Le lien entre le prédiabète et le développement des maladies cardiovasculaires est de plus en plus évident depuis quelques années. Des études récentes ont marqué le prédiabète comme un état toxique propice au développement des complications micro et macrovasculaires avant même d’évoluer vers un diabète vrai.
S. Khensal, K. Benmohammed, Service d’Endocrinologie, CHU Abdesselam Benbadis, Constantine.
Date de soumission : 01 Octobre 2020.
Résumé : Le lien entre le prédiabète et le développement des maladies cardiovasculaires est de plus en plus évident depuis quelques années. Des études récentes ont marqué le prédiabète comme un état toxique propice au développement des complications micro et macrovasculaires avant même d’évoluer vers un diabète vrai. En effet le risque global d’atteinte coronarienne, d’accident vasculaire cérébral et de mortalité toute causes, dans le prédiabète, a été estimé par plusieurs méta-analyses entre 10 et 40%. Ces nombreuses études, admettent que ce risque élevé reste significatif même après avoir contrôlé les autres facteurs de risque cardiovasculaires connus, ce qui suggère que l’hyperglycémie en soi (même en dessous du seuil de diabète) joue un rôle délétère non négligeable. Malgré ces constatations alarmantes, l’attitude pratique des professionnels de la santé vis-à-vis du prédiabète demeure timide. Il convient donc de réaffecter les ressources en vue d’intervenir efficacement durant cette phase réversible de la maladie.
Mots clés : prédiabète, complications cardiovasculaires, prévention.
Abstract: The link between pre-diabetes and the development of cardiovascular disease has become increasingly evident in recent years. Recent studies have marked pre-diabetes as a toxic condition conducive to the development of micro and macrovascular complications before even progressing to true diabetes. In fact, the overall risk of coronary heart disease, stroke and all-cause mortality in pre-diabetes has been estimated by several meta-analysis to be between 10-40%. These numerous studies admit that this high risk remains significant even after controlling for other known cardiovascular risk factors, which suggests that hyperglycaemia in itself (even below the diabetes threshold) plays a significant deleterious role. Despite these alarming findings, the practical attitude of healthcare professionals towards pre-diabetes remains timid. It is therefore necessary to reallocate resources in order to intervene effectively during this reversible phase of the disease.
Keywords: prediabetes, cardiovascular complications, prevention.
Introduction
Le prédiabète se définit comme un état toxique « d’hyperglycémie intermédiaire », constituant un important signe avant-coureur de développement du diabète de type 2 (DT2) avec un taux de conversion annuelle allant de 5 à 10% et une proportion similaire de retour à la normoglycémie (1).
Il s’agit d’une situation borderline incluant deux entités cliniques : l’hyperglycémie modérée à jeun (HMJ) et l’intolérance au glucose (IG) et/ou une élévation de l’HbA1C de 5,7 à 6,4%. L’HMJ se manifeste par une glycémie à jeun allant de 1,10 g/l à 1,25 g/l ; l’IG est définie quant à elle par une glycémie 2 heures après une charge orale de 75 g de glucose entre 1,40 g/l et 1,99 g/l (2).
La prévalence du prédiabète est en nette ascension. Dans le monde plus de 400 millions de personnes sont prédiabétiques et ce nombre pourrait passer à 587 millions à l’horizon 2040 (3).
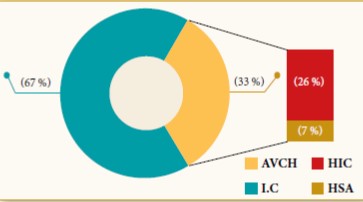
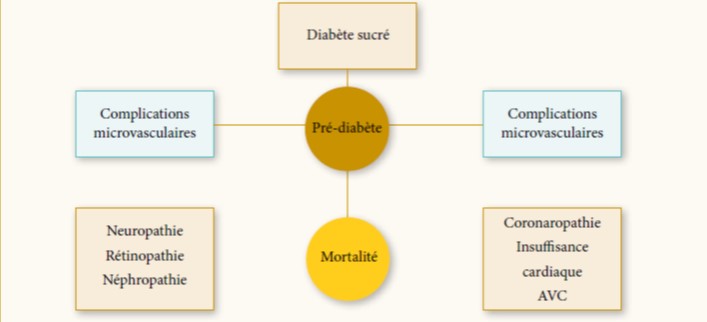
En plus de la dysglycémie, le prédiabète regroupe un cluster d’anomalies métaboliques à savoir : la dyslipidémie, l’hypertension artérielle (HTA), le surpoids ou l’obésité, l’insulinorésistance, le stress oxydatif et l’inflammation, plaçant ainsi le sujet prédiabétique face à un risque accru de développement de DT2, de maladies cardiovasculaires (MCV), de microangiopathies et de mortalité toutes causes (4,5) (figure1). En effet, selon la méta-analyse de Ford et al., le prédiabète augmente le risque de MCV d’environ 20% (6). D’autres études prospectives de cohortes révèlent que ce risque cardio-vasculaire (RCV) serait plus élevé dans l’IG, en effet, dans une méta-analyse de 10 études européennes, enrôlant plus de 22000 sujets, le risque de mortalité par MCV était ∼30% plus élevé chez les personnes atteintes d’IG que dans l’HMJ ou chez les sujets normo-glycémiques (SNG). En revanche, le sur-risque d’accident vasculaire cérébral semble être associée à la fois à l’HMJ et à l’IG (7,8).
