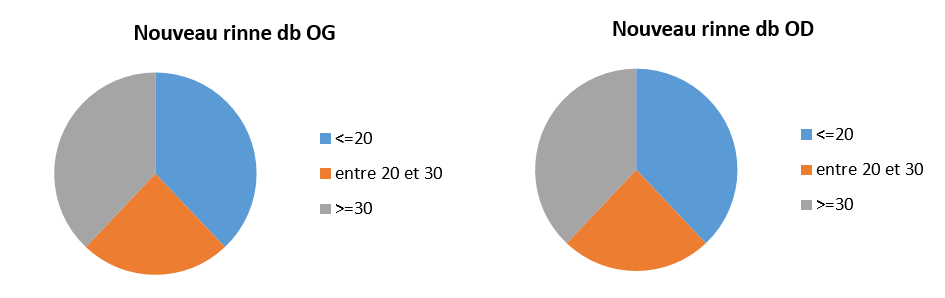
Le cancer colorectal (CCR) est un problème de santé publique dans le monde, et le plus fréquent des cancers digestifs, les études épidémiologiques montrent une grande variabilité de son incidence dans le monde.
Zemmour(¹), N. Ghazi(¹), B. Krelil(2), B. Larbaoui(¹)
(¹) Centre de Lutte Contre le Cancer « Emir Abd El Kader » Misserghine, Oran
(2) Clinique Chirurgicale Aït Idir Ali-CHU Oran
Date de soumission : 29 Janvier 2020.
Résumé : Le cancer colorectal (CCR) est un problème de santé publique dans le monde, et le plus fréquent des cancers digestifs [1], les études épidémiologiques montrent une grande variabilité de son incidence dans le monde. En Algérie, l’incidence de ce type de cancers a considérablement augmenté ces dernières années, cette incidence rejoint celle des pays du Maghreb [2], plusieurs facteurs de risque interviennent dans la genèse de ce cancer comme : l’allongement de l’espérance de vie, l’alimentation riche en graisses et la sédentarité, cependant, la mortalité reste relativement constante vu la sensibilisation de la population, le diagnostic plus précoce et les innovations thérapeutiques majeures, tant dans les méthodes de chirurgie que dans les traitements médicaux.
Mots-clé : cancer colorectal, incidence, alimentation, graisse, sédentarité, sensibilisation.
Abstract: Colorectal cancer is a public health problem worldwide, the most frequent of digestive cancers [1]. Epidemiological studies show a great variability in its incidence worldwide, in Algeria, the incidence of this type of cancer has considerably increased these last years, this incidence is similar to that of the Maghreb countries [2], several risk factors participate in the genesis of this cancer such as: the longer life expectancy, the diet rich in fats and the sedentary lifestyle, however, the mortality remains relatively constant being the sensitization of the population, the earlier diagnosis and the major therapeutic innovations in both surgical methods and medical treatments.
Keywords: colorectal cancer, incidence, diet fat, sedentary lifestyle, awareness.
Introduction
Les cancers du côlon et du rectum sont deux entités souvent associées car leur physiopathogénie est similaire ; et de par leur fréquence, ils représentent le troisième cancer dans le monde. Les études épidémiologiques montrent une grande variabilité de l’incidence du cancer colorectal dans le monde. L’Algérie figure parmi les pays où cette incidence est moyenne (entre 14,8-22,7/100.000 habitants/an) [3]. Le nombre de cas incidents est en augmentation constante vu l’allongement de l’espérance de vie et le changement des habitudes (sédentarité, obésité).
L’adénocarcinome est le type histologique le plus fréquent, il se développe le plus souvent sur un adénome préexistant après plusieurs années de latence, cette carcinogenèse lente et bien connue fait du cancer colorectal un modèle de dépistage et de prévention secondaire.
Les avancées thérapeutiques considérables qui concernent la qualité de la résection chirurgicale même en phase métastatique et les nouveaux traitements médicaux ciblés, ont permis de stabiliser les taux de mortalité de ce cancer.
Fréquence
Le cancer colorectal est le troisième cancer le plus fréquemment diagnostiqué dans le monde avec 1,8 million de cas, soit 10,2% du total [4].
En Algérie, il est le deuxième après le cancer du sein (50% des cancers digestifs) [5] ; et représente 15% des cancers en 2015 [10].
