D. BENLALDJ, M.A. MOUEDEN, F. SEGHIER, Service d’Hémobiologie et banque de sang, CHU Benaouda Benzerdjeb, Oran.
Résumé : La maladie de Von Willebrand (VWD) est la maladie hémorragique constitutionnelle la plus fréquente dans le monde, elle est due à une anomalie du facteur Von Willebrand (VWF) qui est une glycoprotéine multimérique très complexe. Cette maladie est de transmission autosomique dominante, parfois récessive mais peut aussi être acquise. Notre étude a été réalisée au niveau du service d’hémobiologie et banque de sang du CHU Benaouda Benzerdjeb d’Oran. Au totale 53 patients ont été diagnostiqués dont 32 présentent une VWD type 1, et 21 une VWD type 1 probable ; le sexe ratio homme femme est de 0,36. L’âge moyen du diagnostic est de 25,84 ans pour le type 1 ; et 18,28 pour le type 1 probable. La notion de consanguinité et la présence d’antécédents familiaux est retrouvée chez la moitié des patients. Les ecchymoses et les épistaxis suivis des ménorragies et hémorragies buccales sont les signes cliniques les plus fréquemment décrits par les patients. Nous retrouvons une différence significative entre les valeurs moyennes du Bleeding Score, du temps de céphaline TCA, de l’activité VWF cofacteur de la ristocétine VWF:RCo, du taux VWF antigène VWF:Ag et du taux de facteur VIII FVIIIc entre les patients de type 1 et type 1 probable. Le diagnostic de la maladie de Von Willebrand type 1 est problématique, étant sujet à un sur-diagnostic, à un sous-diagnostic ou à un diagnostic erroné. Selon le théorème de Bayes qui est une approche mathématique probabiliste, notre démarche diagnostique nous permet un diagnostic de type 1 avec une probabilité minimum de 80%, et cette probabilité est de 50% pour le type 1 probable.
Mots-clés et Abréviations : Maladie de Von Willebrand, VWD type 1, VWD type 1 probable
Abstract : Von Willebrand disease (VWD) is the most common constitutional bleeding disorder in the world, it is a result of von Willebrand factor abnormality (VWF). The VWF is a very complex multimeric glycoprotein. VWD disease is inherited autosomal dominant sometimes recessive but can also be acquired. Our study was conducted at the heamobiology and blood bank department of Oran University Hospital. A total of 53 patients were diagnosed including 32 patients presenting a VWD type 1 and 21 a Low Von Willebrand, the sex ratio is 0.36. The average age of diagnosis is 25.84 years for type 1 and 18.28 for low Von Willebrand. The consanguinity and the presence of a bleeding family history is found in half of the patients. Bruising and epistaxis followed by menorrhagia and oral haemorrhage are the most common bleeding described by patients. We found a significant difference between the mean values of the Bleeding Score, the partial thromboplastin time (TCA), the VWF cofactor VWF:RCo, the VWF antigen VWF:Ag, and the factor VIII FVIIIc between the patients VWD type 1 and low Von Willebrand. Diagnosis of von Willebrand disease type 1 is difficult because it’s subject to over-diagnosis, under-diagnosis or misdiagnosis. According to Bayes theorem, which is a probabilistic mathematical approach, our diagnostic approach diagnoses VWD type 1 with a minimum probability of 80% and this probability is 50% for low Von Willebrand.
Key-words : Von Willebrand disease, VWD type 1, low Von Wille- brand
Introduction
La maladie de Von Willebrand (VWD) est la maladie hémorragique constitutionnelle la plus fréquente dans le monde, 1% de la population générale est atteinte de ce trouble hémorragique. Elle touche les deux sexes et est causée par un déficit et/ou un dysfonctionnement du facteur Von Willebrand (VWF) [1].
Selon la première enquête épidémiologique sur la prévalence de la VWD réalisée par Rodeghiero et al [2] (population de 1.218 enfants âgés de 11 à 14 ans), la prévalence de la VWD dans la population générale était de 0,82% tous types confondus. Aussi, l’étude de Wemer et al retrouve une prévalence de 1,3%, et cette prévalence n’est pas limitée à un seul groupe ethnique [3].
La VWD peut être cliniquement silencieuse. En effet, en 1998, l’enquête sur la prévalence des sujets symptomatiques atteints de VWD a révélé que les formes symptomatiques étaient moins fréquentes, environ 100 cas pour 1 million d’habitants versus 3-5 cas par millions d’habitants pour les formes sévères [4].
