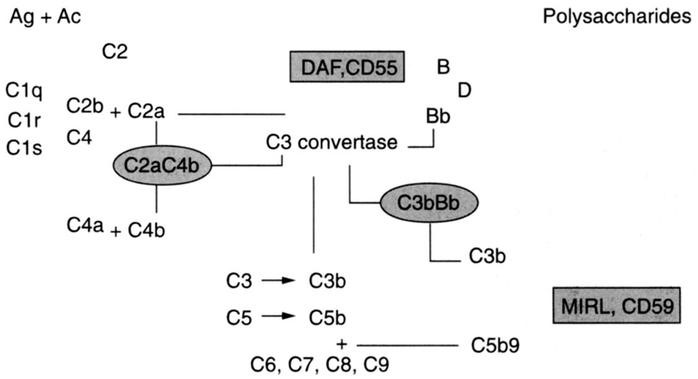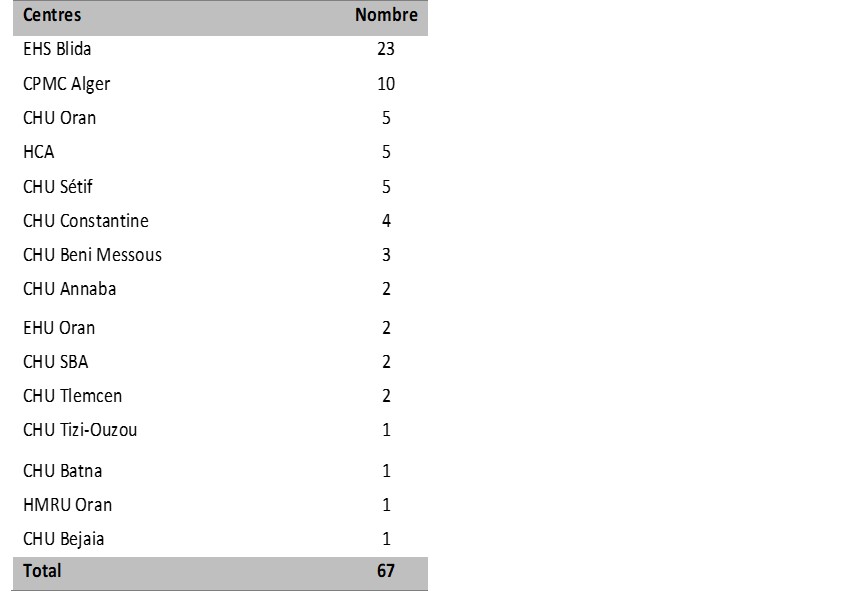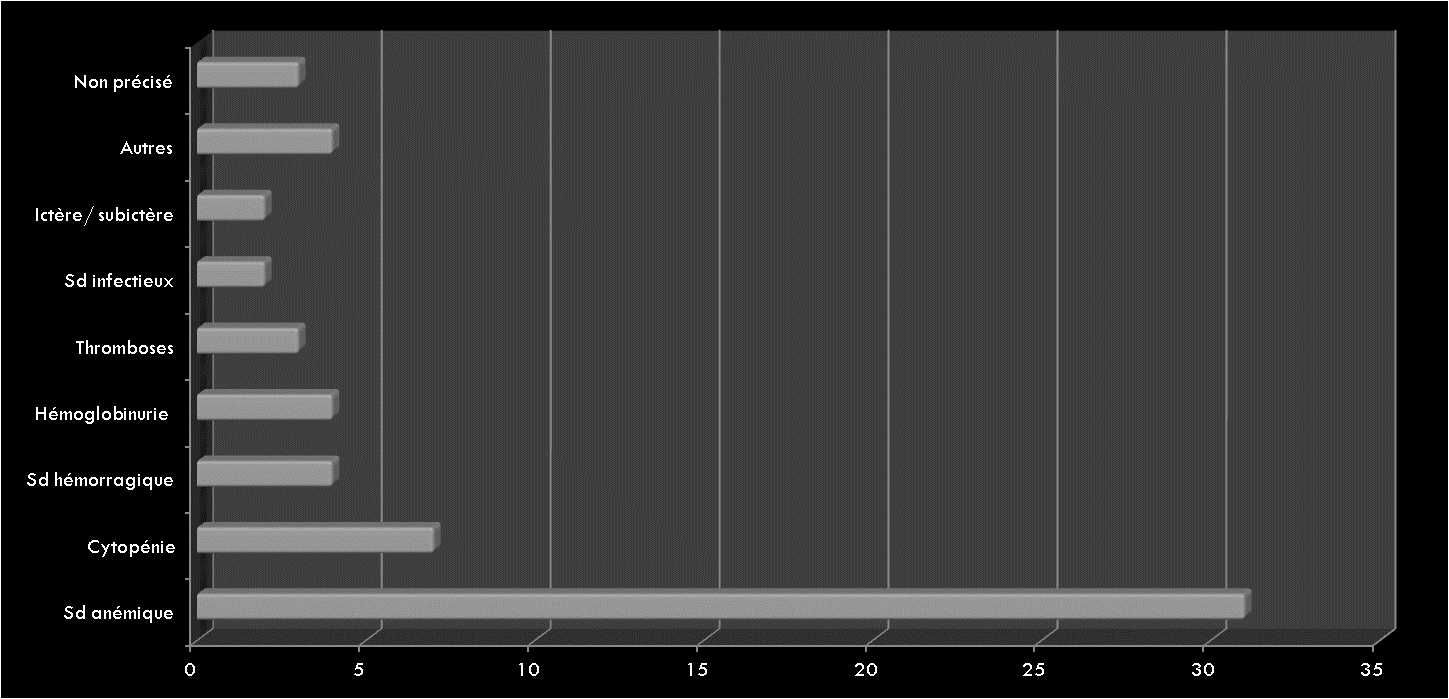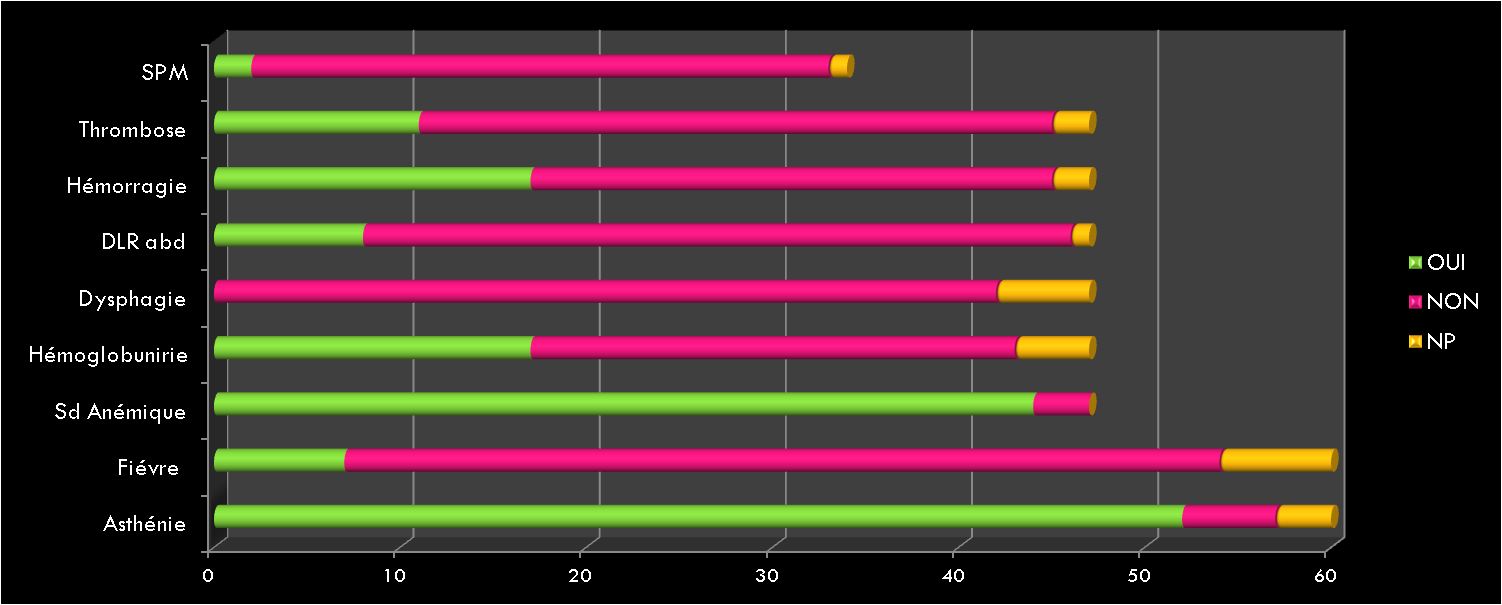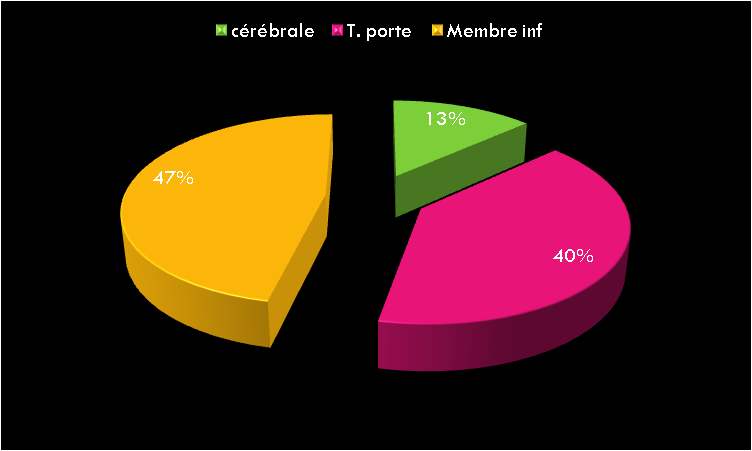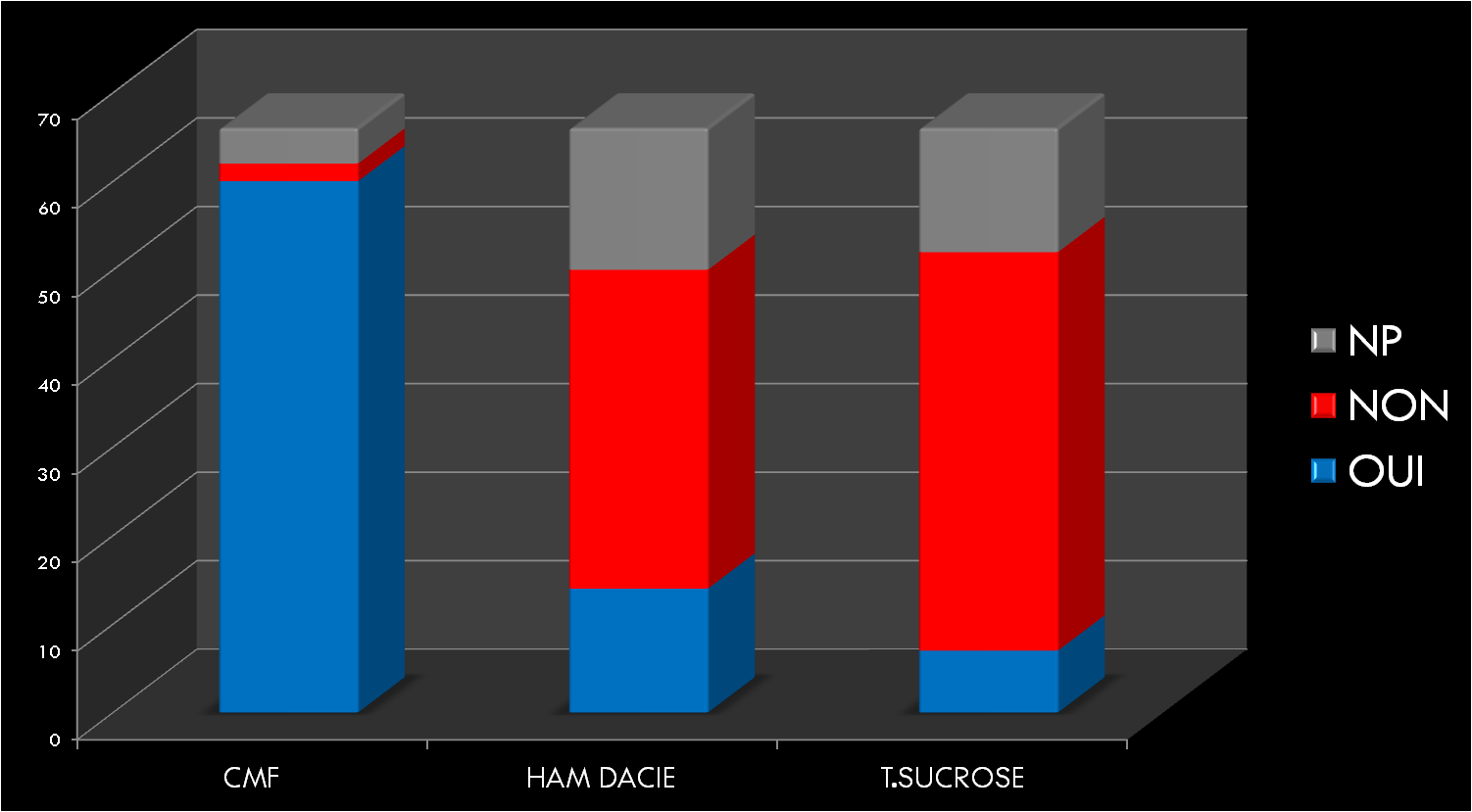
Les Hémorragies digestives basses constituent un symptôme relativement fréquent en consultation pédiatrique. Elles varient entre rectorragie, hématochézie, méléna et saignement occulte.
Ait Idir (1), Z. Benyahia(2),
(1) Service de pédiatrie. Centre de Consultations Spécialisées de l’Armée. Hussein Dey, Alger.
(2) Service de Pédiatrie, Hôpital Central de l’Armée, Ain Naâdja, Alger.
Résumé : Les Hémorragies digestives basses constituent un symptôme relativement fréquent en consultation pédiatrique. Elles varient entre rectorragie, hématochézie, méléna et saignement occulte. La prise en charge est multidisciplinaire et commence toujours par l’appréciation du retentissement sur l’état général avant d’entamer l’enquête étiologique qui est orientée selon l’âge de l’enfant et les données anamnestico-cliniques. C’est ainsi que l’exploration suit un raisonnement logique dans le but de localiser l’origine du saignement et d’établir un plan thérapeutique ciblé. C’est une source de stress parental qui poussent les parents à poser de nombreuses interrogations.