Le cancer du côlon représente 65%, alors que le cancer du rectum constitue 35% des cancers colorectaux [6].
Incidence
L’incidence des tumeurs colorectales peut être répartie selon trois zones géographiques :
Les taux d’incidence les plus élevées sont observés en Australie et en Nouvelle-Zélande (44,8 et 33,2/100.000 habitants chez les hommes et les femmes respectivement ; et les plus faibles en Afrique de l’ouest (4,5 et 3,8/100.000 habitants) [34].
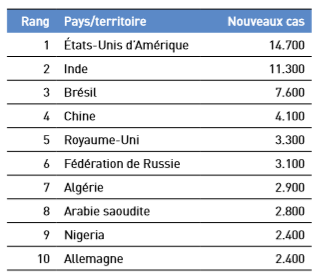
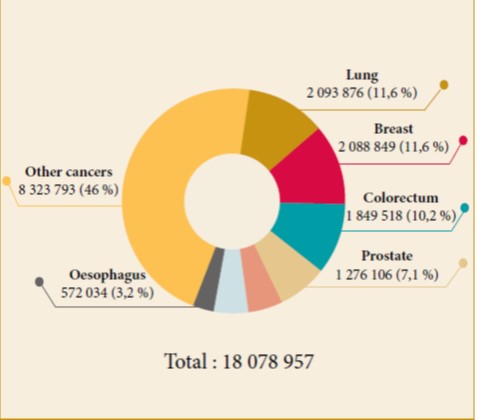
Figure 01 : Nombre de nouveaux cas estimés en 2018 dans le monde [3]
En Asie, les taux d’incidence varient considérablement avec 38.067 nouveau cas dans l’Asie de l’ouest et 736.573 nouveau cas dans l’Asie de l’est [3].
En Amérique, l’incidence est de 97.600 nouveaux cas en Amérique latine et 179.771 nouveaux cas en Amérique du nord [3]. C’est l’un des taux les plus élevés au monde.
En Europe, les taux les plus élevés sont enregistrés dans l’Europe centrale et l’Europe de l’est avec 164.998 nouveaux cas [3].
En France, on évalue l’incidence en 2015 à 43.065 nouveaux cas [7].
En Afrique les taux vont de 6.010 en Afrique centrale à 18.810 nouveaux cas en Afrique du nord [3].
Ces taux sont directement corrélés au niveau de vie et aux habitudes alimentaires des populations.
Figure 02 : L’incidence du cancer colorectal dans le monde [3]
En Algérie
Les cancers digestifs représentent 25% environ des cancers chez l’homme et 17,5% chez la femme [8], le cancer colorectal est le premier des cancers digestifs avec 5.430 nouveaux cas colligés en 2018, loin devant le cancer gastrique avec 2.241 nouveaux cas ; et il est le deuxième cancer dans les deux sexes après le cancer du sein chez la femme et le cancer du poumon chez l’homme [3,9].
Nous avons enregistré en 2015, 3.532 ou 16,9/100.000 habitants chez l’homme et 3.003 ou 14,3/100.000 habitants chez la femme [10]. Ces chiffres placent l’Algérie dans une zone d’incidence moyenne, comparée avec les incidences mondiales.
Chez l’homme, le cancer du colon a une incidence brute de 5,8/100.000 habitants et une incidence standardisée sur l’âge de 8/100.000 habitants ; et le cancer du rectum présente une incidence brute de 5,6/100.000 habitants et une incidence standardisée sur l’âge de 7/100.000 habitants [10].
Chez la femme, le cancer du colon à une incidence brute de 7,7/100.000 habitants et une incidence standardisée sur l’âge de 10/100.000 habitants. Le cancer du rectum présente une incidence brute de 5,4/100.000 habitants et une incidence standardisée sur l’âge de 7,3/100.000 habitants [10].
L’âge moyen de survenue du cancer colorectal en Algérie est de 65 ans [9].
La majorité des cas surviennent à un âge ≥ 50 ans mais 23% des cas surviennent avant 40 ans.
Le sex-ratio est de 1 pour le côlon et de 1,2 pour le rectum [8].