La VWD est liée à une anomalie du VWF qui est une glycoprotéine multimérique très complexe [5]. En hémostase, le VWF a deux fonctions principales : il est responsable de l’attachement des plaquettes au sous-endothélium vasculaire ; ainsi, il permet la formation du clou plaquettaire, et il assure également le transport du facteur VIII (FVIII), sa protection de la protéolyse lui permet ainsi de le concentrer au site de l’hémostase [6]. Plusieurs études ont été menées pour comprendre sa structure et son rôle en hémostase, ce qui a permis de démontrer l’importance du VWF dans les interactions impliquant les plaquettes et le sous endothélium à différentes forces de cisaillement. Le VWF est capable aussi, au-delà de l’hémostase, d’induire des voies de signalisation cellulaire. Il est associé à plusieurs autres processus physiopathologiques tels que l’angiogenèse, l’inflammation, l’apoptose des cellules tumorales et les métastases [7].
La VWD est de transmission autosomique dominante, parfois récessive, mais peut aussi être acquise (premier cas de VWD acquise décrit en 1968)[8].
Cette maladie est très hétérogène dans son expression phénotypique, clinique et biologique et les manifestations hémorragiques sont variées et principalement caractérisées par des hémorragies cutanéomuqueuses. Cette hétérogénéité est à la base de la classification de la VWD. Il existe trois types de la VWD selon la nature du déficit en VWF, le type 1 (déficit quantitatif partiel), le type 3 (déficit quantitatif total) et le type 2 qui révèle le plus de variabilités génétiques (déficit qualitatif) [9].
Le diagnostic de la VWD est basé sur une évaluation de la sévérité des signes cliniques et sur une série des tests biologiques antigéniques et fonctionnels variés, en constante évolution qui imposent la mise en place d’une stratégie de diagnostic adaptée [10]. Mais en pratique, la mesure du VWF antigénique (VWF:Ag) ; et celle de l’activité cofacteur de la ristocétine (VWF:Rco), continuent d’être les tests diagnostiques les plus importants, et ceci malgré l’évolution de la compréhension de la génétique moléculaire de la VWD. Le test génétique en tant qu’élément du diagnostic est limité à certains sous-types [10,11].
Une bonne démarche diagnostique est la clé du dépistage des patients et permet ainsi de leur proposer un suivi et une prise en charge thérapeutique spécialisée pluridisciplinaire et, suivant la sévérité de la maladie, le traitement des patients atteints de la VWD a pour objectif l’arrêt des saignements et leurs préventions en cas de challenges hémostatiques. Il a été relativement inchangé et repose toujours sur la stimulation de la libération de VWF endogène avec de la desmopressine et la perfusion de concentrés de VWF [12,13,14].
Méthodologie
Cette étude a été réalisée au niveau du service d’hémobiologie et banque de sang du CHU d’Oran. Il s’agit d’une étude prospective de 4 ans et demi (janvier 2013 à juin 2017) portant sur des patients adressés des services d’hématologie et de pédiatrie de 10 wilayas de l’ouest Algérien (Oran, Mascara, Relizane, Mostaganem, Sidi Bel Abbes, Saida, Bechar, Tlemcen, Ain Temouchent et Tiaret).
Critères d’inclusion
- La présence d’une histoire hémorragique documentée personnelle et/ou familiale significative.
- Résidence dans l’Ouest Algérien.
Critères diagnostiques
Patients classés en type 1 si les critères suivants sont vérifiés :
- Critère 1 : Hémorragies cutanéomuqueuses significatives (score hémorragique positif ≥ 3).
- Critère 2 : Tests de laboratoire compatibles avec un type 1 (VWF:Rco et / ou VWF:Ag < 30% après deux déterminations).
- Critère 3 : Histoire familiale compatible avec un type 1 de transmission dominante (les traits dominants sontpresque toujours accompagnés d’une histoire familiale positive).
- Critère 4 : VWF:CB/VWF:Ag > 0,6.
- Critère 5 : VWF:Rco/VWF:Ag > 0,6.
- Critère 6 : FVIIIC /VWF:Ag > 0,6.
Patients classés en type 1 probable si les critères 1 ou 3 sont absents et les critères suivants sont vérifiés :
- Critère 2 : Tests de laboratoire VWF:Rco et/ou VWF:Ag [30%-50%] après deux déterminations.
- Critère 4 : VWF:CB/VWF:Ag > 0.6.
- Critère 5 : VWF:Rco/VWF:Ag > 0.6.
- Critère 6 : FVIIIC /VWF:Ag > 0.6.