Par ailleurs, plusieurs auteurs admettent récemment, la présence des trois complications microvasculaires classiques du diabète (neuropathie, rétinopathie et néphropathie) chez les prédiabétiques. En effet, une prévalence plus élevée des neuropathies périphérique et autonome a été signalée chez les prédiabétiques (13% et 11,3% en IG et HMJ, respectivement), par rapport aux sujets normo-glycémiques de même âge (7,4%) (9). La prévalence de la rétinopathie diabétique précoce dans le prédiabète a été estimé à 8%, parfois à des taux plus élevés (13%) en cas de combinaison d’HMJ et d’IG chez les mêmes individus (10). Les résultats de l’étude NHANES ont révélé une prévalence de l’atteinte rénale de l’ordre de 17% dans l’HMJ contre 12% chez les SNG (11).
Bien qu’une partie de ces risques soit rattachée à la progression du prédiabète vers un diabète manifeste, un risque indépendant est toujours présent chez la communauté de prédiabétiques qui n’a pas encore évolué vers un diabète avéré (12). Le grand challenge des cliniciens est d’identifier précocement les sujets prédiabétiques et d’assurer une prise en charge adéquate, ce qui permettra de prévenir non seulement la progression vers le diabète, mais également l’avènement des complications micro et macrovasculaires (13).
Figure 1 : Les principales complications du pré-diabète.
Étiopathogénie
Le prédiabète est un état hétérogène de par la nature du métabolisme d’hyperglycémie et du risque cardiovasculaire. L’IG est caractérisée par une augmentation de l’insulinorésistance des muscles squelettiques, suivie par un hyperinsulinisme compensatoire. L’HMJ, quant à elle, est associée à une insulinorésistance hépatique et une production endogène excessive de glucose. De plus, les facteurs de risque cardiométabolique, notamment l’augmentation de l’indice de masse corporelle (IMC), l’hypertension artérielle (HTA) et l’hypertriglycéridémie, coexistent souvent chez les sujets prédiabétiques et sont responsables de l’athérosclérose et des complications macrovasculaires. En outre, les prédiabétiques présentent une élévation de deux facteurs pro-athérogènes : le fibrinogène et la protéine C-réactive (CRP) (14,15). Ces troubles sont largement reconnus chez les patients DT2, mais leur apparition et leur progression commencent au stade du prédiabète (16) (figure 2).
Les complications macrovasculaires du prédiabète comprennent les MCV, les accidents vasculaires cérébraux (AVC) et les artériopathies périphériques (17,18).
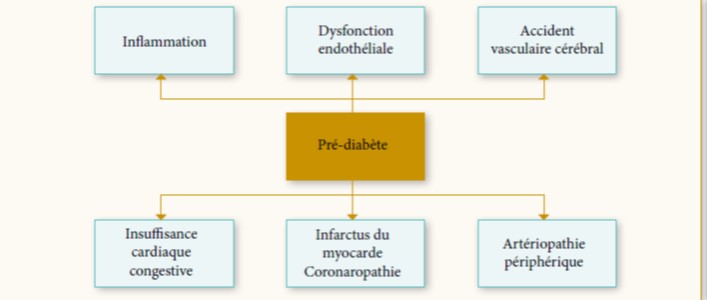
Figure 2 : Les complications macrovasculaires associées au prédiabète
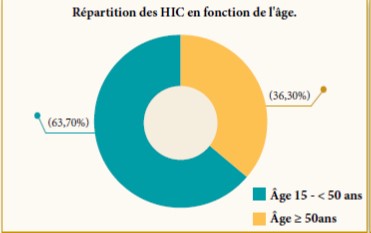
Atteinte coronarienne
Des résultats publiés en 2018, provenant d’enquêtes transversales menées de 1988 à 2014 admettent une similitude surprenante dans la prévalence de l’infarctus du myocarde et des AVC dans le prédiabète par rapport au diabète (19). Le lien entre le prédiabète et la maladie coronarienne a été démontré également par Sen et coll., qui ont mené une étude sur 62 patients hospitalisés pour coronaropathies aiguës et ont découvert que 48,4% de cette population avaient un prédiabète contre 25% seulement qui souffraient de diabète franc (20).
