S. BOUZID1, M. DERGUINI2 ; EPH Hassen BADI, El Harrach, Alger, EPH Bachir MENTOURI, Kouba, Alger
Résumé
Les contraceptions dites réversibles de longue durée d’action ont une efficacité prolongée dans le temps de 3 à 5 ans selon la méthode. Elles sont devenues ces dernières années un sujet d’intérêt considérable en matière de contraception. En effet, ces méthodes, incluant les dispositifs intra-utérins, les systèmes intra-utérins et l’implant sous-cutané, voir injectables, permettent d’obtenir une efficacité contraceptive optimale tout en réduisant de façon majeure les contraintes pour les femmes. Toutes ces contraceptions imposent cependant la participation d’un personnel qualifié pour leur mise en place et leur retrait. Elles peuvent habituellement convenir à toutes les femmes après avoir éliminé quelques rares complications. Pour chaque méthode, les avantages et les inconvénients ptentiels doivent être pris en compte afin de choisir avec chaque femme la méthode la plus adaptée. Une surveillance régulière s’impose toujours, permettant un retrait prématuré en cas d’effets indésirables, d’incident ou de souhait de grossesse. Leur réversibilité est plus ou moins immédiate.
Mots-clés : Contraception, contraception réversible à action prolongée postnatale immédiate (LARC), post-partum, planification familiale, stérilet, implant.
Abstract
Contraceptives known as long-acting reversible contraceptives have a prolonged effectiveness over time from 3 to 5 years depending on the method. They have become a topic of considerable interest in contraception in recent years. In fact, these methods, including intrauterine devices, intrauterine systems, and the subcutaneous implant, or even injectables, allow optimal contraceptive efficacy to be obtained while significantly reducing the constraints for women. All these contraceptives, however, require the participation of qualified personnel for their placement and removal. They can usually be suitable for all women after elimination of the rare complications. For each method, the potential advantages and disadvantages must be considered in order to choose the most suitable method with each woman. Regular monitoring is always necessary, allowing premature withdrawal in the event of undesirable effects, incident, or desire for pregnancy. Their reversibility is more or less immediate.
Keywords : Contraception, immediate postnatal long-acting reversible contraception (LARC), postpartum, family planning, IUD, Implant..
Introduction
La planification de la reproduction est essentielle pour toutes les femmes et surtout pour celles qui ont des problèmes de santé complexes ou qui présentent un risque élevé de complications. La planification de la grossesse peut donner, à ces femmes à haut risque, la possibilité de recevoir des conseils avant la conception, l’adaptation des médicaments et une évaluation des risques liés à des problèmes de santé ayant un impact direct sur le risque de morbidité et de mortalité maternelles. Malgré la nécessité de planifier la grossesse, les femmes médicalement complexes se heurtent à des obstacles en matière d’utilisation de la contraception, notamment systémiques, ainsi qu’une faible utilisation efficace de la contraception notamment du post-partum. Fournir des conseils en matière de contraception et d’options contraceptives, y compris une contraception réversible à action prolongée postnatale immédiate (LARC), est un moyen de surmonter ces obstacles (1).
Définition
A- Qu’est-ce que la contraception réversible à longue durée d’action ?
Les contraceptions réversibles à longue durée d’action sont des contraceptions qui ne dépendent pasdusouvenir de laprise jour- nalière, ou de l’utilisation, lors des relations sexuelles, comme on le ferait avec des méthodes telles que la pilule ou les préservatifs. Elles sont très efficaces pour prévenir la grossesse non désirée.
Elles incluent :
- Les injections contraceptives, qui fonctionnent pendant 12 semaines et peuvent être répétées.
- Les dispositifs installés à l’intérieur de l’utérus, dispositifs intra-utérins (DIU) et systèmes intra-utérins (SIU), qui durent entre 5 et 10 ans et peuvent être remplacés ultérieurement.
- Les implants placés sous la peau, qui durent 3 ans et peuvent ensuite être remplacés.
Les injections, les DIU, le SIU et les implants sont appelés contraceptifs réversibles à longue durée d’action car on peut arrêter de les utiliser si nous décidons d’un projet de grossesse. Ces méthodes sont toutes destinées aux femmes. Actuellement, il n’existe pas de contraceptif réversible à longue durée d’action pour les hommes.
B- Rentabilité
Les méthodes LARC sont plus rentables que les contraceptifs oraux combinés (COC) même à 1 an d’utilisation.
Les DIU, le SIU et l’implant sont plus rentables que les contraceptifs injectables.
L’augmentation de l’utilisation des LARC réduira le nombre de grossesses non désirées.
C- Choix de la méthode LARC
Toutes les méthodes LARC conviennent aux femmes :
- Nullipares,
- Allaitantes,
- Après fausse couche,
- Migraineuses (avec ou sans aura),
- Avec une contre-indication à l’œstrogène,
- Avec le VIH,
- Avec un IMC supérieur à 30,
- Souffrant de diabète.
D- Initiation de méthode
Excluez la grossesse en fonction des antécédents menstruels et sexuels.
- DIU/SIU : À n’importe quel moment du cycle menstruel (SIU **). Immédiatement après l’avortement du premier ou du deuxième trimestre, ou à tout temps après, à partir de 4 semaines post-partum.