Mots clés : Rectorragie, âge, retentissement, endoscopie, étiologie.
Abstract: Low digestive haemorrhages are a relatively common symptom in paediatric consultation. It is represented by rectorragia, haematochezia, melena and occult bleeding. The management is multidisciplinary and always begins with the assessment of the impact on the general condition before starting the etiological survey which is oriented according to the age of the child, the clinical history and physical data. The investigation must follow a rational way in order to find the source of bleeding. This symptom is often an alarming one for parents, and usually paediatricians are asked to see the patient right away.
Key words: Rectal bleeding, age, impact, endoscopy, cause.
Introduction
Rectorragie / Hématochézie : Les hémorragies digestives basses (HDB) sont le reflet d’un saignement distal du tube digestif. Elles se manifestent généralement par des rectorragies et hématochézies. Ce dernier terme est rarement utilisé, il signifie un saignement rouge issu de l’anus, fait de sang non digéré et accompagné de selles. On la confond toujours avec le terme rectorragie qui traduit un saignement rougeâtre d’origine rectale alors qu’une hématochézie ne précise pas le site du saignement. En général et en pratique, l’appellation rectorragie est préférée et plus utilisée [1][2]. Ces deux types de saignement prennent naissance au-delà de l’angle de Treitz qui est la limite de transition entre la partie haute et la partie basse du tube digestif [2].
Méléna : élimination par l’anus de sang noir, épais et fétide, mélangé ou non aux selles. C’est du sang digéré en rapport avec hémorragie localisée au-dessus de l’angle de Treitz.
Saignement occulte fécal : présence de sang dans les selles qui n’est pas visible à l’œil nu et détectable aux bandelettes réactives.
Ces hémorragies digestives sont dites aiguës et sévères quand elles surviennent en moins de 3 jours et accompagnées de signes de retentissement hémodynamique, trouble de conscience et nécessitent une transfusion [3]. En cas d’hémorragie digestive haute massive, le sang peut arriver à l’anus non digéré et donc rouge.