Dans l’étude EPIC-Norfolk, on constate qu’une augmentation de 1% d’HbA1c (dans la fourchette normale) était associée à une augmentation de la mortalité cardiovasculaire sur 10 ans (21). Ces résultats similaires ont été rapportés par l’étude de cohorte de la Paris Prospective Study qui a objectivé un doublement du taux de mortalité par MCV chez les sujets IG par rapport aux SNG. Cette augmentation pourrait être rattachée au fait que la plupart des patients prédiabétiques présentent des caractéristiques du syndrome métabolique : notamment l’obésité androïde, l’hypertriglycéridémie, la diminution du taux de cholestérol HDL et l’HTA. Les composants du syndrome métabolique peuvent souvent être identifiés chez les sujets prédiabétiques plusieurs années avant le diagnostic de DT2 (22).
Dans une autre étude parue en 2013 dans Diabetes care, ayant pour objectif d’apprécier l’impact du prédiabète sur l’athérosclérose coronarienne, des patients atteints d’une coronaropathie ont subi une évaluation angioscopique des principaux troncs artériels. Parmi les 67 cas étudiés, 16 étaient non diabétiques, 28 présentaient un prédiabète et 23 étaient diabétiques. Les plaques d’athérome identifiées sont généralement considérées comme la lésion primaire dans les syndromes coronariens aigus, et la présence de deux plaques ou plus par vaisseau est considérée comme un facteur de risque de survenue ultérieure d’événements cardiaques. Tous les groupes ont été évalués pour le nombre de plaques et l’intensité du signal. Ces deux paramètres étaient plus élevés chez les patients prédiabétiques que chez les non-diabétiques (P=0,02 et P=0,04 respectivement), mais similaire dans les deux groupes de patients prédiabétiques et diabétiques (P=0,44 et P=0,21 respectivement). Dans l’analyse de régression logistique multivariée, on retrouve que le diabète et le prédiabète étaient des prédicteurs indépendants de plusieurs plaques (23).
Dans une étude plus large dirigée par Scicali et al., l’impact du prédiabète sur le score calcique coronaire (CAC) et l’épaisseur intima-media carotidienne (EIMC), ont été comparés chez les patients prédiabétiques et non diabétiques. Sur 272 patients recrutés, les scores calciques et EIMC étaient significativement plus élevés dans le groupe prédiabète (P<0.001 et P<0,001 respectivement) (24). En somme, le prédiabète pourrait potentiellement avoir un impact similaire au diabète sur les maladies coronariennes et l’athérosclérose périphérique.
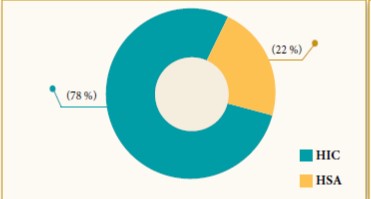
Insuffisance cardiaque diastolique
Bien que des études telles que la Framingham Heart Study aient établi un lien épidémiologique entre le diabète et l’insuffisance cardiaque (IC) (25), il n’y a pas d’association pathophysiologique bien établie entre le prédiabète et l’altération de la fonction cardiaque chez l’homme. Par contre, dans un modèle animal expérimental, Koncsos et al., visant à mieux éclaircir la relation entre le prédiabète et l’IC diastolique en administrant une faible dose de streptozotocine à des rats richement nourris en graisses, ce qui induit un prédiabète caractérisé par une légère élévation de la glycémie à jeun ainsi qu’une augmentation du tissu adipeux viscéral. La conséquence cardiaque de cette perturbation métabolique a été ensuite évaluée. La mesure morphologique et fonctionnelle des paramètres cardiaques évalués par échocardiographie, indique que la masse ventriculaire gauche (VG) ainsi que la partie antérieure du VG et l’épaisseur de la paroi postérieure étaient augmentées chez les rats prédiabétiques (26).
