La spasticité est définie comme un trouble moteur caractérisé par une augmentation vitesse dépendante du réflexe tonique d’étirement associée à une exagération des réflexes tendineux secondaire à une hyperexcitabilité du réflexe d’étirement(Lance et al.1980). C’est une des trois composantes du syndrome pyramidal qui inclut également le déficit moteur et la perte de la sélectivité du mouvement.
M.Ammenouchea, M. Boubirb,
a Professeur en médecine, ancien chef de service de l’EHS Azur Plage-Alger, Enseignant universitaire, Investigateur coordonnateur de l’étude ‘’Spadys’’.
b Medical Advisor, Ipsen
Résumé
Contexte : La spasticité est définie comme un trouble moteur caractérisé par une augmentation vitesse dépendante du réflexe to- nique d’étirement associée à une exagération des réflexes tendineux secondaire à une hyperexcitabilité du réflexe d’étirement(Lance et al.1980). C’est une des trois composantes du syndrome pyramidal qui inclut également le déficit moteur et la perte de la sélec- tivité du mouvement. Ces deux dernières composantes ne sont pas accessibles aux thérapeutiques pharmacologiques, chirurgi- cales ou physiques. À l’inverse, de nombreuses thérapeutiques permettent de réduire le symptôme spasticité ( D. BEN SMAÏL, C. KIEFER, B. BUSSEL ,Neurochirurgie, 2003, 49, 190-198). Parmi les traitements, la toxine botulique, provenant d’une bactérie, Clostridium botulinum, agit en bloquant la transmission neuro-musculaire, entrainant alors une relaxation musculaire. De nom- breux travaux existent sur l’utilisation de la toxine botulinique dans le traitement de la Spasticité. La diminution de la spasticité est certaine avec le plus souvent une diminution de son retentissement fonctionnel (recommandations de bonnes pratiques, traite- ment médicamenteux de la spasticité-AFSSAPS 2009). A la période où le protocole de cette étude a été établi, en Algérie, aucune donnée n’était disponible sur cette complication, son traitement et ses étiologies.Cette étude a fourni des données sur le profil, la démographie et la prise en charge clinique des patients atteints de spasticité quelle qu’en soit la cause, ainsi que sur les modalités planifiées d’utilisation de la toxine botulinique de type A (BoNT-A) et les traitements d’appoint.
Méthodes : Cette étude nationale, multicentrique, observationnelle, descriptive, non interventionnelle transversale, réalisée au- près de 10 centres hospitaliers de prise en charge des patients spastiques a été menée entre novembre 2011 et mars 2013 dans un contexte où peu de données existaient sur le traitement de la spasticité dans la population de patients algériens. Au total, 205 patients âgés de plus de 18 ans présentant une spasticité des membres supérieurs et/ou inférieurs ont été inclus. L’objectif principal était de décrire les caractéristiques démographiques, cliniques et étiologiques d’une population de patients algériens atteints de spasticité et nécessitant un traitement par la toxine botulinique de type A (BoNT-A). Les objectifs secondaires étaient de décrire les méthodes d’administration de la BoNT-A utilisées en pratique clinique courante en Algérie ainsi que les traitements concomitants potentiellement utilisés dans la spasticité.
Résultats : 135 (65%) patients avaient plus de 41 ans et chez 115 (56%) patients, l’Accident Vasculaire Cérébral (AVC) était la cause de la spasticité. Soixante-quatorze (36%) patients avaient une spasticité des membres supérieurs et inférieurs (SMIS), 68 (33%) une spasticité des membres supérieurs seuls (SMS) et 63 (31%) patients une spasticité des membres inférieurs seuls (SMI). L’évaluation par l’échelle modifiée d’Ashworth a été utilisée en routine chez 185 (90%) patients pour les membres supérieurs et chez 200 (98%) patients, pour les membres inférieurs avec une atteinte modérée des muscles des membres supérieurs ou infé- rieurs. L’échelle ROM (Range Of Motion) active a également été utilisée chez 204 (99%) sujets pour les membres supérieurs et 202 (98.5%) pour les membres inférieurs. L’échelle ROM passive a été utilisée pour la totalité des sujets pour les membres supérieurs et inférieurs. Enfin, l’échelle de Tardieu et la Medical Research Council Scale (MRCS) n’étaient pas évaluées en routine. Les patients étaient traités par une approche physique et/ou médicamenteuse, en accord avec les données retrouvées dans la littérature (Hy- man et al.; Ward, 2002;). Les injections de la BoNT-A ont été planifiées pour être réalisées au niveau des membres supérieurs et/ ou inférieurs. Le délai prévu entre la séance d’injection et la prochaine visite d’évaluation était globalement inférieur à 6 semaines chez 173 (85%) des patients.
Conclusion Cette étude, sur une population algérienne de patients atteints de spasticité, a montré que plus des deux tiers des patients avait plus de 41 ans. L’étiologie la plus fréquente de la spasticité était l’AVC ischémique avec une répartition comparable de la spasticité au niveau des membres supérieurs et/ou inferieurs, entrainant un déficit moteur chez la majorité des patients. L’échelle d’Ashworth modifiée ainsi que la ROM (Range of Movement) passive et active étaient les plus utilisées en routine. Des objectifs thérapeutiques ont été fixés chez la majorité des patients afin d’évaluer l’efficacité des injections planifiées de la BoNT-A. Les patients étaient traités par une approche physique et/ou médicamenteuse et les modalités d’administration des injections plani- fiées étaient en accord avec les mentions légales locales du produit ainsi que les données retrouvées dans la littérature.