Les variations de l’incidence
Dans le Monde
Le cancer colorectal a connu une augmentation d’incidence ces dernières années avec un taux variable selon les pays [12]. En Amérique du nord, en Europe et en Australie, l’incidence du cancer colorectal est 20 fois plus élevée que dans les pays en développement d’Afrique ou d’Asie, des variations rapides de l’incidence sont parfois notées. Ainsi, au Japon, l’incidence standardisée sur l’âge est passée de 10,9/100.000 en 1975 à 43,8/100.000 en 1999 ; ceci est attribué à l’occidentalisation du style de vie. Au contraire, aux États-Unis, l’incidence standardisée passe de 64,2/100.000 en 1985 à 49,5/100.000 en 2003 grâce au développement de la colonoscopie et au traitement des polypes [13].
En Algérie
L’Algérie est un exemple de transition épidémiologique, cette transition est marquée par un changement structurel dans le profil épidémiologique de la population, l’augmentation de l’espérance de vie durant ces 50 dernières années, les transformations de l’environnement, les changements des habitudes de la vie (tabagisme, stress, sédentarité, urbanisation) ; ont probablement contribué à l’augmentation de l’incidence de certains cancers [1,9]. Le cancer colorectal a marqué une tendance à l’augmentation durant la période 1986-2014 avec un changement annuel en pourcentage (APC) de +6,7 IC 95% [6,1-7,3] chez l’homme [10], et une tendance à l’augmentation durant la même période avec un APC de +6,5 IC 95% [5,9-7,1] chez la femme [10].
Dans l’estimation des projections d’incidence des cancers colorectaux, on s’attend à un nombre de nouveaux cas estimés à 3.668 en 2020 et 4.450 en 2025 chez l’homme et estimé à 3.128 en 2020 et 4.141 en 2025 chez la femme [10].
Mortalité
La mortalité est relativement plus élevée dans les pays en voie de développement, par défaut de diagnostic précoce et d’accès au traitement.
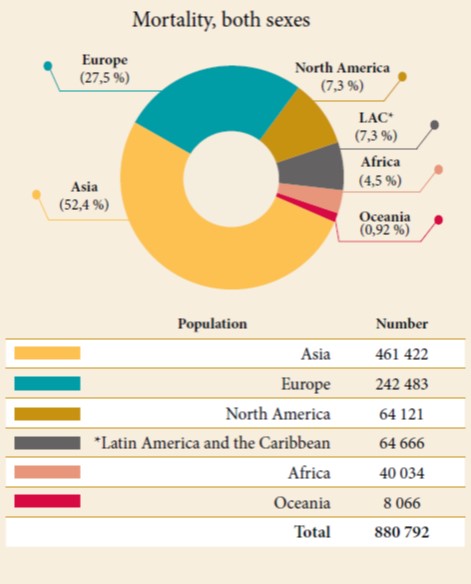
Figure 03 : La fréquence de la Mortalité par CCR dans le Monde [3] Figure 04 : les taux d’incidence et de mortalité standardisée sur l’âge par CCR [3]
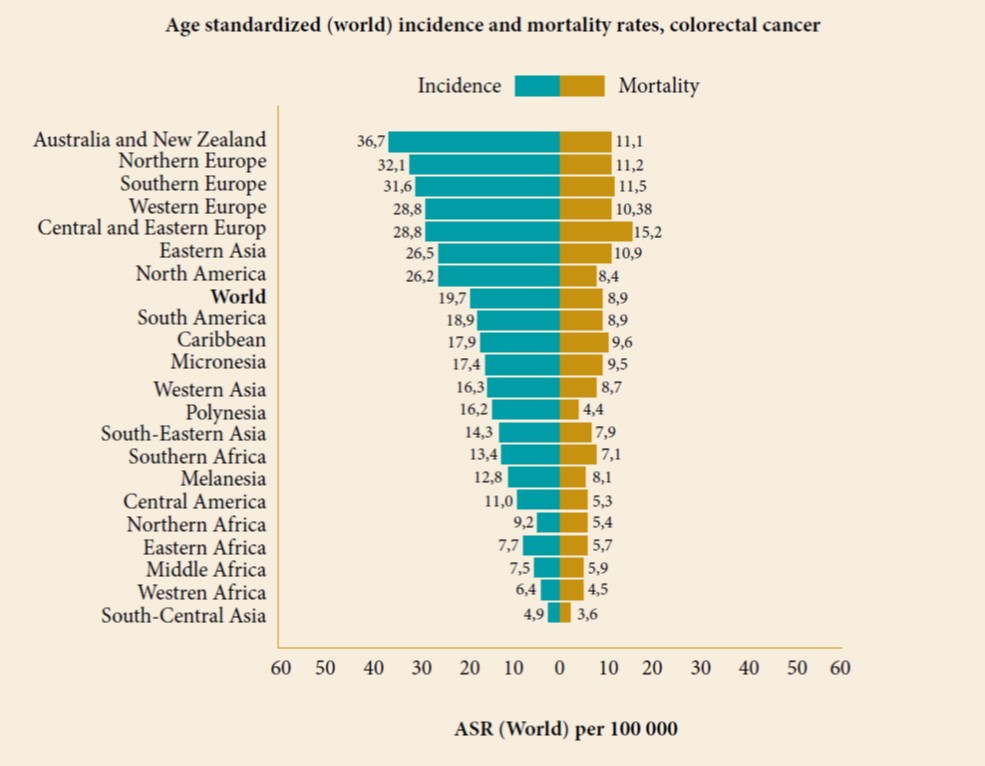
Figure 4 : Les taux d’incidence et de mortalité standardisée sur l’âge par CCR [3]
En Asie, les taux de mortalité vont de 20.418 en Asie de l’ouest à 325.128 en Asie de l’est [3].