Données épidémiologiques
Les HDB représentent un motif non rare de consultation [4]. Habituellement, elles ne font pas l’objet d’une hospitalisation [5]. Contrairement à l’adulte où les HDB sont 5 fois moins fréquentes que les HD hautes et représentent 25% de l’ensemble des hémorragies digestives, il n’y a peu de données épidémiologiques chez l’enfant [6,7]. Aux USA entre 2006 et 2011, le Healthcare Cost and Utilization Project Nationwide Emergency Department a recensé 437.000 enfants de moins de 19 ans ayant consulté pour hémorragie digestive, 30% avaient une HDB, et seulement 11,6% ont été hospitalisés [4][5]. Une autre enquête a révélé qu’uniquement 0,3% d’enfants ayant recouru aux urgences pédiatriques avaient des rectorragies sur 40.000 consultations [4][5]. Une cohorte bangladaise récente a objectivé que seulement 1,5% soit 326 enfants avaient une rectorragie comme maitre symptôme sur 21.533 consultations [6]. Une série égyptienne a révélé que sur 91.000 consultations d’enfants âgés de moins de 18 ans entre Mars 2002 et Février 2004, 194 enfants ont consulté pour rectorragie [7].
Présentation clinique
Le diagnostic est facile quand le saignement est extériorisé mais il est indispensable de l’évoquer dans d’autres situations notamment lors d’un choc hypovolémique ou au contraire lors d’une anémie trainante inexpliquée. Les hémorragies de faible abondance doivent être recherchées notamment par des bandelettes apposées sur les selles [8][9]. Une anamnèse détaillée est de rigueur.
La couleur du sang est liée au siège de l’origine du saignement, à sa proximité de l’orifice anal, à la rapidité du transit et à la formation d’hématine qui lui confère le teint brunâtre. Il est bien établi que la durée de la présence du sang dans la lumière digestive détermine pour beaucoup, la couleur du saignement. Ainsi, après un séjour du sang < 14h, le patient présentera une rectorragie, alors qu’un séjour > 14h donnera lieu à un méléna [2].
L’abondance, de cette hémorragie, sa durée et les constantes hémodynamiques orientent le diagnostic étiologique et aident à évaluer le pronostic vital.
L’examen proctologique confirme un saignement en cours et peut aider à préciser son abondance, tout en sachant qu’un examen normal n’élimine pas le diagnostic positif. Il est réalisé chez un enfant couché soit en décubitus dorsal avec les hanches et les genoux complètement fléchis soit en décubitus latéral gauche. A noter que cet examen doit s’effectuer en la présence des parents tout en expliquant à l’enfant et le rassurant aussi.
Évaluation de la gravité : Le plus souvent – comme déjà cité – le saignement est d’importance modérée et surévalué par des parents anxieux [10,11] . Selon les données de la littérature, la résolution spontanée serait de règle comme chez l’adulte [11]. Cependant, la gravité réside dans les états de troubles hémodynamiques et d’altération de l’état général que peut provoquer un saignement en particulier aigu et abondant (rupture vasculaire) ou chronique et trainant (MICI) qui nécessiterait des mesures de réanimation adaptées [1,11].
Les examens complémentaires
Les examens complémentaires morphologiques, endoscopiques et biologiques sont orientés et ciblés [8,9]. C’est ainsi que les explorations digestives, ne sont pas toujours systématiques et sont programmées selon l’orientation mais aussi en fonction de leur accessibilité. Ces investigations ont pour but de :
- Confirmer le saignement,
- Situer l’origine de l’HDB ou des rectorragies,
- Préciser le caractère aigu ou chronique,
- Établir un bilan de retentissement.
L’endoscopie digestive est la pierre angulaire. Elle peut être à double visée diagnostique et thérapeutique. Selon les recommandations du Groupe Francophone d’Hépato-Gastro-Endoscopie et Nutrition Pédiatrique (GFHGENP), établies en 2002 et réévaluées en 2012, ainsi que de la Société Européenne d’Hépato-Gastro-Endoscopie et Nutrition Pédiatrique (ESPGHAN) 2017 ; la coloscopie est indiquée dans le cadre des HDB en l’absence de lésions ano-périnéales « banales », en l’absence de lésions gastroduodénales, et dans le cas des diarrhées chroniques glairo-sanglantes nécessitant une exploration [12,13]. Cela nous mène à dire qu’en cas de rectorragies, il est indispensable en premier lieu de faire un bon examen proctologique, puis éventuellement compléter par une rectosigmoïdoscopie pour écarter toute lésion anale ou rectale avant d’indiquer la nécessité d’une iléocoloscopie (voir annexes 2) [13,14].
L’HDB est la première indication de vidéocapsule quand l’endoscopie classique n’a pas abouti à la visualisation de la source du saignement. Ce moyen diagnostic est en pleine expansion, mais l’âge reste une contrainte, vu qu’elle ne peut s’utiliser actuellement que chez le grand enfant. D’autre part, son grand inconvénient est l’impossibilité de la pratique des biopsies [15].
L’étude anatomo-pathologique reste primordiale et prend toute son importance surtout dans l’analyse d’une masse endoluminale (polype, polypose, tumeur) [16][17] ; ou pour le diagnostic des Maladie Inflammatoire Chronique Intestinale (MICI) [18].
Pour les examens radiologiques, l’échographie abdominale reste facile d’accès, et demandée le cas échéant en première ligne. L’IRM, la TDM, l’Angio-IRM, l’écho-endoscopie ont considérablement amélioré, par leur sensibilité, le diagnostic et la prise charge, surtout dans les MICI [19], et les tumeurs colorectales.
Par ailleurs, les examens biologiques recherchent un retentissement hématologique (une anémie), un syndrome inflammatoire, ou sont spécifiques (sérologie anti-saccharomyces cerevisiae, sérologie de l’allergie aux protéines de lait de vache).
L’enquête étiologique peut même amener à pratiquer une exploration isotopique, notamment dans le cadre du diverticule de Meckel (DM).
Les étiologies
L’anamnèse, l’examen clinique et le contexte dans lequel évolue la rectorragie permettent habituellement de trouver l’étiologie, mais l’élément clé reste l’âge de l’enfant. Il convient d’éliminer une fausse rectorragie. Quelques substances (médicaments, colorant alimentaire, betteraves), ont tendance à colorer les selles.