Di Pino et al., ont étudié les effets du prédiabète sur la fonction diastolique chez 167 patients avec une HbA1c entre 5,7% et 6,4%. Chez ces patients, ils ont objectivé que la mesure de la vitesse du flux sanguin à travers la valve mitrale avec une mesure de la période diastolique précoce (onde E), témoin du remplissage diastolique précoce et une mesure de la période diastolique tardive (onde A), témoin du remplissage diastolique tardif (P<0,05), le volume supérieur de l’oreillette gauche (VSOG) (P<0,05) ainsi que l’indice de sphéricité (IS) (P<0,05) ; étaient significativement plus faibles par rapport aux sujets témoins. Le rapport E/A, VSOG et IS sont considérés comme premiers signes de dysfonctionnement diastolique dans le prédiabète (27).
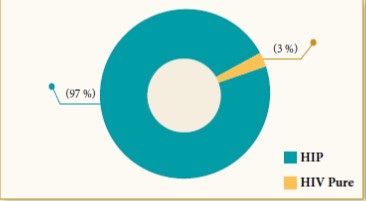
Accident vasculaire cérébral (AVC)
La prévalence du prédiabète chez les patients non diabétiques présentant un AVC ischémique ou accidents ischémiques transitoires (AIT) varie de 23% à 53%( 28). En effet, comparé aux SNG, les sujets prédiabétiques ont un risque accru de maladies cérébrovasculaires (29, 30). L’étude de Tanaka et al., a démontré que le diabète et le prédiabète étaient associés de façon identique au mauvais pronostic, trente jours après la survenue d’un AVC ischémique aigu (31). L’étude de Qiao et al., indique que le taux de glucose après hyperglycémie provoquée par voie orale est un puissant prédicteur d’AVC et de MCV (32).
Par ailleurs, dans l’étude IRIS qui a concerné 3.876 patients non diabétiques présentant une résistance à l’insuline ainsi que des antécédents récents d’AVC ou d’AIT, le traitement par la pioglitazone a réduit de manière significative les risques d’AVC, d’infarctus du myocarde ou de développement de DT2 par rapport au groupe témoin ; par contre, la pioglitazone était également associée à un risque plus élevé de prise de poids, d’œdème et de fracture (33).
Artériopathie oblitérante des membres inférieurs : La prévalence du prédiabète dans l’artériopathie oblitérante des membres inférieurs a été estimé entre 26% et 28%. Cependant, les mécanismes exacts restent à élucider (7).
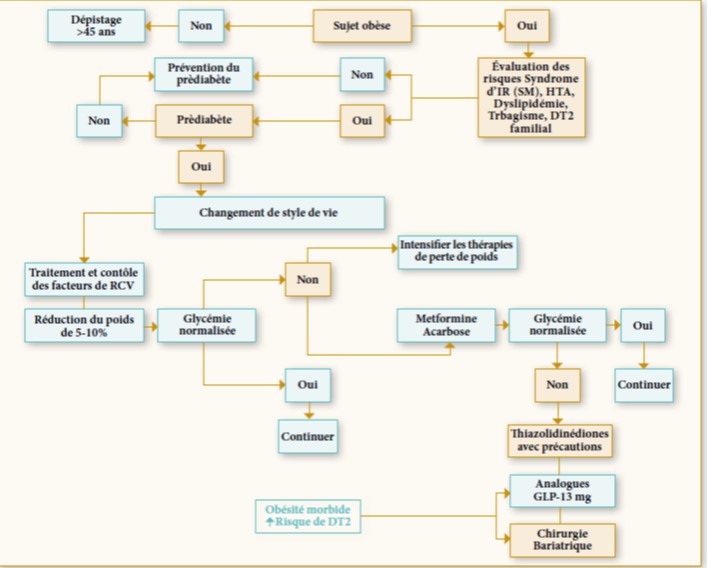
La prévention du diabète aura-t-elle un effet bénéfique sur la réduction du risque de complications cardiovasculaires dans le prédiabète ?
Plusieurs études basées sur les modifications du mode de vie et les traitements pharmacologiques ont prouvé leur efficacité dans la réduction du risque d’évolution vers un DT2 mais également dans la réduction du risque CV dans le prédiabète (tableau 1).
L’étude Da Qing était le premier grand essai contrôlé randomisé qui a apprécié les effets bénéfiques d’intervention sur le mode de vie, sur la prévention du diabète chez les sujets IG. Le suivi à long terme dans cette étude a démontré qu’en plus de la prévention du DT2, il y avait une diminution de la mortalité CV et la mortalité toutes causes après 23 ans de suivi. L’incidence cumulée de la mortalité par MCV était de 11,9% dans le groupe d’intervention versus 19,6% dans le groupe témoin (P=0,033). La mortalité toutes causes était de 28,1% versus 38,4% (P=0,049). L’incidence du diabète était de 72,6% dans le premier groupe contre 89,9% dans le groupe contrôle (P=0,001) (34).