En Amérique, les taux de mortalité les plus bas sont notés aux caraïbes avec 6.258, les taux les plus élevés sont en Amérique du nord avec 64.121 [3].
En Europe, les taux de mortalité varient de 32.659 en Europe du nord à 94.545 en Europe centrale et Europe de l’est [3].
En Afrique, les taux de mortalité vont de 3.801 en Afrique du sud à 12.201 en Afrique de l’est [3].
En Algérie, nous avons comptabilisé 2.984 décès par cancer colorectal en 2018 [3], plaçant ainsi notre pays dans une zone de mortalité moyenne.
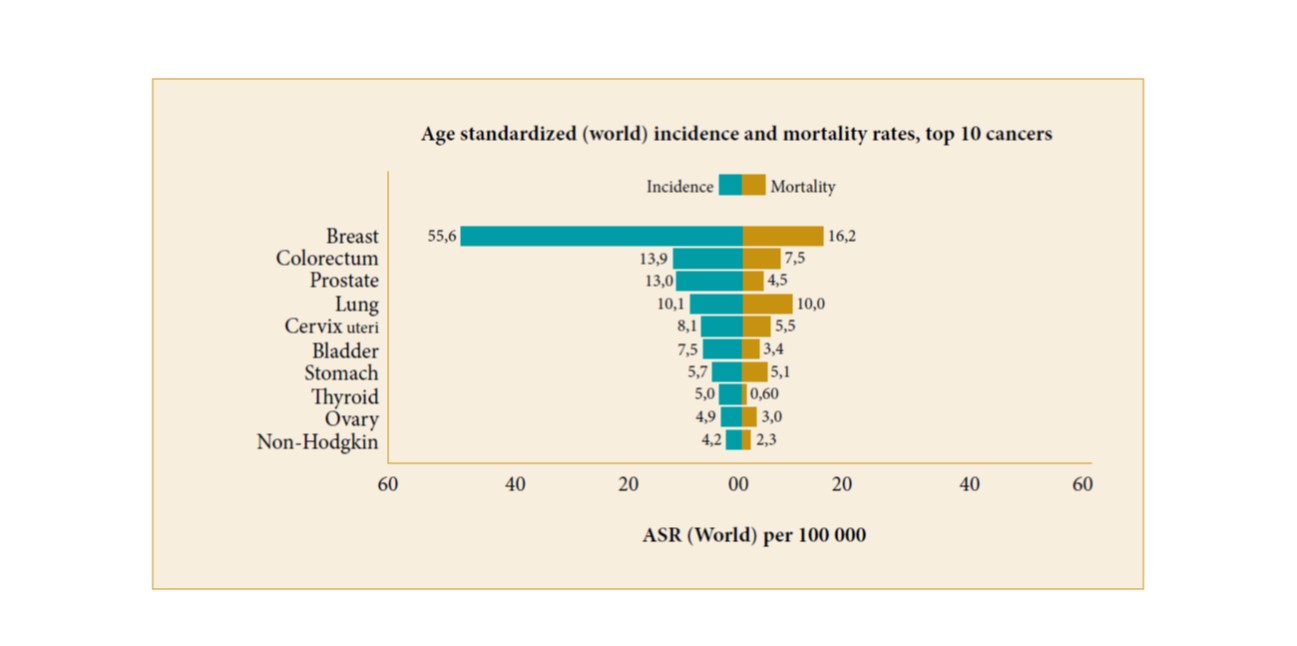
Figure 05 : Incidence et Mortalité standardisée sur l’âge par cancer en Algérie [3]
Les polypes et les lésions pré-néoplasiques
Il existe deux types de polypes : les polypes hyperplasiques, muqueux, les pseudo-polypes inflammatoires, les sous muqueux et les hamartomes qui n’évoluent que rarement vers le cancer, et les polypes dentelés et adénomateux qui augmentent le risque de développer un cancer colorectal [11]. C’est la séquence adénome-cancer, la prévalence des polypes adénomateux dans une population de moins de 50 ans est de 30-40% [12,13], seulement 5% de ces polypes dégénèrent vers un cancer sur 7-10 ans [11]. Pour ce, la participation d’autres facteurs comme les mutations APC, Kras, TP53 paraît indispensable. Le risque de cancer augmente avec le nombre des adénomes, leur taille (> 1cm), et la proportion du contingent villeux, sur 1.000 adénomes, 100 atteignent un volume de 1 cm et 25 évoluent vers le cancer [14].
Les facteurs de risque du cancer colorectal sporadique
Le cancer colorectal est sporadique dans 95% des cas, il est alors sous l’influence des facteurs environnementaux, cependant, plusieurs facteurs sont incriminés dans sa genèse.
- Les facteurs non modifiables