>>> Mots-clés :
Spasticité musculaire, hypertonie musculaire, toxine botulinique type A, toxine botulinique, protéines bactériennes.
Introduction
La spasticité est un trouble moteur résultant d’une lé- sion du cerveau ou de la moelle épinière. Elle est carac- térisée par une augmentation vitesse dépendante du réflexe tonique d’étirement, associée à une exagération des réflexes tendineux secondaire et à une hyperexcita- bilité du réflexe d’étirement. Elle constitue, avec le défi- cit moteur et la perte de la sélectivité du mouvement, les trois composantes du syndrome pyramidal. Les pa- tients développent une spasticité généralement après un accident vasculaire cérébral (AVC) ou un traumatisme crânien. On l’observe également chez des personnes atteintes de sclérose en plaques, de paralysie cérébrale. De nombreuses approches thérapeutiques permettent de réduire les symptômes de la spasticité (Hyman et al., 2000; Li and Francisco, 2015; Ward, 2002;).
La prise en charge de la spasticité n’est pas standardisée et nécessite l’établissement d’une liste d’objectifs person- nalisés pour chaque patient, après avoir évalué les diffé- rentes composantes du trouble moteur et la gêne fonc- tionnelle. La stratégie thérapeutique est fondée sur cette approche par objectifs. Tout malade atteint de spasticité ne nécessite pas systématiquement de traitement (Cau- lin et al., 2009).
La BoNT-A est une neurotoxine provenant de la bac- térie Clostridium botulinum. Elle inhibe la libération d’acétylcholine au niveau de la jonction neuromus- culaire, empêchant ainsi le muscle de se contracter et permettant ainsi de réduire la spasticité, aussi bien des membres supérieurs que des membres inférieurs (Blac- kie and Lees, 1990; Elston, 1992; Poewe et al., 1998). La toxine botulinique de type A est recommandée car il existe une preuve scientifique établie de son effet sur la réduction locale de la spasticité après injection intra- musculaire (Grade A). Elle peut être utilisée en trai- tement de première intention de la spasticité lorsque l’objectif est focal ou multifocal (Recommandations de bonne pratique, traitement médicamenteux de la spasti- cité, AFSSAPS 2009).
Le traitement médicamenteux doit être le plus souvent envisagé comme n’étant qu’une composante d’un pro- gramme thérapeutique devant associer, à des degrés divers : kinésithérapie qui demeure le traitement de base pour tout patient spastique, ergothérapie, appareillage, auto-rééducation, chirurgie orthopédique et neurochi- rurgie (Recommandations de bonne pratique, traitement médicamenteux de la spasticité, AFSSAPS 2009).
A la période où le protocole de cette étude a été établi, Il y avait peu de données sur l’étiologie de la spasticité, les traitements et le profil des patients algériens spastiques.
Une compréhension des caractéristiques cliniques des sujets souffrant de spasticité des membres supérieurs et/ ou inférieurs et nécessitant une BoNT-A ainsi que les traitements d’appoint était nécessaire pour établir des sous-groupes spécifiques de patients.
Cette étude a fourni des données sur les sujets atteints de spasticité des membres, sur l’étiologie ainsi que sur les modalités planifiées d’utilisation de la BoNT-A et les traitements concomitants.
En 2011, l’année où le protocole de cette étude a été éta- bli, la seule BoNT-A disponible était l’abobotulinum- toxinA.
Matériels et méthodes
Schéma de l’étude
Cette étude, nationale, multicentrique, observation- nelle, descriptive, non interventionnelle transversale a été conduite de Novembre 2011 à mars 2013, auprès de 10 centres hospitaliers de prise en charge des patients spastiques. 205 patients ont été enrôlés. La décision de prescrire et les modalités d’injection (muscles cibles, préparation et dose) de la BoNT-A ont été prises préala- blement et indépendamment de la décision d’inclure le patient, conformément aux pratiques cliniques usuelles du centre hospitalier participant et telles que décrites dans les informations produit approuvées en Algérie. La période d’inclusion était de 18 mois, et la durée de par- ticipation à l’étude de chaque patient était d’une visite seulement.
Cette étude a été approuvée par les autorités règle- mentaires locales ainsi que par le comité d’éthique et conduite en accord avec la Déclaration de Helsinki et les directives des bonnes pratiques cliniques.
Population d’étude
Les sujets de plus de 18 ans, naïfs de tout traitement à la BoNT-A, ayant donné leur consentement écrit et éclairé, ont été inclus s’ils présentaient une spasticité des membres supérieurs et/ou inférieurs et dont la première injection de la BoNT-A était préalablement prévue. Les patients n’étaient pas éligibles en cas de grossesse ou de participation à une autre étude clinique.
Objectifs de l’étude
L’objectif primaire de l’étude consistait à décrire les caractéristiques cliniques, démographiques et étiolo- giques d’une population de patients algériens atteints de spasticité et nécessitant un traitement par la toxine botulinique de type A (BoNT-A).