Dans le Diabetes Prevention Program (DPP), l’intervention intensive sur le mode de vie a non seulement réduit de façon considérable le risque de DT2 (réduction de 58% dans le groupe modification du mode de vie et de 31% avec la metformine par rapport au placebo), mais également réduit l’incidence de l’HTA. Rappelons que la prévalence de l’HTA dans la cohorte DPP était au départ d’environ 30%, celle de l’hypertriglycéridémie de 29% et l’hypercholestérolémie de 44% dans les trois groupes de comparaison (placebo, metformine, modification de mode de vie). Après 3 ans de suivi, l’utilisation des traitements pharmacologiques était réduite de 27 à 28% pour l’hypertension et 25% de moins pour l’hyperlipidémie dans le groupe d’intervention intensive sur le mode de vie par rapport aux groupes placebo et metformine. Au contraire, la prévalence de l’hypertension a augmenté d’environ 40% dans les bras placebo et metformine. Ainsi, l’intervention conçue pour prévenir le DT2 semble également avoir évité l’augmentation de l’incidence de l’HTA. Par ailleurs, on a également rapporté une réduction significative (P<0,001) du phénotype pro-athérogène avec une augmentation des taux de cholestérol HDL et une réduction des taux de triglycérides et du LDL pendant environ 3 ans de suivi (35). Outre l’impact des modifications du mode de vie sur les FDR[1] de MCV, il semblait intéressant d’analyser la possibilité de prévenir la survenue des MCV avec la prévention du DT2. Cette question a été examinée par le Diabetes Prevention Program Outcome Study (DPPOS). L’analyse des modèles de régression dans le DPPOS a montré que les personnes dont la glycémie est revenue à la normale présentaient une réduction à long terme de 56% de l’incidence du diabète, associée à une diminution du risque de MCV par rapport à celles qui sont restées dysglycémiques (36).
Dans le même sillage, les résultats de l’essai PREDIMED (PREvención con DIeta MEDiterránea), multicentrique contrôlé randomisé pour la prévention primaire des MCV, ont montré une réduction de 40% de l’incidence du DT2 chez les participants soumis à un régime méditerranéen supplémenté en huile d’olive extra vierge par rapport au groupe contrôle (37). D’autres rapports sur le régime méditerranéen ont montré des résultats concordants sur le profil cardiométabolique. Il a été démontré que le régime de style méditerranéen entraîne une plus grande perte de poids ainsi qu’une amélioration des marqueurs inflammatoires par rapport aux conseils généraux sur le mode de vie (14kg contre 3kg, p<0,001) (38).
Conclusion
Bien que le diabète soit un facteur de risque indéniable de complications cardiovasculaires redoutables, il est désormais établi que ces risques précèdent le diabète et sont déjà évidents au stade de prédiabète. Cette période intermédiaire offre la possibilité d’intervenir pendant la phase réversible de la maladie, occasion à saisir par l’ensemble des praticiens et des patients afin d’éviter la progression vers le diabète de type 2 et ses complications. Identifier et intervenir auprès des populations prédiabétiques à risque, supposées « normales » nécessite une éducation et une sensibilisation accrue grâce à des programmes d’hygiène de vie simples et bien structurés, fondés sur des données probantes et largement diffusées. Ceci pourrait avoir un impact impressionnant sur l’économie de santé.