L’âge : le cancer colorectal est rare avant 45 ans, son incidence augmente avec l’âge [15]. 95 % des cancers du côlon et du rectum sont diagnostiqués après 50 ans et 46% après 70 ans, un cancer colorectal précoce est lié à des facteurs génétiques [16], L’âge moyen au diagnostic est de 69,5 ans pour les hommes et de 72,8 pour les femmes [17].
Les facteurs génétiques : Les syndromes héréditaires comptent pour 5% des cancers colorectaux [2], il s’agit de :
- la polypose adénomateuse familiale qui est rare (moins de 5% de toutes les tumeurs colorectales) [12], où le risque de cancer en cas de mutation du gène suppresseur APC est de 100% surtout chez le sujet jeune [18].
- Le syndrome de Lynch ou syndrome HNPCC (hereditary non polyposis colorectal cancer) où le risque de cancer chez les porteurs de la mutation de la réparation aux mésappariements MMR (mismatchrepair) de l’ADN est de 80% [2]. Il se caractérise volontiers de cancer colorectal plus précoce (âge < 45 ans) du côlon droit, parfois des cancers multiples synchrones ou métachrones (20%) [19].
Les facteurs familiaux : les parents de premier degré d’un patient atteint d’un cancer colorectal ont un risque élevé de développer un cancer colorectal, cela semble indiquer une possible anomalie génétique, ces cas représentent au minimum 20 % des cancers colorectaux sporadiques [14].
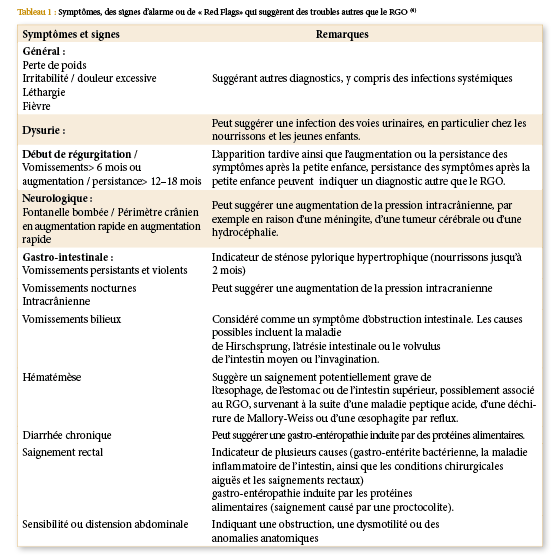
Les maladies inflammatoires : RCH et maladie de Crohn présentent un risque 8-30 fois plus élevé de développer un cancer précoce du colon et du rectum [12], une méta-analyse sur 194 études que les taux d’incidence du cancer colorectal sur une colite ulcérative correspondent à une probabilité cumulative de 2% à 10 ans, 8% à 20 ans et 18% à 30 ans [20] ; et 60% à 40 ans [21]. Les facteurs de risque incluent la durée, l’étendue, une histoire familiale de cancer colorectal [22].