Les antécédents hémorragiques personnels et familiaux des patients sont quantifiés par un questionnaire standardisé (ISTH-BAT) [15], ce qui nous permet de calculer pour chaque patient un score hémorragique qui varie entre 0 (pas de saignement spontané, pas d’hémorragie après chirurgie, extraction dentaire et accouchement), et 52 (saignement grave et important nécessitant une transfusion pour chaque symptôme du questionnaire).
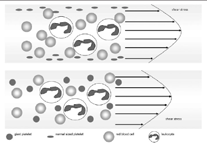
Pour chaque patient nous avons réalisé des tests de dépistage (figure 1) qui incluent un hémogramme, un bilan standard de coagulation (Temps de Quick [TQ], Temps de Céphaline Activateur [TCA] et dosage du fibrinogène. Des tests spécifiques qui incluent un dosage chronométrique du FVIIIc par méthode en un temps sur automate STA compact, dosage de l’activité du facteur Von Willebrand cofacteur de la ristocétine VWF : Rco ; par technique d’agglutination sur lame de SIEMENS, un dosage du VWF:Ag ; par méthode immunologique basée sur la turbidimétrie sur automate STA compact et un dosage immunoenzymatique de la capacité de liaison du facteur Willebrand au collagène :
Asserachrom VWF : CB de STAGO. Notre laboratoire participe à un programme international de contrôle externe de qualité : IEQAS (International External Quality Assessment Scheme), sous le parrainage de la fédération mondiale de l’hémophilie et cela depuis 2011.
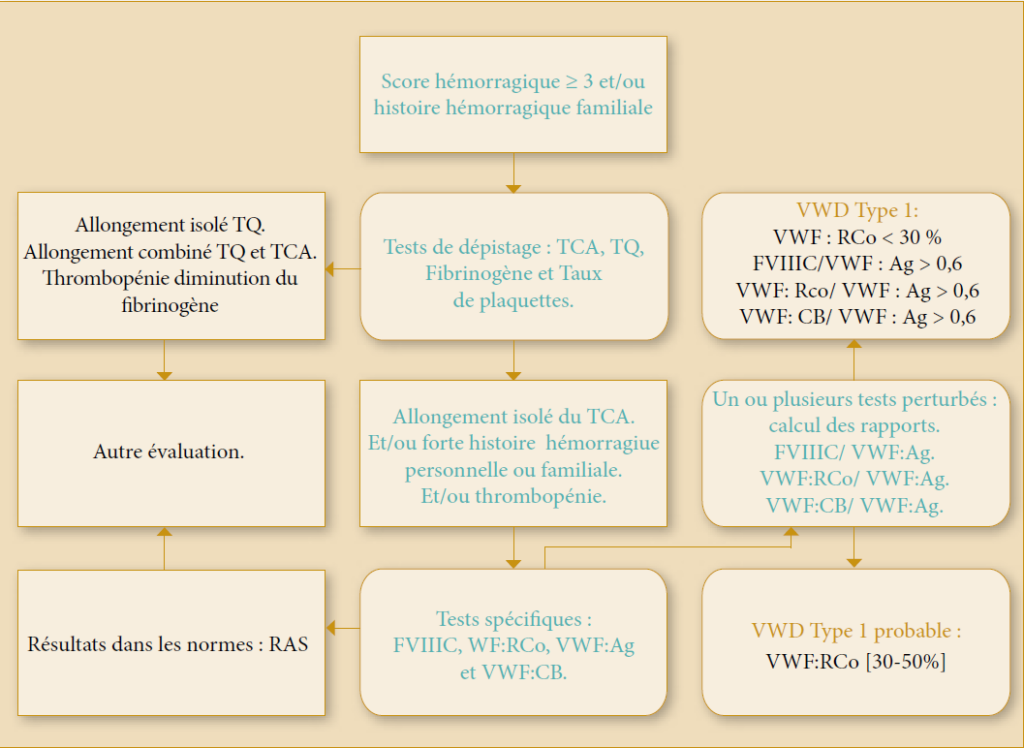
Figure 1 : Arbre décisionnel pour diagnostique de la maladie de Von Willebrand type 1, au service d’hémobiologie du CHU d’Oran.
Résultats
Au total, 53 patients ont été diagnostiqués, dont 32 présentent une VWD type 1, et 21 une VWD type 1 probable, le sexe ratio homme femme est de 0,36. L’âge moyen du diagnostic est de 25,84 ans pour le type 1 ; et 18,28 pour le type 1 probable.
La notion de consanguinité est retrouvée chez la moitié des patients de type 1, et chez seulement 9,5% des patients de type 1 probable.