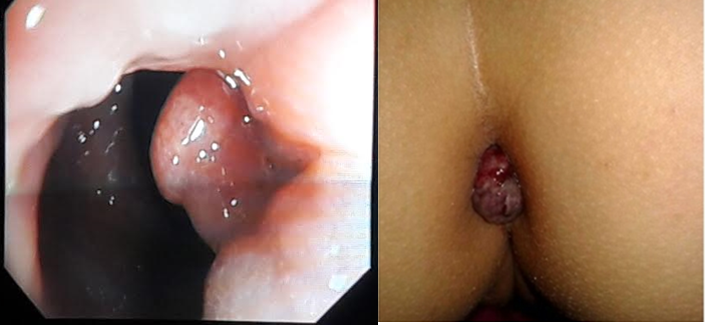
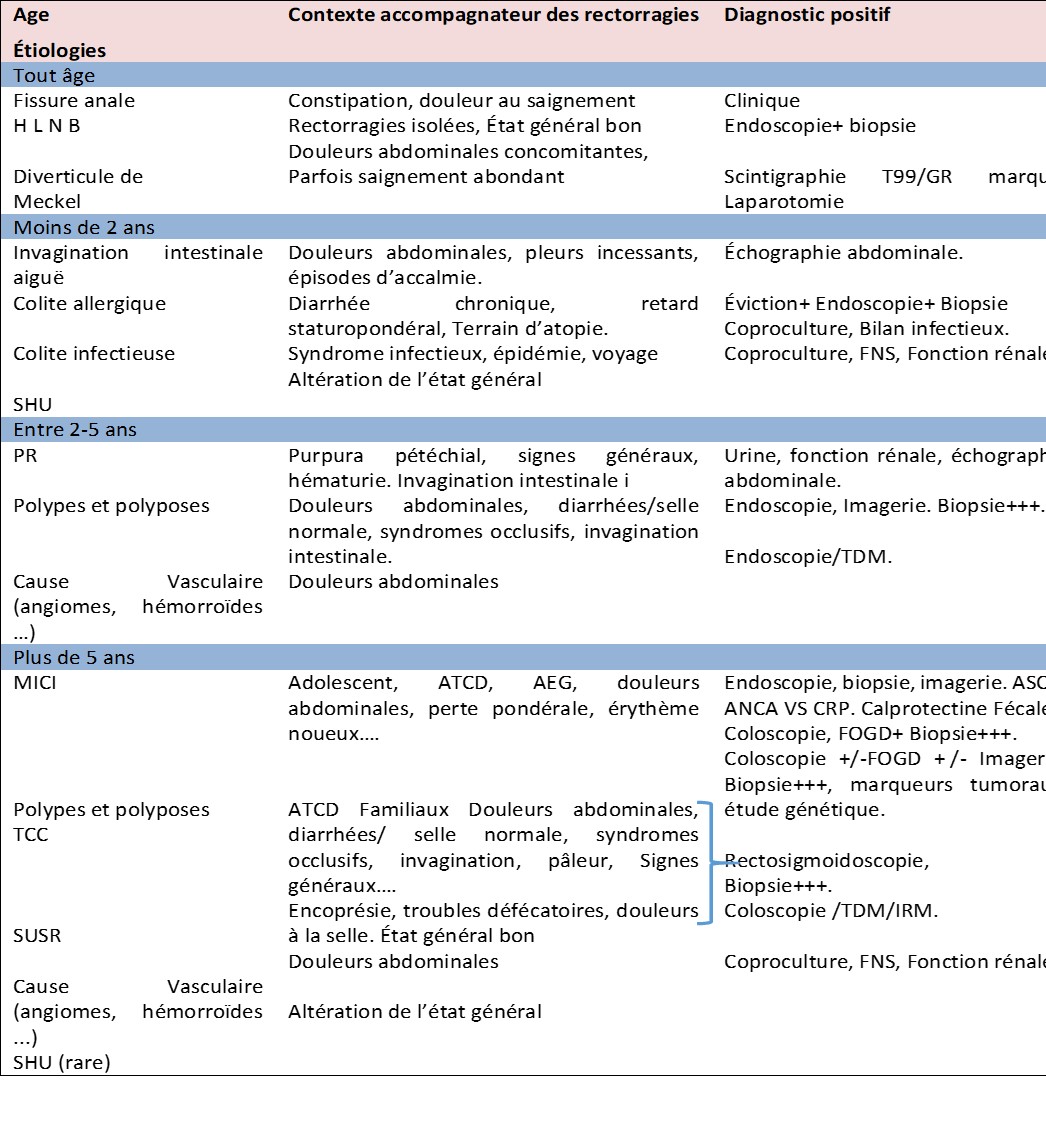
Les étiologies (tableau 1) sont nombreuses et varient selon des différents âges pédiatriques et sont relativement distinctes de celles qui touchent l’adulte [4]. Si chez ce dernier, les diverticules sont la principale cause, les polypes colorectaux (figures 1), les colites chroniques et les fissures anales constituent les causes les plus répandues en pédiatrie [6,11] .
Figures 1:Polype hyperplasique pédiculé rectal chez un enfant de 5 ans. Polype juvenile accouché chez un enfant de 6 ans.
Quel que soit l’âge, les causes traumatiques, les fissures anales (figure 2), les causes infectieuses et l’hypertrophie lymphoïde nodulaire bénigne (figure 3) (HLNB) sont communément retrouvées [9].
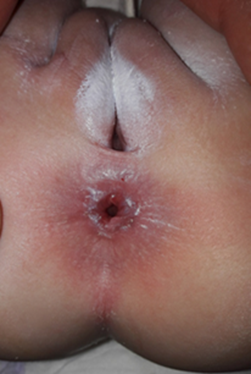 Figure 2: Fissure anale chez un nourrison de 6 mois secondaire à la constipation.
Figure 2: Fissure anale chez un nourrison de 6 mois secondaire à la constipation.