- Les facteurs modifiables
Les habitudes alimentaires : on estime que 70% des cancers colorectaux pourraient être évités par une intervention nutritionnelle, une alimentation très calorique et riche en graisses animales (viandes rouges) et pauvres en fibres et légumes augmente le risque de cancer colorectal [23].
L’étude de la prévalence du cancer colorectal dans certaines populations (notamment les migrants) a montré un changement de l’incidence du cancer colorectal en fonction du nouveau mode de vie dès la 2ème génération.
L’alcool : Une méta-analyse réalisée à partir de 8 cohortes nord-américaines et européennes a montré qu’au-delà de 45 g d’alcool/j, le RR de développer un cancer colorectal est de 1,41 ; IC 1,16-1,72 [24].
Le tabac : 12% des cancers colorectaux sont imputables au tabagisme, la consommation de tabac est en effet liée à l’apparition de mutations dans l’ADN de la muqueuse digestive [25].
Niveaux de risque :
|
Niveau |
Moyen |
Elevé |
Très élevé |
|
Population concernée |
Population générale · 50-74 ans · Asymptomatique |
Antécédents personnels de maladie inflammatoire chronique intestinale (MICI) · Maladie de Crohn · Rectocolite hémorragique |
Prédisposition héréditaire · Polypose Adénomateuse Familiale (PAF) · Cancer colorectal héréditaire non polyposique (syndrome de Lynch) |
Tableau : les principaux facteurs de risque du cancer colorectal [26].
Les facteurs protecteurs du cancer colorectal : On estime que 70% des cancers colorectaux pourraient être prévenus par une intervention nutritionnelle [8].
Aspirine et AINS : 09 études cas-témoin sur 13 ont montré que la consommation régulière et continue d’aspirine et/ou d’AINS est associée à une diminution significative du risque de cancer et de polypes recto-coliques, l’amplitude de cette diminution est de l’ordre de 40% [27].
Par ailleurs, une étude cas-contrôle est menée sur 10.280 patients et 102.800 contrôles et l’usage continu (≥ 5 ans) de faibles doses d’aspirine est associé à une réduction de 27% du risque de CCR (OR 0,73 ; IC 0,54-0,99) [28].
Calcium : Une supplémentation de 3g de carbonate de calcium (6 cps de Calperos®) réduit le risque de rechute d’adénome (risque ajusté de 0,81 ; IC 0,67-0,99) [29].
Sulindac et PAF : 100-400 mg/j de Sulindac entraînent une diminution du nombre et de la taille des polypes chez des patients atteints de PAF [29].
Vitamines : selon plusieurs études la supplémentation en β-carotène, vitamine C et E et sélénium n’ont pas montré d’effet protecteur [30]. Une étude récente portant sur 521.330 hommes et femmes entre 25-70 ans suivis pendant 12 ans dans 10 pays européens a montré qu’un modèle de mode de vie sain composé de cinq facteurs : un poids normal, une activité physique régulière, pas de tabac, avec une consommation modérée d’alcool et un régime alimentaire riche en fibres et pauvre en graisses animales a permis de réduire le risque de cancer colorectal avec un HR de 0,63 ; IC 0,54-0,74 p<0,0001 ; quand tous ces facteurs sont réunis [31].
La survie du cancer colorectal
Le dépistage des lésions précancéreuses par le test immunologique a permis le diagnostic précoce dans les cancers coliques. En Algérie, pays à faible risque, un dépistage de masse ne se justifie pas, cependant un dépistage individuel pour les personnes à risque élevé est recommandé [5].
La survie standardisée sur l’âge à 5 ans est de 63% en 2015 en France [32].