On retrouve des antécédents familiaux chez plus de la moitié des patients 62,50% et 57,15% pour le type 1 et type 1 probable respectivement.
Fréquence des signes cliniques
Les ecchymoses et les épistaxis suivies des ménorragies et hémorragies buccales sont les signes cliniques les plus fréquemment décrit par les patients.
| Type 1 (%) | Type 1 probable (%) | |
| Ecchymose | 78 | 75 |
| Épistaxis | 75,6 | 80 |
| Hémorragie buccal | 30,10 | 35 |
| Hémorragie après coupure | 6 | 5 |
| Hémorragie après chirurgie | 0 | 5 |
| Circoncision | 6 | 0 |
| Extraction dentaire hémorragique | 42,5 | 15 |
| Ménorragie | 51 | 40 |
Résultats du bilan de dépistage
| Tests VWD type 1 (n = 32) VWD type 1 probable (n = 21) |
| TP (%) Moyenne 96,76 95,60 p=0,416 Maximum 100 100 Minimum 83 82 Écart type 4,96 6,24 |
| TCA (ratio) Moyenne 1.41 1.10 p<0,0005 Maximum 3.00 1.60 Minimum 1.00 0,95 Écart type 0,52 0,145 |
| Fibrinogène (g/l) Moyenne 2.97 3.40 Maximum 4,00 4,20 p=0.003 Minimum 1,99 2,04 Écart type 0,58 0,56 |
| Plaquettes (G/L) Moyenne 269 272 p=0,876 Maximum 400 537 Minimum 142 185 Écart type 62,46 83,43 |
| Hémoglobine (g/dl) Moyenne 11,36 12,28 p=0,006 Maximum 15,60 14,80 Minimum 5,70 10,00 Écart type 2,23 1.33 |
On retrouve une différence significative entre les valeurs moyennes du TCA, fibrinogène et hémoglobine entre es patients de type 1 et type 1 probable.
Résultats des tests spécifiques et spécialisés
| Tests VWD type 1 (n = 32) VWD type 1 probable (n = 21) |
| VWF:RCo (%) Moyenne (Écart type) 23,24(9,03) 37.6(6,24) p<0,0005 Maximum 40 4,84 Minimum 3 30 |
| VWF:Ag (%) Moyenne (Écart type) 29,48 (13,65) 47,65(10,06) p<0,0005 Maximum 59 68 Minimum 3 32 |
| FVIIIc (%) Moyenne (Écart type) 46,21(32,9) 70,35(30,30) p=0,002 Maximum 144 130 Minimum 2 26 |
| VWF:CB (%) Moyenne (Écart type) 57,96 65,45 p=0,212 Maximum 140 95 Minimum 9 45 Ecart type 29,65 18,66 |
| Score hémorragique Moyenne (Écart type) 7,76(3,39) 5,85(285) p=0,007 Maximum 20 11 Minimum 2 0 |
| VWF:RCo/VWF:Ag Moyenne (Écart type) 0,93 (0,49) 0,81(0,125) p<0,0005 Maximum 3,25 1,03 Minimum 0,60 0,61 |
| FVIIIc/VWF:Ag Moyenne (Écart type) 1,54 (0,802) 1,51(0,69) p=0,85 Maximum 4,5 3,42 Minimum 0,4 0,47 |
| VWF:CB /VWF:Ag Moyenne (Écart type) 2,05 (1,13) 1,39(0,49) p 0,001 Maximum 5,6 2,56 Minimum 0,95 0,82 |
On retrouve une différence significative des valeurs moyennes du VWF:RCo, VWF:Ag, FVIIIc, TCA, Bleeding Score, VWF:RCo /VWF:Ag et VWF:CB /VWF:Ag entre les patients de type 1 et type 1 probable.
Corrélation de Pearson entre les résultats des différents tests diagnostiques chez les patients de type 1
| VWF : RCo | VWF : Ag | FVIIIc | VWF : CB | TCA ratio | BS | Taux Hb |
| VWF : RCo | 0,908 p< 0,0005 | 0,639 p< 0,0005 | 0,447 p= 0,029 | -0,76 p<0,0005 | -0,536 p=0,001 | 0,566 p= 0,001 |
| VWF : Ag | 0,822 p<0,0005 | 0,547 p=0,006 | -0,804 p<0,0005 | -0,529 p=0,002 | 0,523 p=0,002 | |
| FVIIIc | 0,368 p=0,076 | -0,818 p<0,0005 | -0,382 p= 0,028 | 0,365 p=0,037 | ||
| VWF : CB | -0,568 p=0,004 | -0,571 p= 0,004 | 0,103 p=0,61 | |||
| TCA ratio | 0,524 p=0,002 | -0,16 p=0,37 | ||||
| BS | -0,503 p= 0,003 |
On constate une forte corrélation entre VWF:RCo et VWF:Ag, entre TCA ratio et VWF:RCo, TCA ratio et VWF:Ag, TCA ratio et FVIIIc aussi entre VWF:Ag et FVIIIc.