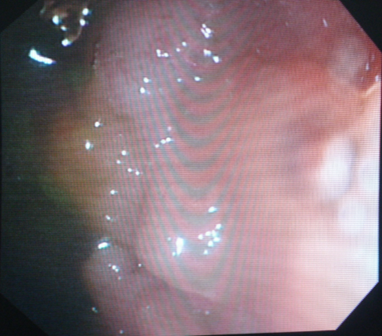
Figure 3 : HNLB chez un garçon de 12 ans.
Pour le nourrisson (< 2ans), l’invagination intestinale aiguë, le colites allergiques (figure 4), les duplications digestives, la diarrhée infectieuse, l’HLNB et le DM, restent les causes les plus fréquentes [21].
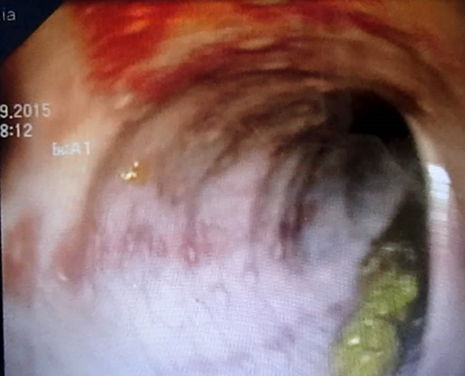
Figure 4 : colite allergique chez un nourrisson de 6 mois suivi pour allergie aux proteins de lait de vache.
Cette ultime étiologie est considérée comme la plus fréquente anomalie congénitale du tractus gastro-intestinal [11,19].
Chez l’enfant de 2-5 ans, les fissures anales, les colites infectieuses, les polypes et l’HLNB restent des causes les plus classiques [3].
Concernant le grand enfant, dominent les polypes et polyposes, les MICI, (la maladie de Crohn, rectocolite hémorragique, colites indéterminées) (figure 5) ; les malformations vasculaires et le syndrome de l’ulcère solitaire du rectum (SUSR) [20,21].
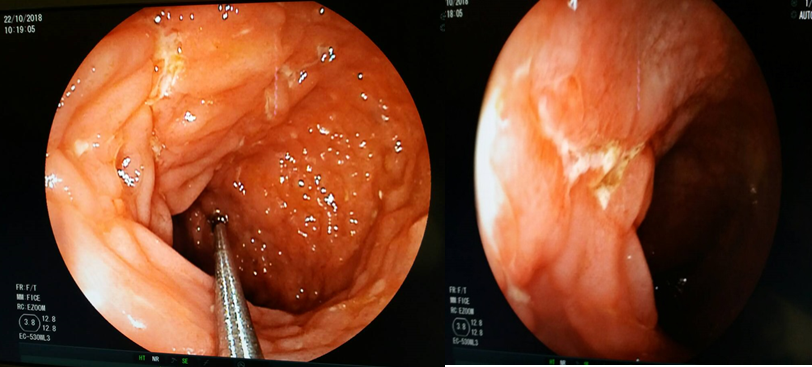
Figure 5 : maladie de Crhon chez un adolescent de 14 ans .Remarquez l’aspect congestif de la muqueuse colique et les ulcerations longitudinales.
Les tumeurs colorectales (TCC) peuvent se voir, mais restent très peu fréquentes et rentrent le plus souvent dans un cadre familial, héréditaire ou syndromique (figure 6) [22,23].
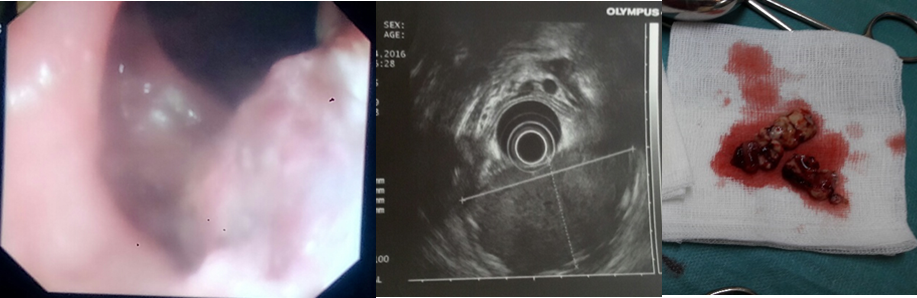
Figure 6 : Aspect endoscopique et echo-endoscopique d’une tumeur neuro-endocrine rectale très friable chez un enfant de 10 ans.