Dans l’étude internationale sur la survie « CONCORDE 3 » publiée dans « The Lancet » qui a concerné 322 registres de cancer de population de 71 pays dont 3 registres algériens : Sétif, Annaba et Tlemcen la survie nette standardisée à 05 ans en Algérie est de 74,2% pour le cancer du colon et 67,3% pour le cancer du rectum [10], comparé aux 67,4% notés aux USA en 2008. Ceci nous renseigne sur l’état de la prise en charge diagnostique et thérapeutique du cancer colorectal et les efforts diagnostiques et thérapeutiques à développer dans notre pays [5].
Conclusion
L’incidence du cancer colorectal est très variable dans le monde, elle tend à l’augmentation dans les pays en développement et se stabilise dans les pays industrialisés.
Il est recommandé de réduire les facteurs exposants comme la sédentarité, l’obésité, les viandes rouges et favoriser légumes, fruits, fibres et activité physique régulière et aussi promouvoir le test immunologique dans les pays à forte incidence et la résection des lésions précancéreuses [33], la formations de chirurgiens en cancérologie digestive, la discussion de tous les cas de cancer colorectal en RCP avant le début de tout traitement et l’acquisition des dernières thérapeutiques innovantes permettra d’aboutir aux objectifs du plan national cancer.
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références :
- Cancer Estimation of incidence and survival in Algeria 2014: Hamdi Cherif M et al, J cancer Res Ther 2015, 3(9) :100-104
- Profil Épidémiologique et Anatomopathologique du Cancer Colorectal, à propos de 36 cas ; M.S. Belhamidi et al ; Pan African Medical Journal. 2018 ; 30 :159 doi :10.11604/pamj.2018.30.159.15061
- International Agency for Research on Cancer 2018
- Communiqué de presse n°263 12 septembre 2018 ; centre international de recherche sur le cancer, OMS.
- Épidémiologie des cancers digestifs en Algérie, L. Abid Hôpital de Bologhine-Alger, cours intensifs de cancérologie digestive, FFCD-SAMGEED-septembre 2016.
- Étude clinico-épidémiologique du cancer colorectal dans le CHU de Tlemcen, Dendane Aek Oussama, Ghellai Abdelhak, mémoire de fin d’étude 2013-2014.
- INCa 2015: http://www.e-cancer.fr/ressources/cancers_en_France.
- Épidémiologie des cancers digestifs en Algérie, leçons à tirer en vue des priorités de santé publique, L. Abid ; cours intensifs e cancérologie digestive FFCD-SAHGEED-septembre 2016
- Aspects anatomo-histo-pathologiques du cancer colorectal ; Bouchouk meriem et Bougheriou lydia, Biochimie et Biologie Moléculaire, Universitaire de Bejaïa thèse soutenue le 19 juin 2017.
- Épidémiologie des Cancers Digestifs en Algérie ; Pr M. Hamdi Cherif, 8ème Congrès National d’Oncologie Médicale.
- Épidémiologie, prise en charge et suivi des polypes colorectaux ; T. Koessler et al Rev Med Suisse 2016 ; 12 ; 982-8.
- Cancer Colorectal : état des connaissances et données disponibles pour le développement d’une politique de santé en Belgique, Marina Puddu, Jean Tafforeau ; IPH/EPI reports nr 2006-023.
- Épidémiologie du cancer colo-rectal ; R. Lambert IARC Lyon Cancero digest Vol.1N°1-2009-2-6.
- Cancer colorectal et polymorphisme de l’Apo lipoprotéine E, Benbadis sarah, Bendjelloul ramzi Sciences Biologiques, Université des frères Mentouri Constantine ; p13.
- Prise en charge des cancers colorectaux des sujets âgés, Aparicio T, Mitr Y E, Cunma et al. Gastroenterol clin biol. 2005; 29: 1014- 23. PubMed. Google Scholar
- HAS, Colorectal Cancer
- Profil épidémiologique et anatomopathologique du cancer colorectal, à propos de 36 cas ; M.S. Belhamidi et al. Pan African Medical Journal. 2018; 30:159 doi:10.11604/pamj.2018.30.159.15061
- Colorectal cancer screening; Levin B, Cancer 1993.72-1056-1060,
- Détection and management of hereditary non polyposis colorectal cancer (Lynch syndrome) Ramsoekh D et al ; Aliment Pharmacol Ther 2007.
- The risk of colorectal cancer in ulcerative colitis a meta-analysis ; J. Eaden, K. Abrams & J. Mayberry ; Gut 2001 Apr 48(4) 526-535.