On retrouve une corrélation moyenne entre VWF:RCo et le FVIIIc, VWF:CB, BS, taux d’hémoglobine ; entre VWF:Ag et VWF:CB, BS, taux d’hémoglobine ; entre VWF:CB et BS, TCA ratio ; entre BS et taux d’hémoglobine.
On retrouve une mauvaise corrélation entre FVIIIc le taux d’hémoglobine et BS.
On ne retrouve pas de corrélation entre taux d’hémoglobine, VWF:CB et TCA ratio.
Comparaison entre les moyennes du VWF:RCo, VWF:Ag et le score hémorragique des sujets de groupe sanguin O et non O chez les patients de type 1
| Groupe sanguin O Groupe sanguin non O |
| VWF:RCo (%) Moyenne 29,06 23,87 p<0,0005 Maximum 48 35 Minimum 5 4 Écart type 9,97 8,57 |
| VWF:Ag (%) Moyenne 34,94 30,55 p=0,132 Maximum 54 48 Minimum 3 3 Écart type 11,40 12,09 |
| FVIIIc (%) Moyenne 66,76 46,77 p=0,052 Maximum 144 80 Minimum 6 2 Écart type 39,32 28,68 |
| VWF:CB (%) Moyenne 59,77 59,13 p=0,939 Maximum 140 105 Minimum 9 15 Écart type 29,37 33,16 |
| Score hémorragique Moyenne 6,35 8,33 p=0,04 Maximum 13 20 Minimum 2 3 Écart type 3,64 4,89 |
On retrouve une différence significative entre la moyenne des valeurs du VWF:RCo et le score hémor- ragique entre les sujets de groupe sanguin O et non O.
Discussion
Le diagnostic de la maladie de von Willebrand type 1 est problématique, étant sujet à un sur-diagnostic, à un sous-diagnostic ou à un diagnostic erroné [16]. Notre démarche diagnostique (figure 1) est basée sur une série de tests de dépistage réalisés à chaque fois que le score hémorragique est ≥ 3 ; avec ou sans histoire hémorragique familiale, suivis par des tests spécifiques de première ligne.
Selon le théorème de Bayes qui est une approche mathématique probabiliste du diagnostic du type 1 de la VWD publié en 2008 par Tosseto et al [17], notre démarche diagnostic nous permet un diagnostic de type 1 avec une probabilité minimum de 80% ; et cette probabilité est de 50% pour le Type 1 probable. Nous avons choisi d’inclure le VWF:CB comme test de première ligne pour compléter le dosage de l’activité VWF:RCo.
Selon l’étude Favaloro et al [18], le plasma de VWD type 1 a été mal identifié en type 2 dans 11% des cas et celui de type 2 mal identifié en type 1 ou type 3 dans 20% des cas, il a été démontré que les laboratoires qui réalisent le VWF: RCo seul, sans le VWF:CB, sont susceptibles de faire ce genre d’erreurs avec un risque multiplié par 3 voire par 6 ; et le plasma normal a été mal identifié comme plasma VWD dans 5% des cas, les laboratoires qui ont effectué le VWF: RCo sans VWF:CB étaient 10 fois plus susceptibles de faire une telle erreur que ceux qui ont effectué le VWF:CB.
Les taux d’erreur de diagnostic sont considérablement réduits lorsque les tests sont complets et incluent le VWF:CB. Une autre étude de Favaloro et al 2017 [16], démontre que si les laboratoires n’utilisent pas le VWF:CB, la VWD de type 2M continuera à être sous diagnostiquée et/ou mal diagnostiquée comme étant un VWD de type 2A ou type 1.
Les types 2A, 2B et PT-VWD continueront à sous diagnostiqués, ou bien à être diagnostiqués à tort comme une VWD de type 1 ou TPI [16].
Le VWF:CB détecte mieux que le VWF:RCo l’absence des multimères de hauts poids moléculaires et par conséquent, il devrait être rajouté au panel de tests utilisés dans le diagnostic de la VWD [19]. Baronciani et al recommandent également l’utilisation du VWF:CB comme test de diagnostic en plus du VWF:RCo et VWF:Ag pour distinguer non seulement le type 2A du type 2M mais aussi le type 2A du type 2B, et recommandent également de l’utiliser comme test de dépistage des patients avec anomalie de l’hémostase primaire [20].
