiles de la courbe de croissance, avec un AO = AC.
Une petite taille familiale est parfois aussi appelée petite taille génétique parce que l’on soupçonne que la cause est due à des facteurs génétiques encore inconnus
Cette petite taille familiale est parfois associée à un développement pubertaire retardé qui va accentuer le retard statural.
Pour ce type de petite taille, il n’y a pas encore de traitement approuvé. Le traitement par hormone de croissance doit donc être considéré comme une thérapie expérimentale. Aux États-Unis, un traitement par hormone de croissance à haute dose est parfois possible lorsque l’on satisfait à des conditions très strictes. Les résultats du traitement sont très variables.
Retard de croissance idiopathique
C’est une petite taille chez un enfant en bonne santé. Le diagnostic est retenu après avoir cherché
toutes les causes possibles.
A la différence de la petite taille familiale, la taille de l’enfant ne se situe pas dans la taille cible. L’AO est égal à l’AC, et il n’y a pas de retard pubertaire.
Traitement
Le traitement d’un retard de croissance est le traitement de la cause lorsque cela est possible car ce n’est pas toujours le cas tel qu’on le voit dans les maladies constitutionnelles.
L’hormone de croissance recombinante a ouvert de nouvelles perspectives thérapeutiques ; l’enregistrement en Algérie est acquis pour : le déficit en GH, le syndrome de Turner, l’insuffisance rénale chronique et le retard de croissance intra- utérin.
Conclusion
Le retard statural est un motif de consultation fréquent en pédiatrie, les étiologies sont nombreuses.
Une démarche diagnostique bien conduite est nécessaire afin de pouvoir poser le bon diagnostic et
proposer par conséquent le traitement adéquat en fonction de l’étiologie.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts
Références
- Michael Conn.- Growth Disorders. In: P. Michael Conn – Pediatric Endocrinology, a practical clinical guide, Second edition 2013, New York: 3-137
- Brauner, Conduite pratique devant une anomalie de la croissance, Encycl. Med. Chir. 2008,4-005-A-10: 14p
- F. Backeljauw and al. Disorders of growth hormone/insulin-like growth factor secretion and action. in: Mark A. Sperling, Pediatric Endocrinology, chapter 10, Elsevier 4th edition 2014.
- Adamsbaum. Age osseux et diagnostic des troubles de la croissance. Encycl. Med. Chir. 2002 30-480-A-20: 8p
- Alan D Rogol and al. Diagnostic approach to children and adolescents with short stature. Up to date 2019
- G. Murray, P.E. Clayton. Disorders of growth. in: T. Mehul Dattani, Brook’s clinical pediatric endocrinology, John Willey 7th edition 2020: 199-234.
- Growth Hormone Research Society. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. J Clin.Endocrinol. Metab 2000; 85: 3990–3993
- Wagner, I.V., Paetzold, C., Gausche, R. et al. Clinical evidence based cutoff limits for GH stimulation tests in children with a backup of results with reference to mass spectrometry. Eur. J. Endocrinol 2014; 171: 389–397.
- Clayton, P.E., Cianfarani, S., Czernichow, P. et al. Management of the child born small for gestational age through to adulthood: a consensus statement of the International Societies of Pediatric Endocrinology and the Growth Hormone Research Society. J. Clin. Endocrinol. Metab 2007; 92: 804–810.
- Cohen, P., Rogol, A.D., Deal, C.L. et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab. 2008: 4210–4217
[1] Abréviations : AO = âge osseux ; AC = âge chronologique