Les objectifs secondaires consistaient à décrire les mé- thodes d’administration de la BoNT-A planifiées en pra- tique clinique courante en Algérie ainsi que les traitements concomitants potentiellement utilisés dans la spasticité.
Les critères d’évaluations primaires comprenaient :
- La description des données démographiques ;
- La description des lésions ayant entraînées la spastici- té des membres supérieurs et/ou inférieurs, l’apparition de spasticité, l’étiologie et la stabilisation de la lésion ;
- Description des déficits physiques associés, des troubles cognitifs, des facteurs confondants(chirurgie antérieure sur le membre à traiter, comorbidités médi- cales, contractures fixes sur le membre à traiter, hyperac- tivités des antagonistes, douleur associée à la non-spas- ticité du membre à traiter, autre), du profil de posture des membres supérieurs et inférieurs (y compris la main et le pied) ;
- Les scores sur les échelles utilisées dans la pratique courante pour évaluer la spasticité (telles que l’échelle modifiée de Tardieu, la Medical Research Council Scale et la ROM) ;
- La description de la dystonie, la présence ou pas de douleur liée à la spasticité des membres supérieurs et/ ou inférieurs et les réactions associées ;
- La description des objectifs prévus : objectif principal, objectifs secondaires ;
- La description des évaluations planifiées pour appré- cier les résultats.
Les critères d’évaluation secondaires consistaient en :
- La description des traitements concomitants (médi- camenteux et physiques) ;
- La description des modalités d’injections planifiées de la BoNT-A (volume de reconstitution, muscles cibles, dose, nombre de points d’injection prévu par muscle cible).
Recueil des données
Toutes les données recueillies en rapport avec les objec- tifs de l’étude ont été collectées lors de l’unique visite prévue dans le cadre de l’étude.
Analyse statistique
L’évaluation statistique a été réalisée avec le logiciel SAS® (Statistical Analysis System).
Des statistiques descriptives (n, moyenne, écart type [SD], médiane, valeur minimale, valeur maximale) ou des comptes de fréquence ont été présentés. Des inter- valles de confiance à 95 % ont été également fournis.
Les données continues ont été décrites au moyen de leur n, moyenne, écart type (SD), médiane, valeur minimale et valeur maximale.
Les données nominales ont été décrites via le nombre et le taux de patients dans chaque catégorie.
Résultats
Critères d’évaluation primaires :
1. Données démographiques
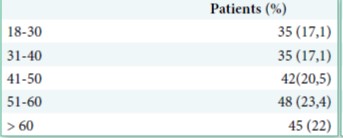
Table 1 : Résumé des caractéristiques démographiques
Catégorie d’âges
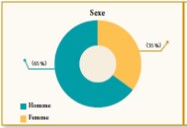
Sexe

Parmi les 205 patients inclus, 112 (55%) patients étaient des hommes. 135 patients soit 65% avaient plus 41 ans.
2. Caractéristiques cliniques
- La lésion causale (Graphes 1,2,3) était cérébrale acquise chez 178 (87%) patients, avec une prédominance d’AVC ischémique chez 115 (56%) patients. Chez 154 (75%) pa- tients, la spasticité était survenue depuis plus de 12 mois.
- Soixante-quatorze (36%) patients avaient une spas- ticité des membres supérieurs et inférieurs (SMIS), 68 (33%) une spasticité des membres supérieurs seuls (SMS) et 63 (31%) patients une spasticité des membres inférieurs seuls (SMI).
Graphe 1 : Etiologie principale de la lésion causale de spasticité des membres supérieurs chez les sujets inclus.
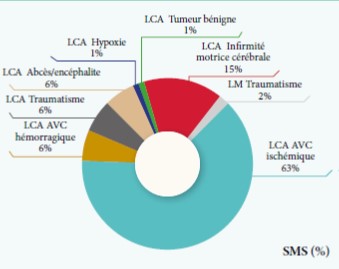
SMS= Spasticité du Membre Supérieur ; LCA=Lésion cérébrale acquise ; LM= Lésion médullaire ; ME= Maladie évolutive
Graphe 2 : Etiologie principale de la lésion causale de spasticité des membres inférieurs chez les sujets inclusts inclus.
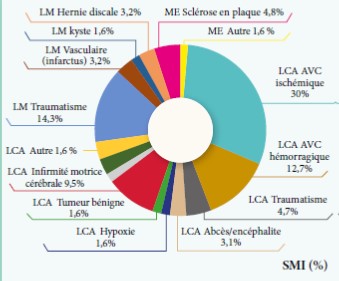
SMI= Spasticité du Membre Inférieur ; LCA=Lésion cérébrale acquise ; LM= Lésion médullaire ; ME= Maladie évolutive
Graphe 3 : Etiologie principale de la lésion causale de spasticité des membres supérieurs & Inferieurs chez les sujets inclus
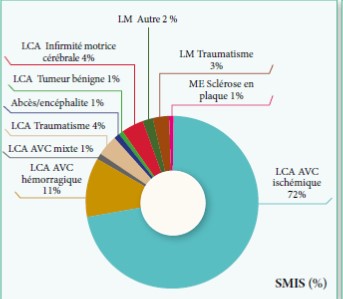
SMIS= Spasticité des Membres Inférieurs et Supérieurs ; LCA=Lésion cérébrale acquise ; LM= Lésion médullaire ; ME= Maladie évolutive
- Les lésions causales étaient stabilisées chez 186 (91%)
3. Description des déficits physiques associés, des troubles cognitifs, des facteurs confondants, du pro- fil de posture des membres supérieurs et inférieurs :
- Le déficit physique le plus courant dû à la spasticité était le déficit moteur touchant les membres supérieurs chez 187 (91%) patients et les membres inférieurs chez 202 (98%) patients. De plus, 167 (81%) des sujets ne présentaient pas d’atteinte sensitive des membres supérieurs seuls et 160 (78%) sujets d’atteinte sensitive des membres inférieurs seuls ; un déficit moteur et sensitif au niveau des membres
supérieurs seuls et des membres inférieurs seuls était pré- sent chez 32 (16%) et 25 (12%) patients, respectivement.