Caractéristiques générales des patients
Au total 53 patients ont été diagnostiqués dont 32 patients présentent une VWD de type 1, et 21 patients une VWD de type 1 probable ; le sexe ratio homme/femme est de 0,36 ; cette nette prédominance féminine pourrait s’expliquer par le nombre plus élevé de femmes qui consultent en particulier en raison du chalenge hémostatique que représente les ménorragies et les accouchements.
Les formes les plus sévères de la VWD sont découvertes plus tôt dans la vie et le diagnostic du type 1 est en général tardif, ce qui concorde avec les résultats de notre population, avec une moyenne d’âge de diagnostic de 25,84 ans pour le type 1 et 18,28 pour le type 1 probable.
La notion de consanguinité est retrouvée chez la moitié des patients de type 1 et chez seulement 9,5% des patients type 1 probable. En effet, dans le type 1 probable, la probabilité d’existence d’une mutation génétique et assez faible, ce type peut être le résultat de combinaisons de facteurs modulants le taux plasmatique du VWF.
Dans le type 1, la présence d’une histoire familiale facilite le diagnostic. En effet selon l’approche Bayésienne de Tossetto et al, l’histoire familiale est importante à considérer dans le calcul de la probabilité d’une VWD encore plus importante que le score hémorragique [17], dans notre étude nous retrouvons des antécédents familiaux chez plus de la moitié des patients de type 1 et de type 1 probable, soit 62,50% et 57,15% respectivement.
Fréquence des signes cliniques
Les ecchymoses et les épistaxis suivies de ménorragies sont les signes cliniques les plus fréquemment décrits par les patients.
Nous retrouvons une différence significative de fréquence des signes cliniques avec l’étude italienne pour les saignements post opératoires, les saignements après coupure ; tandis que l’étude italienne décrit des hématomes, des hémarthroses et des hémorragies intracrâniennes dans le type 1.
| Type 1(%) n = 32 | Type 1 probable (%) n = 21 Type 1 n = 944 Italie | |
| Ecchymose | 78 | 75 0 |
| Épistaxis | 75,6 | 80 56 |
| Hémorragie buccal | 30,10 | 35 30 |
| Hémorragie après coupure | 6 | 5 36 |
| Hémorragie après chirurgie | 0 | 5 20 |
| Circoncision | 6 | 0 0 |
| Extraction dentaire hémorragique | 42,5 | 15 31 |
| Hémorragie intracrânienne | 0 | 0 0,5 |
| Ménorragie | 51 | 40 31 |
| Hématome | 0 | 0 14 |
| Hémarthrose | 0 | 0 2 |
Il existe une différence significative entre les valeurs moyennes du TCA des type 1 et type 1 probable p<0.0005, cette différence est liée au taux de FVIIIC qui est plus bas dans le type 1 (p=0.002), de même que le taux de VWF:Ag et VWF:RCo sont plus bas dans le type 1 (p<0.0005). Le taux de fibrinogène (p=0.003) est plus bas dans le type 1. Le taux d’hémoglobine est aussi plus bas chez les patients de type 1 (p=0.006) ; ce résultat est conforté par la moyenne plus élevée du score hémorragique dans le type 1 (p=0.007), qui expose ces patients à un risque d’anémie plus élevé que pour le type 1 probable. Nos résultats sont comparables aux résultats du taux d’hémoglobine (10,6 g/dl) du type 1 de l’étude tunisienne (p=0.057) [22]. Les ratios et VWF:RCo/ VWF:Ag et VWF:CB/VWF:Ag sont plus bas dans le type 1 (p<0.0005 et p=0.001). Chez les patients de type 1 de l’étude tunisienne, le ratio VWF:RCo/VWF:Ag est de 0,97 ; très proche de nos résultats (0,93) p=0.707 [22].
Corrélation de Spearman entre les résultats des différents tests diagnostiques chez les patients de type 1
On retrouve une bonne corrélation positive entre VWF:RCo et VWF:Ag (r=0.908, p<0.0005) puisque dans le type 1, le ratio VWF:RCo/VWF:Ag est en général proche de 1.
On retrouve une corrélation moyenne entre VWF:RCo et le FVIIIC (r=-0.639, p<0.0005), cette corrélation est meilleure entre le VWF:Ag et le FVIIIC (r=0.822 p<0.0005). En effet le taux de FVIIIC est plus proche du taux de VWF:Ag que VWF:RCo et ce dernier est toujours plus abaissé.