Tableau 1 : Principales étiologies des rectorragies chez l’enfant et signes cliniques évocateurs (nouveau-né exclu) [2,6,9,24,25]
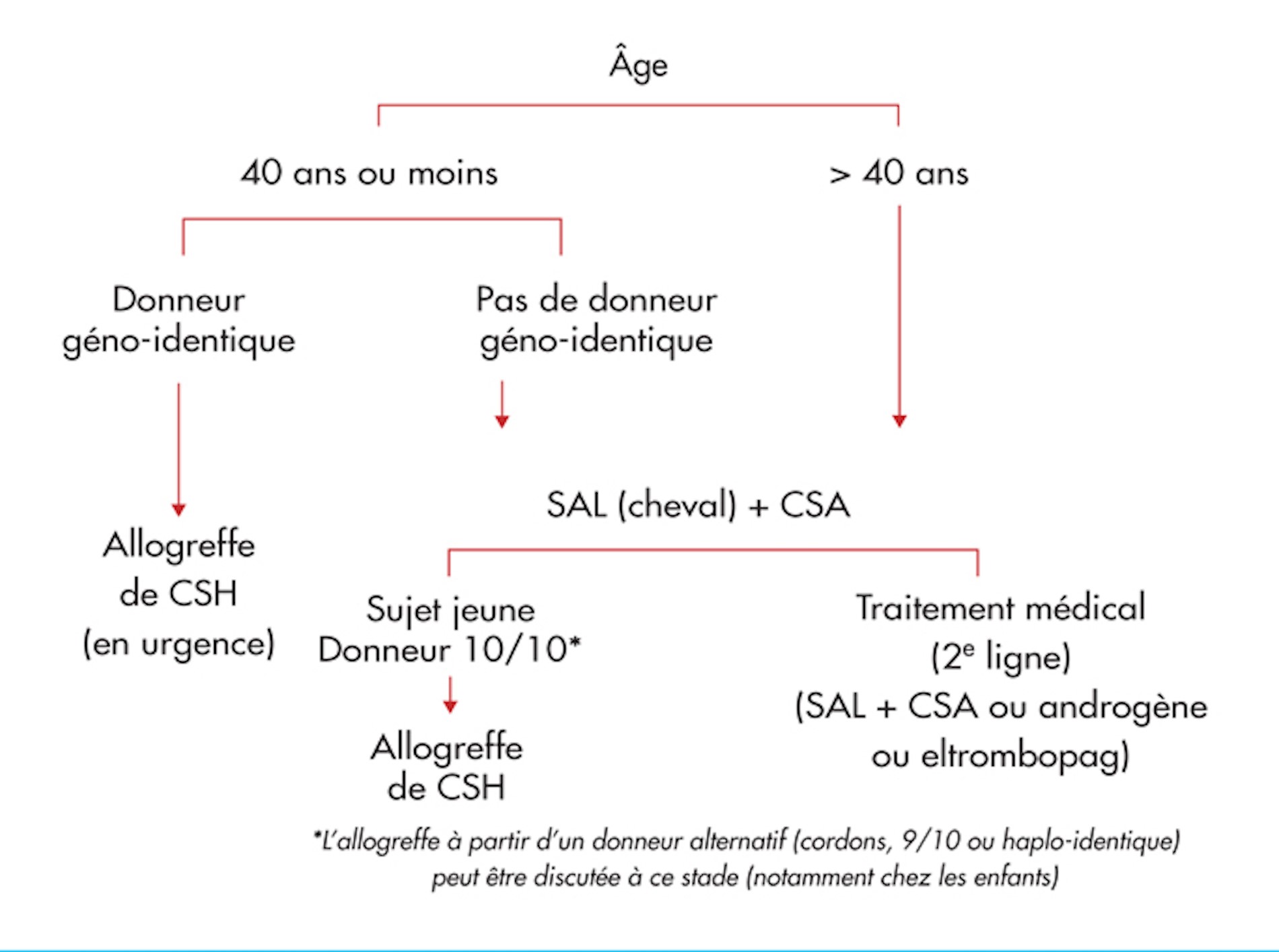
Conduite à tenir devant une rectorragie de l’enfant (nouveau-né non inclus)
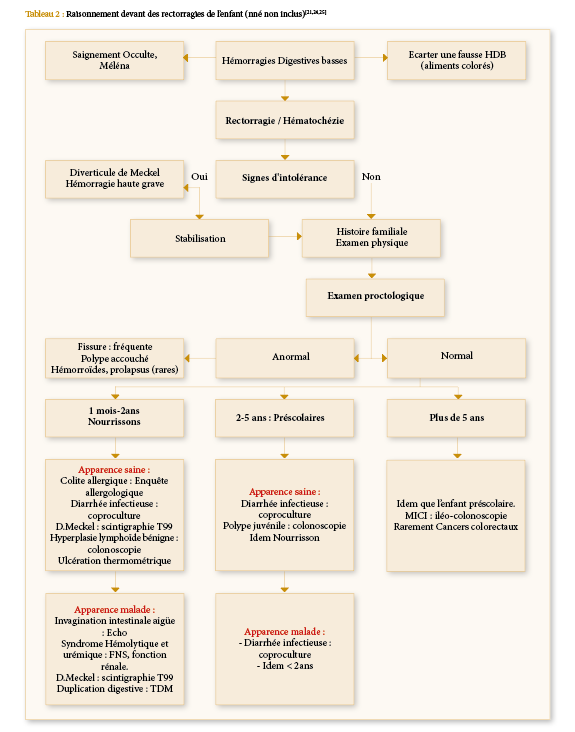
Devant toute rectorragie, un raisonnement se base sur l’âge, les signes cliniques, l’examen physique, l’évolution et les examens paracliniques. L’algorithme suivant récapitule l’approche diagnostique et l’enquête étiologique.
Tableau 2: Raisonnement devant des rectorragies de l’enfant (nné non inclus)[21,24,25]
Conclusion
Les rectorragies chez l’enfant constituent un symptôme non rare et pourvoyeur de stress parental. L’interrogatoire, l’examen clinique sont indispensables pour le raisonnement du clinicien notamment l’examen proctologique. L’endoscopie est essentielle mais non systématique dans l’enquête étiologique.
En l’absence de diagnostic endoscopique, d’autres méthodes de diagnostic peuvent être utilisées, avec des degrés variables de spécificité et sensibilité.
Les causes sont généralement âge-dépendantes et différentes de celle de l’adulte. Chez les moins de 5 ans, prédominent les infections, les fissures anales, la proctocolite allergique, l’invagination intestinale, le diverticule de Meckel et les malformations vasculaires, l’HLNB.
Chez le plus de 5 ans, sont essentiellement retrouvés les polypes et les MICI. Une collaboration multidisciplinaire impliquant le gastro-pédiatre, le radiologue, l’endoscopiste, le pathologiste et le chirurgien infantile est indispensable afin de localiser la source du saignement et d’assurer une prise en charge adaptée.
Date de soumission : 04 Décembre 2019.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
- E. Padilla and W. Moses, “Lower Gastrointestinal Bleeding & Intussusception,” Surg. Clin. NA, vol. 97, no. 1, pp. 173–188, 2017.