- Un problème de communication a été observé chez 48 (23%) patients et 12 (6%) des patients avaient un trouble visuel. Vingt et un (10%) patients présentaient un déficit cognitif et 31 (15%) patients des troubles psychiatriques.
- La comorbidité médicale étant signalée comme le principal facteur confondant pour l’apparition de la spasticité, a été rapportée chez 45,9% des sujets (n = 94), suivie de contractures fixes sur le membre supérieur chez 31,7% des sujets (n = 65).
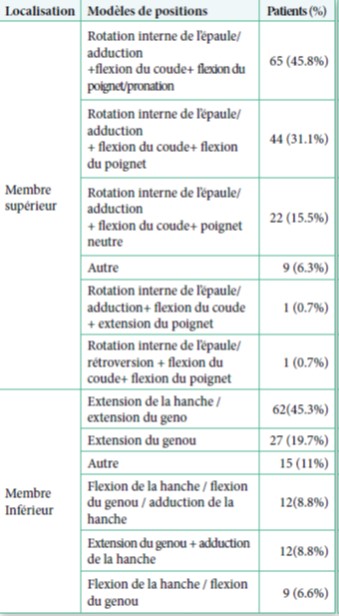
- Les modèles de positions (Table 2) des membres spastiques les plus fréquents rapportés par les investigateurs sont pour le membre supérieur la rotation interne de l’épaule / Adduc- tion + Flexion du coude + Flexion du poignet / Pronation chez 65 sujets (45,8%), et l’extension de la hanche / genou dans le membre inférieur chez 62 sujets (45,3%).
Table 2 : Modèles de positions des membres spastiques
4. Scores sur les échelles utilisées dans la pratique courante pour évaluer la spasticité :
- L’échelle d’Ashworth modifiée a été planifiées en rou- tine chez 185 (90%) patients pour les membres supé- rieurs et chez 200 (98%) patients, pour les membres inférieurs. Globalement, pour les membres supérieurs et inférieurs, les scores étaient majoritairement de 2 ou 3 (table 3).
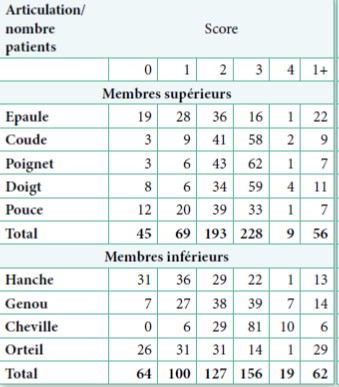
- L’échelle ROM active a également été utilisée chez 204 (99%) sujets pour les membres supérieurs et 202 (98.5%) pour les membres inférieurs. L’échelle ROM passive a été utilisée pour la totalité des sujets pour les membres supérieurs et inférieurs. Enfin, l’échelle de Tardieu et la MRCS n’étaient pas évaluées en routine.
5. Description de la dystonie, la présence ou pas de douleur liée à la spasticité des membres supé- rieurs et/ou inférieurs et les réactions associées :
- Une dystonie associée du membre supérieur et infé- rieur a été observée chez 6 et 4 patients, respectivement.
-Des douleurs des membres supérieurs dues à la spasti- cité ont été observées chez 47 (33% ; 63 données man- quantes) patients et au niveau des membres inférieurs chez 16 (12% ; 68 données manquantes).
- Les réactions associées n’étaient pas évaluées en rou- tine pour 32 (15.6%) patients. Elles étaient présentes chez 18/171 (10.5% ; 2 données manquantes).
La plupart des sujets de l’étude n’avaient pas de réactions associées (153 ; 89.5%).
- Description des objectifs prévus : un objectif prin- cipal, des objectifs secondaires
- Pour la spasticité des membres supérieurs, un objectif principal a été défini chez 142 (69%) patients et consistait en : amélioration de la fonction passive et active pour 86 (60%) et 52 (37%) sujets, respectivement et douleur pour 5 (3.5%) sujets. Des objectifs secondaires ont été défini chez 142 (69%) et consistaient en : amélioration de la fonction passive et active pour 126 (89%) et 58 (41%) sujets, respectivement et douleur associée pour 31 (22%) sujets.
- Pour la spasticité des membres inférieurs, un objectif principal a été défini chez 137 (67%) et consistait en : amélioration de la fonction passive et active pour 120 (88%) et 15 (11%) sujets, respectivement et douleur pour 2 (1.5%) sujets. Des objectifs secondaires ont été défini chez 136 (66%) et consistaient en : amélioration de la fonction passive et active pour 101 (74%) et 84 (62%) sujets, respectivement, réactions associées chez 14 (10%) sujets et douleur associée pour 13 (10%) sujets.