Il existe une corrélation moyenne entre VWF:RCo et le VWF:CB (r=-0.447, p=0.029), et entre le VWF:Ag et le VWF:CB (r=0.547, p=0.006).
Cette corrélation a été également décrite par l’équipe du professeur Hariti [23]. En effet ce test permet d’explorer les multimères de haut poids moléculaire, auxquels le VWF:RCo et VWF:Ag sont beaucoup moins sensibles. On note une bonne corrélation négative entre TCA ratio et VWF:RCo (r=-0.76 p<0.0005), VWF:Ag (r=-0.804, p<0.0005), FVIIIC (r=-0.818, p=0.004). Le TCA est
un test de dépistage qui dépend du taux de FVIIIc, lui-même lié aux taux de VWF.
Une corrélation moyenne négative existe entre le VWF:RCo, VWF:Ag, VWF:CB, de même le taux d’hémoglobine et le score hémorragique (respectivement r=-0.536, p=0.001 ; r=-0.529, p=0.002 ; r=-0.571, p=0.004 ; r=-0.503, p=0.003).
En effet un taux bas de VWF n’est pas toujours synonyme de saignement et l’histoire hémorragique personnelle est influencée par différents facteurs (âge, sexe).
On note également une corrélation moyenne entre VWF:RCo, VWF:Ag, et le taux d’hémoglobine (respectivement r=0.566, p=0.001 ; r=0.523, p=0.002). Une VWD n’est pas toujours synonyme d’anémie en plus dans le type 1 les formes modérées sont prédominantes.
Comparaison entre les moyennes du VWF:RCo, VWF:Ag et le score hémorragique des sujets de groupe sanguin O et Non O chez les patients de type 1
Pour les patients de groupes sanguins non O la valeur moyenne du VWF:RCo est plus basse de 5,19% par rapport au groupe sanguin O (p<0.0005) ; et VWF:Ag est plus basse de 4,39% (p>0.05).
On retrouve aussi dans notre étude une différence de 2 points pour le score hémorragique entre les sujets de groupe sanguin O et non O (p=0.04).
On constate clairement chez nos patients que dans le type 1, les sujets de groupe non O ont des taux plasmatiques de VWF plus bas et un score hémorragique plus élevé.
L’étude de Tosseto et al sur 204 patients démontre que les différences de VWF:RCo ou VWF:Ag dans le groupe sanguin O contre non O, n’est pas significative et n’influence pas le diagnostic (p>0,1) [24].
Limite
La première des difficultés que nous avons rencontrées lors de notre étude est le recrutement des patients, étant donné le caractère labile du taux de VWF et la nécessité de répéter les tests diagnostiques. Aussi, dans un contexte économique difficile, la disponibilité des réactifs reste problématique puisque le diagnostic de VWD repose sur une large gamme d’examens de laboratoire et donc le coût d’un diagnostic reste élevé.
Conclusion
La fréquence élevée de la VWD type 1 dans le monde, et le nombre limité de patients diagnostiqués en Algérie, reflètent un problème de dépistage du fait de l’inexistence de recommandations nationales ou de guidelines pour le diagnostic de la VWD. De plus, ce dernier est complexe du fait de la fluctuation du taux plasmatique de VWF, et de la grande série de tests fonctionnels nécessaires. En effet, les critères de diagnostic de cette maladie font l’objet de révisions régulières et les stratégies diagnostiques sont en constante évolution.
La VWD reste sous diagnostiquée dans notre pays en comparaison avec l’hémophilie et les autres déficits rares en facteur de la coagulation, de plus il n’existe pas de registre national de la VWD et les publications nationales sont peu nombreuses et ne décrivent que de petites séries. Cette maladie pose un réel problème de santé publique, compte tenu de la fréquence des épisodes hémorragiques, de leur retentissement sur la qualité de vie des patients et leur entourage, et sur la société.
Références
- Zimmerman, TS, Ratnoff, OD, Powell, AE. Immunologic differentiation of classic haemophilia (factor VIII deficiency) and von Willebrand’s disease. With observations on combined deficiencies of antihemophilic factor and proaccele- rin (factor V) and on an acquired circulating anticoagulant against antihemo- philic factor. Journal of Clinical Investigation. 1971; 50:244–254.
- Rodeghiero F., Castaman, G. et Dini, E. Epidemiological investigation of the pre- valence of von Willebrand’s disease. Blood 1987; 69(2): 454-9.