- Epidemiology and risk factors in colorectal dysplasia and cancer in ulcerative colitis ; Edward V et al, Gastroenterol Clin N An 35(2006) 517-531.
- Risk for colorectal cancer in ulcerative colitis, changes, causes and management strategies ; , Lakatos P.L ; Lakatos L ; World J Gastroenterol 2008 Jul 7, 14(25) :3937-47.
- Epidémiologie des cancers colorectaux dans la région de Marrakech durant la période 1995-2008 ; Hakima El ouarradi ; thèse soutenue en 2010
- Alcohol intake and colorectal cancer, a pooled analysis of 8 cohort studies Cho E et al Annales Intern 2004 Apr 20, 140 (8) : 603-13.
- Cigarettes and Alcohol in relation with Colorectal Cancer; The Singapore-Chinese health study, Tsong WH; J Br 2007, mar 12 96 (5) : 827 : 1-7.
- Les principaux facteurs de risque du cancer colorectal www.hopital-dcss.org
- AINS et chimio-prévention du cancer colorectal ; R. Benamouzig et Stanislas Chaussade Aspirine, Hépato-Gastro & Oncologie Digestive Vol 10, N° – Nov 2003 411-4.
- Aspirine, AINS et risque de CCR ; J.M.M revue francophone des laboratoires vol 2015 n° 476 p 15 (Nov 2015).
- Quelle chimio-prévention du cancer colorectal prescrire en 2012, Robert Benamouzig, post’U (2012) 1-6.
- Colon cancer preventive agents and the present status of chemoprevention. Half. E, Arber. N ; Expert Opin Phamaco-ther 2009; 10 :211-9.
- Combined impact of healthy lifestyle factors on colorectal cancer ; a large European cohort study Krasimira Alexandrova et al BMC Medicine 2014 ;12 ;68 published on line 2014 Oct 10.
- Institut national du cancer mise à jour 05/07/2019.
- L’incidence du cancer colorectal selon le sexe et le site anatomique ; institut national de santé publique au Québec vol 01 : 2011 constituent des facteurs pouvant diminuer l’incidence à long terme.
- Un dépistage du cancer colorectal est-il justifié en Algérie ? ; L. Abid, santé mag janvier 2016.



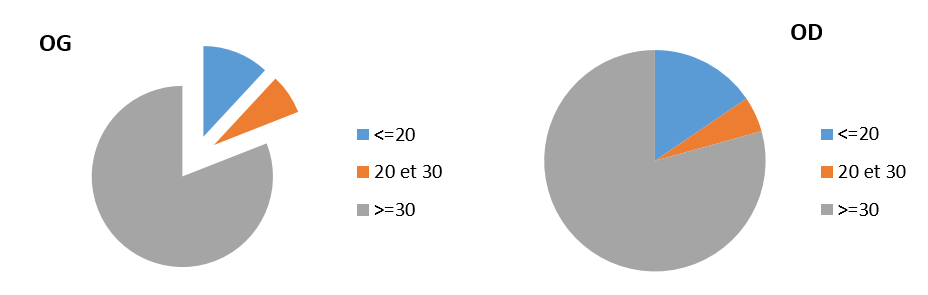
 Graphe 2 b : Conduction osseuse de l’oreille gauche
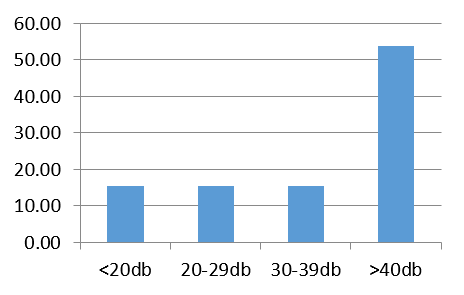
Graphe 2 b : Conduction osseuse de l’oreille gauche Graphe 3 : Résultats audiométriques
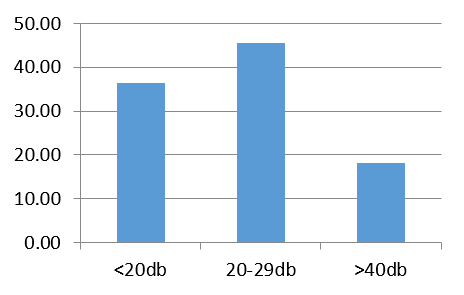
Graphe 3 : Résultats audiométriques