7. Description des évaluations planifiées pour apprécier les résultats
Les investigateurs prévoyaient l’évaluation de l’injection de la BoNT-A chez 204 (99.5%) des patients.
La majorité des patients devaient être évalués en ≤ 6 semaines (85,2%, n = 73). L’évaluation clinique (98,5%, n = 201) et la mesure formelle des résultats (92,2%, n =
188) étaient les méthodes souhaitées d’évaluation. L’évaluation des objectifs a été généralement prévue en notant simplement les objectifs atteints (47,34%, n =
89). « L’évaluation de la facilité de soins par le soignant »
était principalement prévue pour l’évaluation de la fonc- tion passive (9,6%, n = 18) et la «vitesse de marche sur 10 mètres» était prévue pour l’évaluation de la fonction active (15,4%, n = 29). L’échelle d’Ashworth / Ashworth modifiée (93,6%, n = 176) et la goniométrie étaient cou- ramment prévues pour évaluer le déficit (61,7%, n = 116). Les réactions associées devaient être évaluées par une analyse de la marche (2,7%, n = 5). D’autres évaluations des résultats ont été prévus par l’impression générale du bénéfice / Satisfaction des patients (82,5%, n = 155).
Critères d’évaluations secondaires
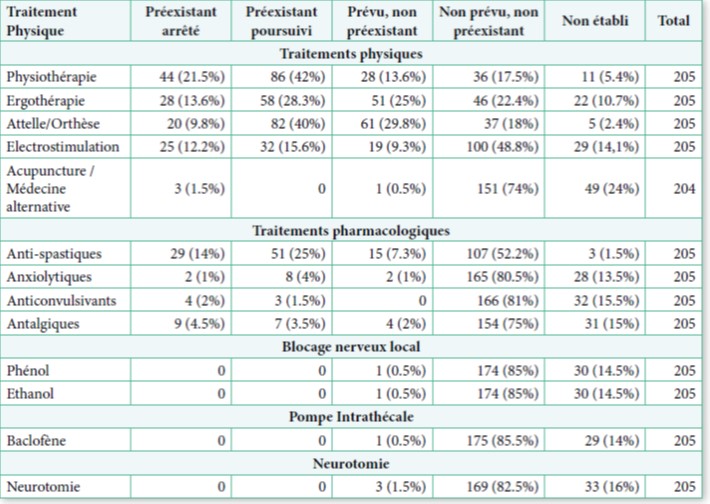
Traitements concomitants
Les traitements de la spasticité (préexistants poursui- vis et arrêtés ou prévus) étaient essentiellement phy- siques, incluant principalement de la physiothérapie
et de l’ergothérapie. Des traitements médicamenteux étaient également dispensés (table 4).
Table 4 : Traitements concomitants de la spasticité.
Description des injections planifiées de la Toxine Botulinique type A
Le volume moyen de reconstitution prévu était de 2.5mL. Le délai prévu pour l’évaluation était globalement inférieur à 6 semaines chez 173 (85% des patients). La médiane du nombre total d’unitésprévues de la BoNT-A injectée était de 500 [400 ; 500] pour les SMS, 800 [500 ; 1000] pour les SMI et 500 [350 ; 600] pour les SMIS.
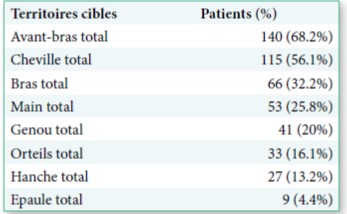
Les territoires d’injections planifiées de la BoNT-A sont détaillés dans la table 5. Les muscles cibles les plus cou- ramment planifiés pour l’injection de la BoNT-A étaient dans l’avant-bras – reportés chez 68,3% des patients (n = 140), et dans la cheville – reportés chez 56,1% des patients (n = 115).
Les muscles cibles les plus fréquemment prévus pour l’administration de la BoNT-A planifiées dans l’avant- bras étaient le fléchisseur commun superficiel – chez 44,4% des sujets (n = 91), le rond pronateur – chez 42,9% des sujets (n = 88) et le fléchisseur radial du carpe
- chez 40,5% des sujets ( n = 83) et à la cheville étaient le soléaire – chez 41,0% des sujets (n = 84), le Lateralis
gastrocnemius – chez 46,3% des sujets (n = 95) et le Me- dialis gastrocnemius – chez 46,8% des sujets, (n = 96).
Table 5 : Répartition des Territoires cibles planifiés pour l’injection de la BoNT-A par % de patients
Concernant le nombre de points d’injection prévu par muscle cible, la moyenne été de 6 [1;12] au niveau des membres supérieurs et de 5 [1;18] au niveau des membres inférieurs. Le nombre de points varie selon les muscles cibles et le type de spasticité.
La durée de participation à l’étude de chaque patient donné étant d’une visite seulement (Visite d’injection de la BonT-A). Il n’y a eu aucune visite de suivi et aucune donnée de sécurité n’a été collectée sur le CRF (Case Report Form). Seule l’intention de traiter avec de la BoNT-A a été recueillie dans cette étude.