Tableau 1 : principaux essais contrôlés randomisés d’intervention dans le prédiabète (39)
|
Étude |
Intervention |
Nombre de sujets |
Population étudiée |
– Réduction du risque de DT2 – Réduction du risque cardio-vasculaire (RCV) |
|
Da Qing (34)
|
Régime alimentaire et exercice |
577 |
IG, âge moyen 46 ans, IMC 26 |
– DT2 : 31 à 46% après 6 ans – Réduction de l’incidence de la mortalité par MCV et de la mortalité toutes causes |
|
Finnish DPS (40)
|
Régime alimentaire et exercice |
522 |
IG, âge moyen 55 ans, IMC 31 |
DT2 : 58% après 3,2 ans |
|
STOP-NIDDM (41)
|
Acarbose |
1428 |
IG, âge moyen 55 ans, IMC 31 |
DT2 : 25% après 3,3 ans |
|
DPP (35)
|
Régime alimentaire et exercice |
3234 |
IG, âge moyen 54 ans, IMC 34 |
DT2 : – Metformine 31%, – Mode de vie 58% après 2 ans RCV : Après 3 ans de suivi : diminution de l’incidence des facteurs de risques des MCV (HTA, phénotype lipidique pro athérogène) |
|
Xendos (42)
|
Orlistat + régime et exercice |
3305 |
21% avec IG, âge moyen 43 ans, IMC> 30, |
DT2 : – Groupe entier – 37%, IG 45% après 4 ans |
|
DREAM (43)
|
Rosiglitazone |
5269 |
IG et / ou HMJ âge moyen 54,7 ans, IMC 30,9 |
DT2 : 62% après environ 3 ans |
|
IDDP-1 (44)
|
Modifications du mode de vie et modifications de la metformine ou du mode de vie |
531 |
IG, âge moyen 46 ans, IMC 25,8 |
DT2 : – Régime alimentaire et exercice : 28,5%, – Metformine 26,4%, – Régime alimentaire + exercice+ metformine : 28,2% après 30 mois |
|
ACT-NOW (45)
|
Pioglitazone |
602 |
IG, âge moyen 53 ans, IMC 33 |
DT2 : 72% avec la pioglitazone sur 2,4 ans |
|
CANOE (46)
|
Association rosiglitazone et metformine vs placebo |
207 |
IG, âge moyen 50 ans, IMC 31,3 |
DT2 : 26% dans le groupe combiné après 3,9 ans |
|
IDDP-2 (47)
|
Modifications du mode de vie ou pioglitazone + modifications du mode de vie |
407 |
IG, âge moyen 45,3 ans, IMC 25,9 |
DT2 : 28% dans le groupe modification de mode de vie seulement |
|
Navigator (48)
|
Nataglinide et modifications du mode de vie ou Valsartan et modifications du mode de vie |
9306 |
IG, âge moyen 63,7 ans, IMC 30,5 |
DT2 : – Nataglinide aucun, – Valsartan 14% |
IG : intolérance au glucose, HMJ : hyperglycémie modérée à jeun, DT2 : diabète de type 2, IMC (kg /m²) : indice de masse corporelle MCV : maladie cardiovasculaire
Liens d’intérêts : Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
- Tabák AG, Herder C, Rathmann W, Brunner EJ, Kivimäki M. Prediabetes: a high-risk state for diabetes development. Lancet (London, England). 2012;379(9833):2279-90.
- Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2020. Diabetes care. 2020;43(Suppl 1): S14-s31.
- International Diabetes Federation. IDF Diabetes Atlas 9th ed Disponible sur : wwwdiabetesatlas.org. 2019; consulté le : 19 juin 2020.
- Agarwal A, Hegde A, Yadav C, Ahmad A, Manjrekar PA, Srikantiah RM. Assessment of oxidative stress and inflammation in prediabetes-A hospital based cross-sectional study. Diabetes & metabolic syndrome. 2016;10(2 Suppl 1): S123-6.
- Perreault L, Færch K. Approaching pre-diabetes. Journal of diabetes and its complications. 2014;28(2):226-33.
- Ford ES, Zhao G, Li C. Pre-diabetes and the risk for cardiovascular disease: a systematic review of the evidence. Journal of the American College of Cardiology. 2010;55(13):1310-7.
- Mahat RK, Singh N, Arora M, Rathore V. Health risks and interventions in prediabetes: A review. Diabetes & metabolic syndrome. 2019;13(4):2803-11.
- Glucose tolerance and cardiovascular mortality: comparison of fasting and 2-hour diagnostic criteria. Archives of internal medicine. 2001;161(3):397-405.
- Ziegler D, Rathmann W, Dickhaus T, Meisinger C, Mielck A. Prevalence of polyneuropathy in pre-diabetes and diabetes is associated with abdominal obesity and macroangiopathy: the MONICA/KORA Augsburg Surveys S2 and S3. Diabetes care. 2008;31(3):464-9.
- The prevalence of retinopathy in impaired glucose tolerance and recent-onset diabetes in the Diabetes Prevention Program. Diabetic medicine: a journal of the British Diabetic Association. 2007;24(2):137-44.
- Echouffo-Tcheugui JB, Narayan KM, Weisman D, Golden SH, Jaar BG. Association between prediabetes and risk of chronic kidney disease: a systematic review and meta-analysis. Diabetic medicine: a journal of the British Diabetic Association. 2016;33(12):1615-24.