- Mushtaq and S. El-hadi, “Rectal bleeding in children e causes and investigations,” Paediatr. Child Health (Oxford)., vol. 24, no. 11, pp. 491–500, 2014.
- Barnert and H. Messmann, “Management of lower gastrointestinal tract bleeding,” vol. 22, no. 2, pp. 295–312, 2008.
- Osman, M. Djibré, D. Da Silva, and C. Goulenok, “Management by the intensivist of gastrointestinal bleeding in adults and children,” pp. 1–17, 2012.
- Pant, M. Olyaee, T. J. Sferra, R. Gilroy, O. Almadhoun, and A. Deshpande, “Brief report Emergency department visits for gastrointestinal bleeding in children: results from the Nationwide Emergency Department Sample 2006 -2011,” Curr Med Res Opin, 2014.
- Alam, I. Kmd, N. Mohammad, and M. Nooruzzaman, “Per Rectal Bleeding in Children: Experiences in the Department of Paediatric Surgery in BSMMU,” pp. 20–25, 2017.
- A. El-khayat, M. A. El-hodhod, F. Z. Abd, and A. M. Hamdy, “Rectal bleeding in Egyptian children,” Ann. Trop. Paediatr., vol. 26, pp. 337–344, 2006.
- Benhamou and C. Dupont, “Diagnostic des hémorragies digestives du nourrisson et de l’enfant,” EMC – Tratado de Medicina. pp. 1–8, 2007.
- Aroulandom, J. Lemale, and H. Chappuy, “Diagnostic des hémorragies digestives du nourrisson et de l’enfant,” vol. 13, no. 18, pp. 1–10, 2019.
- R. Fleisher, “Rectal Bleeding in the Pediatric Emergency Department,” no. June, pp. 1252–1258, 1994.
- Sahn and S. Bitton, “Lower Gastrointestinal Bleeding in Children,” Gastrointest. Endosc. Clin. NA, vol. 26, no. 1, pp. 75–98, 2016.
- B. et al Dabadie, “Evolution des indications de la coloscopie chez l ’enfant en 2012. Mise au point,” Arch. Pédiatrie, vol. 19, pp. 1247–1251, 2012.
- Mougenot, J. Cardey, and P. Vannerom, “Endoscopie digestive pédiatrique,” vol. 8, no. 13, pp. 1–10, 2019.
- Hepatology et al., “Paediatric Gastrointestinal Endoscopy: European Society for Paediatric Nutrition and European Society of,” vol. 64, no. 1, pp. 133–153, 2017.
- Recommendations de la SFED, “Indications et techniques de la vidéocapsule endoscopique de l’intestin grêle chez l’enfant,” Acta Endoscopica, vol. 46, pp. 63–67, 2016.
- HMA, “Pathology and genetics of hereditary colorectal cancer,” Pathology, vol. 50, no. 1, pp. 49–59, 2018.
- Tavano et al., “Pathologie tumorale intestinale de l’enfant,” vol. 4, no. 4, pp. 1–10, 2019.
- Viala, C. Jung, N. Belarbi, D. Berrebi, and J. Hugot, “Maladies inflammatoires chroniques intestinales de l’enfant : maladie de Crohn , rectocolite hémorragique,” EMC – Pediatría, vol. 11. Elsevier, pp. 1–10, 2019.
- Gallinet and F. Sauvat, “Diverticule de Meckel et pathologie du canal omphalomésentérique,” vol. 12, no. 16, pp. 1–6, 2019.
- Sun, T. Hull, and G. Ozuner, “Facteurs de risque et caractéristiques cliniques du prolapsus rectal chez le sujet,” J. Chir. viscérale, vol. 151, no. 6, pp. 1–6, 2014.
- Ait Idir, A. Tibouk, S. Kordjani, and N. Zidane, “Le syndrome de l’ulcère solitaire du rectum chez l’enfant : une entité pas si fréquente. Cas clinique et mise au point,” Batna J Med Sci, vol. 6, no. 1, pp. 65–67, 2018.
- Tougeron, “Carcinogenèse colorectale, données fondamentales,” Colloids Surfaces A Physicochem. Eng. Asp., vol. 9, no. 3, pp. 1–15, 2019.
- Bonnet and R. Guimbaud, “Polyposes et cancers colorectaux familiaux,” Colloids Surfaces A Physicochem. Eng. Asp., vol. 10, no. 4, pp. 1–7, 2019.
- A. Lane and I. D. Sugarman, “Investigation of rectal bleeding in children,” Paediatr. Child Health (Oxford)., vol. 20, no. 10, pp. 465–472, 2010.
- Shaoul, “Practical Algorithms in Pediatric Gastroenterology,” 2014.


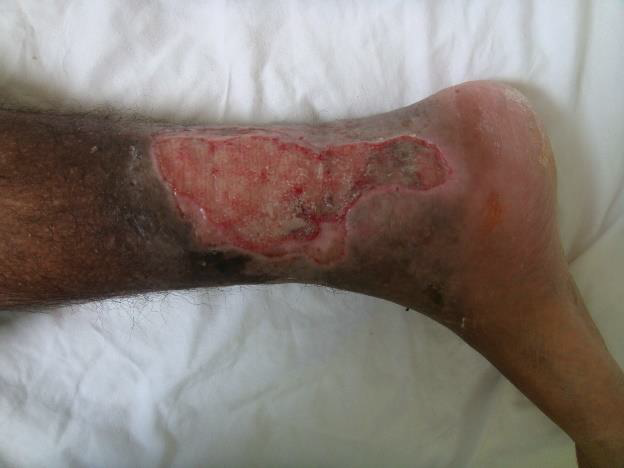 Figure 02 : Ulcère pré malléolaire droit. Patient âgé de 32 ans « de notre série ».
Figure 02 : Ulcère pré malléolaire droit. Patient âgé de 32 ans « de notre série ».