Discussion
La spasticité est une affection neurologique fréquente chez l’adulte et chez l’enfant, dans laquelle certains muscles sont en permanence contractés. Elle survient à la suite d’une lésion cérébrale ou médullaire. Les causes les plus fréquentes de la spasticité sont la paralysie céré- brale, l’AVC, la sclérose en plaque (Hyman et al., 2000; Li and Francisco, 2015; Ward, 2002; Wissel et al., 2013). L’AVC ischémique, quatrième cause de décès dans le monde, constitue actuellement la cause la plus impor- tante de spasticité (Disa et al., 2004; Munter et al., 2002; CL Watkins et al., 2002). Néanmoins, sa prévalence est difficile à établir. En fonction des études, la prévalence de la spasticité post-AVC varie entre 19% et 92% (Li and Francisco, 2015; Ward, 2012; Wissel et al., 2013). Dans notre population d’étude, l’AVC ischémique est la cause la plus fréquente de spasticité mais les données ne per- mettent pas d’établir sa prévalence dans la population al- gérienne. Les patients inclus dans cette étude présentent une spasticité modérée. L’évaluation clinique de la spas- ticité des patients a été réalisée à l’aide de l’échelle modi- fiée d’Ashworth ce qui constitue la méthode de choix dans la pratique courante en Algérie.Il existe d’autres méthodes d’évaluation notamment l’échelle de Tardieu qui est plus spécifique à la spasticité et tient compte de la posture et de la vitesse d’étirement (Balci, 2018; Ben Smaïl et al., 2003a; Ward, 2002) mais son utilisation ne faisait pas partie de la pratique courante en Algérie à l’époque ou l’étude a été réalisée.
Lors de cette étude, des objectifs thérapeutiques ont été fixés par les investigateurs avec une description des éva- luations planifiées pour apprécier les résultats du traite- ment BoNT-A. La Goal Attainment Scaling (GAS), une méthode qui permet d’écrire des échelles d’évaluation personnalisées n’a pas été utilisée. Il est établi que l’éta- blissement d’objectifs thérapeutiques individualisés qui ont de l’importance pour le patient et sa famille ont plus de chances d’être atteints (Turner-Stokes L et al. BMJ Open 2013; 3: e002771).
La prise en charge de la spasticité associe des traitements physiques et médicamenteux (Ben Smaïl et al., 2003a; Chang et al., 2013; Ward, 2002). Le traitement physique est primordial dans la prise en charge et doit être débuté
au plus vite chez tous les patients, afin de prévenir et réduire la gêne fonctionnelle et les complications qui en découlent (Chang et al., 2013; Esquenazi et al., 2017; Ward, 2002). En accord avec la littérature, près de 80% de nos patients ont bénéficié, bénéficient ou bénéficie- ront d’un traitement physique et notamment de la phy- siothérapie, de l’ergothérapie et des orthèses (Hyman et al., 2000; Ward, 2002; CL Watkins et al., 2002).
Il est reconnu que lorsque les moyens physiques de- viennent insuffisants, une intervention médicamenteuse est nécessaire (Ward, 2002). On distingue les agents antispastiques à action centrale (Baclofène®, agonistes Alpha-2 et anticonvulsivants) ou à action périphérique (Dantrolène®). Le Baclofène® constitue le traitement de première ligne chez l’adulte (Chang et al., 2013).
Les agonistes Alpha-2 sont rarement utilisés seuls, à cause de leurs effets indésirables (Esquenazi et al., 2017; Kamen et al., 2008; Nance et al., 1985; Rabchevsky and Kitzman, 2011; Stewart et al., 1991). Le Dantrolène® est le seul antispastique approuvé par la FDA. Un traite- ment antispastique a été de mis en place chez presque la moitié de nos patients. Néanmoins, la pompe intrathé- cale de Baclofène qui est largement recommandée dans les spasticités sévères(Chang et al., 2013; Esquenazi et al., 2017; Pin et al., 2011; Simon and Yelnik, 2010) n’est prévue que chez 0.5% de nos patients, mais il est à rappeler qu’ils ne présentent pas une forme sévère de la maladie. L’injection locale de phénol ou d’alcool ainsi que la neurotomie ne sont prescrits que dans 1% et 1.5% des patients, respectivement.
Ce faible taux de prescription concorde avec les données présentes dans la littérature (Hyman et al., 2000; Ward., 2002., Chang et al., 2013 ). Le traitement à la BoNT-A offre une nouvelle approche dans la prise en charge de la spasticité. La BoNT-A est devenue le traitement de choix et peut être utilisée en traitement de première intention, lorsque l’objectif est focal ou multifocal (Re- commandations de bonne pratique, traitement médica- menteux de la spasticité, AFSSAPS 2009). Chez l’adulte, la plupart des résultats proviennent d’études concernant des patients ayant fait un AVC, mais son utilisation peut être envisagée quelle que soit l’étiologie de la spasticité (Caulin et al., 2009; Ianieri et al., 2018; Mémin et al., 1992). En fonction des muscles concernés, les doses utilisées varient de 400 à 1 500 unités de la BoNT-A ce qui correspond aux doses planifiées dans cette étude. Le nombre de sites d’injections dépend de la structure et de la taille du muscle ; le nombre prévu de points d’injec- tion par segment, par membre et par muscle ont été éta- blis pour notre population. L’avant-bras, le bras, la main,
la hanche, la jambe et la cheville sont les zones privilégiées de traitement, ce qui correspond aux données de la littéra- ture (Hyman et al., 2000; Li, S., and Francisco., 2015)..