- Werner EJ, Broxson EH, Tucker EL, Giroux DS, Shults J, Abshire TC. Preva- lence of von Willebrand disease in children: a multi-ethnic study. J Pediatr.1993; 123: 893-898.
- Joint WHO / ISTH Meeting, Londres 1998.
- Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Bio- chem. 1998; 67:395–424.
- Ruggeri, Z.M. Von Willebrand factor, platelets and endothelial cell interac- tions. J Thromb Haemost 2003; 1(7):1335-42.
- Lenting PJ, Casari C, Christophe OD, Denis CV. von Willebrand factor: the old, the new and the unknown. J Thromb Haemost 2012; 10: 2428–37.
- Simone JV, Cornet JA, Abildgaard CF. Acquired von Willebrand’s syndrome in systemic lupus erythematosus. Blood. 1968 Jun;31(6):806-12.
- Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006; 4(10):2103–2114.
- Rodeghiero F. von Willebrand disease: still an intriguing disorder in the era of molecular medicine. Haemophilia 2002; 8(3): 292-300.
- V. Siguret, A.-S. Ribba, D. Meyer. Diagnostic biologique de la maladie de Willebrand. Annales de Biologie Clinique, Novembre-Décembre 1997. Vol.55, N°6, p: 601-6.
- Kaufmann JE, Vischer UM. Cellular mechanisms of the haemostatic effects of desmopressin (DDAVP). J Thromb Haemost. 2003;1(4):682–689.
- Federici AB. Highly purified VWF/FVIII concentrates in the treatment and prophylaxis of von Willebrand disease: the PRO. WILL Study. Haemophilia. 2007 Dec;13 Suppl 5:15-24.
- James PD, Lillicrap D. von Willebrand disease: clinical and laboratory les- sons learned from the large von Willebrand disease studies. Am J Hematol. 2012 May; 87 Suppl 1: S4-11.
- Rodeghiero, F., Tosetto, A., Abshire, T., Arnold, D. M., Coller, B., James, P., Neunert, C., Lillicrap, D. And On Behalf Of The ISTH/SSC Joint VWF And Perinatal/Paediatric haemostasis subcommittees working group 2010, ISTH/ SSC bleeding assessment tool: A standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. Journal of Thrombosis and Haemostasis, 8: 2063–2065.
- Favaloro EJ. Utility of the von Willebrand factor collagen binding assay in the diagnosis of von Willebrand disease. Am J Hematol. 2017 Jan;92(1):114-118.
- Alberto Tosetto, Giancarlo Castaman, and Francesco Rodeghiero. Evidence- based diagnosis of type 1 Von Willebrand disease: a Bayes theorem approach. Blood, 15 April 2008 volume 111, number 8.
- Favaloro EJ, Bonar RA, Mohammed S, Arbelaez A, Niemann J, Freney R, Meiring M, Sioufi J, Marsden K. Type 2M von Willebrand disease – more often misidentified than correctly identified. Haemophilia. 2016 May;22(3): e145-55.
- A. Casonato, E. Pontara, A. Bertomoro, F. Sartorello. Von Willebrand factor col- lagen binding activity in the diagnosis of von Willebrand disease: an alternative to ristocetin co-factor activity? British Journal of Haematology, 2001, 112, 578±583
- Baronciani L, Federici AB, Cozzi G, Canciani MT, Mannucci PM. von Wille- brand factor collagen binding assay in von Willebrand disease type 2A, 2B, and 2M. J Thromb Haemost 2006; 4: 2088–90.
- Federici AB, Mannucci PM, Castaman G et al. The 20-year (1978–98) natu- ral history of von Willebrand disease in Italy: a multicentre retrospective analy- sis on diagnosis and therapy in 1234 patients. Haemophilia 2000; 6: 9.
- Ben Lakhal F, El Borgi W, Gouider E, Meddeb B, Ben Salah N, Hafsia R. Monocentric study of Willebrand’s disease in Tunisia: assets and difficulties. Tunis Med. 2015 Oct;93(10):628-32.
- Ferhat-Hamida MY, Boukerb H, Hariti G. Contribution of the collagen bin- ding activity (VWF:CB) in the range of tests for the diagnosis and classification of von Willebrand disease. Ann Biol Clin (Paris). 2015 Jul-Aug;73(4):461-8.
- A. Tosetto, F. Rodeghiero, G. Castaman et al. Impact of plasma von Wille- brand factor levels in the diagnosis of type 1 von Willebrand disease: results from a multicentre European study (MCMDM-1VWD). Journal of Thrombosis and Haemostasis, 5: 715–72