- Zand A, Ibrahim K, Patham B. Prediabetes: Why Should We Care? Methodist DeBakey cardiovascular journal. 2018;14(4):289-97.
- Færch K, Vistisen D, Johansen NB, Jørgensen ME. Cardiovascular risk stratification and management in pre-diabetes. Current diabetes reports. 2014;14(6):493.
- Isordia-Salas I, Galván-Plata ME, Leaños-Miranda A, Aguilar-Sosa E, Anaya-Gómez F, Majluf-Cruz A, et al. Proinflammatory and prothrombotic state in subjects with different glucose tolerance status before cardiovascular disease. Journal of diabetes research. 2014; 2014:631902.
- Bembde AS. A study of plasma fibrinogen level in type-2 diabetes mellitus and its relation to glycemic control. Indian journal of hematology & blood transfusion: an official journal of Indian Society of Hematology and Blood Transfusion. 2012;28(2):105-8.
- Nyenwe EA, Dagogo-Jack S. Metabolic syndrome, prediabetes and the science of primary prevention. Minerva endocrinologica. 2011;36(2):129-45.
- Selvin E, Lazo M, Chen Y, Shen L, Rubin J, McEvoy JW, et al. Diabetes mellitus, prediabetes, and incidence of subclinical myocardial damage. Circulation. 2014;130(16):1374-82.
- Sánchez E, Betriu À, López-Cano C, Hernández M, Fernández E, Purroy F, et al. Characteristics of atheromatosis in the prediabetes stage: a cross-sectional investigation of the ILERVAS project. Cardiovasc Diabetol. 2019;18(1):154.
- Ali MK, Bullard KM, Saydah S, Imperatore G, Gregg EW. Cardiovascular and renal burdens of prediabetes in the USA: analysis of data from serial cross-sectional surveys, 1988-2014. The lancet Diabetes & endocrinology. 2018;6(5):392-403.
- Sen K, Mukherjee AK, Dharchowdhury L, Chatterjee A. A study to find out the proportion of prediabetes in patients with acute coronary syndrome in a medical college of Kolkata. Journal of the Indian Medical Association. 2008;106(12):776-8.
- Khaw KT, Wareham N, Bingham S, Luben R, Welch A, Day N. Association of hemoglobin A1c with cardiovascular disease and mortality in adults: the European prospective investigation into cancer in Norfolk. Annals of internal medicine. 2004;141(6):413-20.
- Milman S, Crandall JP. Mechanisms of vascular complications in prediabetes. The Medical clinics of North America. 2011;95(2):309-25, vii.
- Kurihara O, Takano M, Yamamoto M, Shirakabe A, Kimata N, Inami T, et al. Impact of prediabetic status on coronary atherosclerosis: a multivessel angioscopic study. Diabetes care. 2013;36(3):729-33.
- Scicali R, Giral P, Gallo A, Di Pino A, Rabuazzo AM, Purrello F, et al. HbA1c increase is associated with higher coronary and peripheral atherosclerotic burden in non-diabetic patients. Atherosclerosis. 2016; 255:102-8.
- Mahmood SS, Wang TJ. The epidemiology of congestive heart failure: the Framingham Heart Study perspective. Global heart. 2013;8(1):77-82.
- Koncsos G, Varga ZV, Baranyai T, Boengler K, Rohrbach S, Li L, et al. Diastolic dysfunction in prediabetic male rats: Role of mitochondrial oxidative stress. American journal of physiology Heart and circulatory physiology. 2016;311(4):H927-h43.
- Di Pino A, Mangiafico S, Urbano F, Scicali R, Scandura S, D’Agate V, et al. HbA1c Identifies Subjects with Prediabetes and Subclinical Left Ventricular Diastolic Dysfunction. The Journal of clinical endocrinology and metabolism. 2017;102(10):3756-64.
- Pan Y, Chen W, Wang Y. Prediabetes and Outcome of Ischemic Stroke or Transient Ischemic Attack: A Systematic Review and Meta-analysis. Journal of stroke and cerebrovascular diseases: the official journal of National Stroke Association. 2019;28(3):683-92.
- Smith NL, Barzilay JI, Shaffer D, Savage PJ, Heckbert SR, Kuller LH, et al. Fasting and 2-hour postchallenge serum glucose measures and risk of incident cardiovascular events in the elderly: The Cardiovascular Health Study. Archives of internal medicine. 2002;162(2):209-16.