Il a été montré des effets bénéfiques de l’injection de la BoNT-A tant au niveau des muscles des membres supérieurs qu’inférieurs. Néanmoins, le protocole de cette étude ne prévoyait pas l’évaluation des effets de la BoNT-A sur la spasticité. Il est recommandé d’évaluer en consultation les résultats entre 3 et 6 semaines après la première injection (Caulin et al., 2009). Enfin, dans cette étude, la spasticité chez la majorité des patients datait de plus de 12 mois alors que certaines études montrent une amélioration de l’efficacité des traitements de la spasticité post-AVC, lors de la prise en charge pré- coce (Hyman et al., 2000, Simon and Yelnik., 2010).
Conclusion
Aucune donnée épidémiologique algérienne antérieure n’était disponible sur la prise en charge de la spasticité au moment de la mise en place du protocole. Cette étude donne un aperçu sur le profil et la prise en charge des patients algériens spastiques nécessitants un traitement à la BoNT-A.
La majorité (65%) des sujets inclus dans cette étude avaient plus 41 ans et étaient principalement des hommes avec un sex-ratio de 1.2
La lésion causale était cérébrale acquise chez la majorité des patients, avec une prédominance d’AVC ischémique. Chez la plupart des patients, la spasticité était survenue depuis plus de 12 mois.
Les déficits moteurs ont été signalés comme déficit phy- sique majeur pour les membres supérieurs et inférieurs avec une répartition globalement équitable de la spasti- cité entre les membres inférieurs, supérieurs et les deux membres à la fois. Les déficits sensoriels ont été rare- ment signalés. Les déficits de perception, de communi- cation, cognitifs, d’humeur, psychiatriques et comporte- mentaux étaient moins fréquents chez ces sujets.
Pour la spasticité des deux membres, les objectifs thé- rapeutiques étaient définis chez la majorité des patients comme étant une amélioration de la fonction passive pour les membres supérieurs et la fonction active pour les membres inférieurs.
Les échelles MAS et ROM actif/passif ont été évaluées chez la plupart des sujets pour les deux membres, l’échelle de Tardieu et le MRCS n’étaient pas pour leur part systématiquement évalué pour les deux membres.
La dose injectée médiane prévue de la BoNT-A adminis- trée était de 500 unités. Les points d’injection médians prévus par muscle cible étaient de 6 pour le membre
supérieur et de 5 pour le membre inférieur. Les muscles cibles les plus couramment planifiés pour l’injection de la BoNT-A étaient dans l’avant-bras et dans la cheville.
Cette étude a montré que la prise en charge des pa- tients atteints de spasticité est en accord avec les don- nées retrouvées dans la littérature. L’évaluation clinique correcte de la spasticité et des éventuelles gênes qu’elle occasionne est un élément fondamental pour une prise en charge individualisée du sujet spastique. Elle permet d’entreprendre la thérapeutique adéquate et d’optimiser ainsi les chances de succès de celle-ci.
Remerciements
Les auteurs souhaitent remercier tous les sites participants, les inves- tigateurs principaux, les investigateurs ainsi que le coordinateur de l’étude, les patients et l’équipe soignante qui ont contribué à l’étude Spadys.
Les auteurs remercient également Clinica Group Algérie d’avoir four- ni un soutien scientifique à la rédaction de la publication de l’étude Spadys sponsorisée par Ipsen conformément aux directives de bonnes
pratiques de publication.
Investigateurs de l’étude Spadys
Ammenouche M ; Aichoune S; Amri Z ; Aouichet L; Arbaoui S ; Bedjaoui M ; Benmouffok M; Bensaber O; Boukara Z ; Guechi A ; Habhoub R ; Ha- madi A ;Kaced H ; Korchi A ; Lemai S; Maougal R ; Marniche B ; Morsi L ; Negli S ; Nouacer Z; Nouar A ; Nouara M; Toumi N ; Seffouh L.
Financement
L’étude Spadys a été sponsorisée par Ipsen.
Conflit d’intérêt
Pr Ammenouche a été rémunéré en tant qu’investigateur coordonna- teur de l’étude Spadys.
M.Boubir est un employé d’Ipsen.
Références
Balci, B.P. (2018). Spasticity Measurement. Noro Psikiyatr Ars 55, S49–S53.
Ben Smaïl, D., Kiefer, C., and Bussel, B. (2003a). [Clinical evaluation of spasticity]. Neurochirurgie 49, 190–198.
Recommandations de bonnes pratiques, traitement médicamenteux de la spasticité-AFSSAPS 2009
Blackie, J.D., and Lees, A.J. (1990). Botulinum toxin treatment in spas- modic torticollis. J. Neurol. Neurosurg. Psychiatry 53, 640–643.
Bohannon, R.W., and Smith, M.B. (1987). Interrater reliability of a modified Ashworth scale of muscle spasticity. Phys Ther 67, 206–207. C L Watkins, M J Leathley, J M Gregson, A P Moore, T L Smith and A K Sharma. (2002). Prevalence of spasticity post stroke. Clin Rehabil, 16: 515.
Caulin, P.C., Bergmann, P.J.-F., Bannwarth, P.B., Debrix, M.I., Dessi,
D.F., Gerson, D.M., Girard, D.L., Goichot, P.B., Larrumbe, D.J.-P., Lievre, M.M., et al. (2009). COMITE DE VALIDATION. 16.
Chang, E., Ghosh, N., Yanni, D., Lee, S., Alexandru, D., and Mozaffar (2013).
A Review of Spasticity Treatments: Pharmacological and Interventional Approaches. Crit Rev Phys Rehabil Med 25, 11–22. Disa K. Sommerfeld, PT, MSc; Elsy U.-B. Eek, PT, MSc; Anna-Karin Svensson, PT, MSc; Lotta Widén Holmqvist, PT, PhD; Magnus H. von Arbin, MD, PhD (2004). Spasticity After Stroke Its Occurrence and Association With Motor Impairments and Activity Limitations. Stroke, 35:134-140.
Hyman et al., 2000; Li and Francisco, 2015; Ward, 2002; Wissel et al., 2013 Elston, J.S. (1992). The management of blepharospasm and hemifacial spasm. J. Neurol. 239, 5–8.
Esquenazi, A., Lee, S., Mayer, N., Garreta, R., Patel, A., Elovic, E., Koelbel, S., Francisco, G., Reuter, I., and PROS World Group (2017). Patient Registry of Spasticity Care World: Data Analysis Based on Physician Experience. Am J Phys Med Rehabil 96, 881–888.
Hyman, N., Barnes, M., Bhakta, B., Cozens, A., Bakheit, M., Krec- zy-Kleedorfer, B., Poewe, W., Wissel, J., Bain, P., Glickman, S., et al. (2000). Botulinum toxin (Dysport) treatment of hip adductor spas- ticity in multiple sclerosis: a prospective, randomised, double blind, placebo controlled, dose ranging study. J. Neurol. Neurosurg. Psy- chiatry 68, 707–712.
Ianieri, G., Marvulli, R., Gallo, G.A., Fiore, P., and Megna, M. (2018). “Appropriate Treatment” and Therapeutic Window in Spasticity Treat- ment with IncobotulinumtoxinA: From 100 to 1000 Units. Toxins (Basel) 10.
Kamen, L., Henney, H.R., and Runyan, J.D. (2008). A practical over- view of tizanidine use for spasticity secondary to multiple sclerosis, stroke, and spinal cord injury. Curr Med Res Opin 24, 425–439.
Lance, J.W. (1980). The control of muscle tone, reflexes, and move- ment: Robert Wartenberg Lecture. Neurology 30, 1303–1313.
Lance, J.W. (1990). What is spasticity? Lancet 335, 606.
Li, S., and Francisco, G.E. (2015). New insights into the pathophysio- logy of post-stroke spasticity. Front Hum Neurosci 9, 192.
Turner-Stokes L et al. BMJ Open 2013; 3: e002771
Mémin, B., Pollak, P., Hommel, M., and Perret, J. (1992). [Treatment of spasticity with botulinum toxin]. Rev. Neurol. (Paris) 148, 212–214.
Muntner Paul, PhD; Elizabeth Garrett, PhD; Michael J. Klag, MD, MPH; Josef Coresh, MD, PhD. (2002). Trends in Stroke Prevalence Between 1973 and 1991 in the US Population 25 to 74 Years of Age. Stroke.33:1209-1213
Nance, P.W., Shears, A.H., and Nance, D.M. (1985). Clonidine in spi- nal cord injury. Can Med Assoc J 133, 41–42.
Pin, T.W., McCartney, L., Lewis, J., and Waugh, M.-C. (2011). Use of intrathecal baclofen therapy in ambulant children and adolescents with spasticity and dystonia of cerebral origin: a systematic review. Dev Med Child Neurol 53, 885–895.
Poewe, W., Deuschl, G., Nebe, A., Feifel, E., Wissel, J., Benecke, R., Kessler, K.R., Ceballos-Baumann, A.O., Ohly, A., Oertel, W., et al. (1998). What is the optimal dose of botulinum toxin A in the treat- ment of cervical dystonia? Results of a double blind, placebo control- led, dose ranging study using Dysport. German Dystonia Study Group. J. Neurol. Neurosurg. Psychiatry 64, 13–17.
Rabchevsky, A.G., and Kitzman, P.H. (2011). Latest approaches for the treatment of spasticity and autonomic dysreflexia in chronic spinal cord injury. Neurotherapeutics 8, 274–282.
Simon, O., and Yelnik, A.P. (2010). Managing spasticity with drugs. Eur J Phys Rehabil Med 46, 401–410.
Stewart, J.E., Barbeau, H., and Gauthier, S. (1991). Modulation of lo- comotor patterns and spasticity with clonidine in spinal cord injured patients. Can J Neurol Sci 18, 321–332.
Ward, A.B. (2002). A summary of spasticity management–a treat- ment algorithm. Eur. J. Neurol. 9 Suppl 1, 48–52; dicussion 53-61.
Ward, A.B. (2012). A literature review of the pathophysiology and onset of post-stroke spasticity. Eur. J. Neurol. 19, 21–27.
Wissel, J., Manack, A., and Brainin, M. (2013). Toward an epidemio- logy of poststroke spasticity. Neurology 80, S13-19.
Télécharger le PDF de cet article