- Roquer J, Rodríguez-Campello A, Cuadrado-Godia E, Giralt-Steinhauer E, Jiménez-Conde J, Dégano IR, et al. Ischemic stroke in prediabetic patients. Journal of neurology. 2014;261(10):1866-70.
- Tanaka R, Ueno Y, Miyamoto N, Yamashiro K, Tanaka Y, Shimura H, et al. Impact of diabetes and prediabetes on the short-term prognosis in patients with acute ischemic stroke. Journal of the neurological sciences. 2013;332(1-2):45-50.
- Qiao Q, Pyörälä K, Pyörälä M, Nissinen A, Lindström J, Tilvis R, et al. Two-hour glucose is a better risk predictor for incident coronary heart disease and cardiovascular mortality than fasting glucose. European heart journal. 2002;23(16):1267-75.
- Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, et al. Pioglitazone after Ischemic Stroke or Transient Ischemic Attack. The New England journal of medicine. 2016;374(14):1321-31.
- Li G, Zhang P, Wang J, An Y, Gong Q, Gregg EW, et al. Cardiovascular mortality, all-cause mortality, and diabetes incidence after lifestyle intervention for people with impaired glucose tolerance in the Da Qing Diabetes Prevention Study: a 23-year follow-up study. The Lancet Diabetes & Endocrinology. 2014;2(6):474-80.
- care ADAJD. Impact of intensive lifestyle and metformin therapy on cardiovascular disease risk factors in the diabetes prevention program. Diabetes care. 2005;28(4):888-94.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet (London, England). 2002;360(9349):1903-13.
- Salas-Salvadó J, Guasch-Ferré M, Lee CH, Estruch R, Clish CB, Ros E. Protective Effects of the Mediterranean Diet on Type 2 Diabetes and Metabolic Syndrome. The Journal of nutrition. 2015;146(4):920s-7s.
- Esposito K, Maiorino MI, Bellastella G, Chiodini P, Panagiotakos D, Giugliano D. A journey into a Mediterranean diet and type 2 diabetes: a systematic review with meta-analyses. BMJ open. 2015;5(8):e008222.
- Brannick B, Dagogo-Jack S. Prediabetes and Cardiovascular Disease: Pathophysiology and Interventions for Prevention and Risk Reduction. Endocrinology and metabolism clinics of North America. 2018;47(1):33-50.
- Tuomilehto J LJ, Eriksson JG, et al… Finnish Diabetes Prevention Study Group. N Engl J Med. 2001; ; 344:1343–1350. [PubMed: 11333990.
- Chiasson JL JR, Gomis R, Hanefeld M, Karasik A, Laakso M.. STOP-NIDDM Trial Research Group: Acarbose treatment the risk of cardiovascular disease hypertension in patients with impaired glucose tolerance: the STOP-NIDDM trial. Jama. 2003;; 290:486–494. [PubMed: 12876091].
- Torgerson JS HJ, Boldrin MN, Sjöström L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes care. 2004;27:155–161. [PubMed: 14693982].
- Investigators DDRAwrarMT. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. Lancet (London, England). 2006;368:1096–1105. [PubMed: 16997664].
- Ramachandran A SC, Mary S, Mukesh B, Bhaskar AD, Vijay V, Indian Diabetes Prevention Programme (IDPP). The Indian Diabetes Prevention Programme shows that lifestyle modification metformin prevent type 2 diabetes in Asian Indian subjects with impaired glucose tolerance (IDPP-1). Diabetologia. 2006; 49:289–297. [PubMed: 16391903].
- Tripathy D SD, Banerji M, et al. Diabetes Incidence and Glucose Tolerance after Termination of Pioglitazone Therapy: Results from ACT NOW. The Journal of clinical endocrinology and metabolism. 2016;101:2056–2062. [PubMed: 26982008].
- Zinman B HS, Neuman J, et al. Low-dose combination therapy with rosiglitazone and metformin to prevent type 2 diabetes mellitus (CANOE trial): a double-blind randomised controlled study. Lancet (London, England). 2010;376:103–111. [PubMed: 20605202].
- Ramachandran A SC, Mary S, et al. Pioglitazone does not enhance the effectiveness of lifestyle modification in preventing conversion of impaired glucose tolerance to diabetes in Asian Indians: results of the Indian Diabetes Prevention Programme-2 (IDPP-2). Diabetologia. 2009; 52:1019–1026. [PubMed: 19277602].
- Group NS. Effect of nateglinide on the incidence of diabetes and cardiovascular events. N Engl J Med. 2010;362:1463–1476. [PubMed: 20228402].
[1] FDR = Facteurs de risque