- Injection de progestatif seul : Jusqu’au cinquième jour du cycle menstruel inclusivement sans le besoin d’administrer un contraceptif supplémentaire. À tout autre moment du cycle, utiliser une barrière contraceptive pendant 7 jours après l’injection. Immédiatement après l’avortement du premier ou du deuxième trimestre, ou à tout temps après et à tout moment du post-partum.
- Implant : si la femme présente une aménorrhée ou si elle a plus de 5 jours depuis les règles, commencer à utiliser la contraception barrière pendant les 7 premiers jours (1).
E- Indications pour le post-partum
(Recommandations de la Société de Médecine Materno-Foetale)
- Il est recommandé que LARC soit proposée aux femmes les plus exposées au risque d’effets indésirables sur leur santé à la suite d’une future grossesse ;
- Il est recommandé aux femmes qui envisagent de placer leur dispositif intra-utérin immédiatement après l’accouchement d’être informées sur le fait que, bien que les taux d’expulsion soient supérieurs à ceux d’une insertion retardée, les avantages semblent compenser le risque d’expulsion, car les taux de maintien à long terme sont plus élevés ;
- Il est recommandé de le placer en postnatal immédiat après une grossesse à haut risque (1) ;
- Chez l’adolescente.
Les méthodes contraceptives réversibles à action prolongée (DIU et des implants) devraient être proposées à toutes les adolescentes en tant qu’options contraceptives de première intention (2).
Le Dispositif Intra Utérin (DIU)
Il s’agit d’un dispositif inséré dans la cavité utérine par un médecin ou une sage-femme. Il assure une contraception sur plusieurs années (5 à 10 ans en fonction des modèles).
Le DIU apparaît comme étant un des moyens de contraception réversibles les plus fiables avec un taux d’échec de 1.1 % dans la première année d’utilisation (3).
Il existe 2 types de DIU :
- Au cuivre
- Hormonal = SIU DIU au cuivre
Le DIU au cuivre peut avoir plusieurs formes (la majorité en forme de T) et contient généralement un fil de cuivre de 380 mm2 enroulé autour de la tige principale. Il existe en plusieurs formes et en plusieurs tailles (modèles « short »). Son efficacité contraceptive varie de 5 à 10 ans en fonction des modèles.

Le DIU hormonal, appelé également SIU (Système intra-uté- rin) avec un corps contenant un réservoir de lévonorgestrel (LNG), qui se libère chaque jour, la durée varie entre 3 et 5 ans selon les modèles.

A- Mode d’action des DIU
Le DIU ralentit la progression des spermatozoïdes qui pénètrent plus difficilement dans la glaire cervicale et diminue leurs capacités à féconder (4). Il rend l’endomètre impropre à la nidation par une réaction inflammatoire aseptique, qui réduit la capacité d’implantation d’un éventuel œuf fécondé (5). Spécifiquement pour le SIU, il réduit le développement de l’endomètre et inhibe parfois l’ovulation (6).
B- Contre-indications
- Grossesse ;
- Saignements génitaux anormaux inexpliqués, déformation ou malformation de la cavité utérine, dont les déformations par des fibromes ;
- Infection génitale en cours (cervicite, vaginite, endométrite, salpingite), tuberculose génito-urinaire
- Âge < 25 ans, partenaires multiples ;
- Dysplasie cervicale, cancer de l’endomètre ;
- Maladie trophoblastique ;
- Allergie au cuivre ;
- Post-partum ou post-abortum septique < 3 mois ;
- Post-partum physiologique < 4 semaines après accouche- ment (7) ;
- Thrombophlébite évolutive ou embolie pulmonaire évolutive ;
- Pathologie hépatique.
Le DIU convient aux femmes :
- Nullipares (8)
- Qui allaitent (9) ;
- Infectées par le VIH (10) ;
- Ayant un antécédent de GEU. Il y a lieu de noter que les grossesses extra-utérines (GEU) sont moins importantes que sans contraception (11) ;
- Diabétiques (12) ;
- De plus, contrairement aux idées reçues, il n’est pas nécessaire d’instaurer un traitement antibiotique lors de (13) et la prise d’AINS n’est pas contre-indiquée ;
- Les DIU sont utilisables chez la femme en post-partum que la femme allaite ou non : à partir de 4 semaines après l’accouchement* ;
- Après avoir évalué et écarté un risque infectieux (Chlamydia trachomatis et Neisseria gonorrhoeae**) avant la pose (14).
C- Complications
- Liées à l’insertion;
- Expulsion;
- Difficultés d’insertion avec douleur;
- Perforation utérine (15).
D- Effets secondaires et inconvénients
- Règles plus abondantes avec le DIU au cuivre;
- Aménorrhée avec le SIU hormonal;
- Spottings;
- Saignements en dehors de la période de règles durant la « période d’adaptation » qui dure de 3 à 6 mois;
- Kystes fonctionnels ovariens;
- Maladie inflammatoire pelvienne (MIP);
- Déplacement hors de l’utérus (dans la cavité péritonéale);
- Grossesse.
La contraception injectable
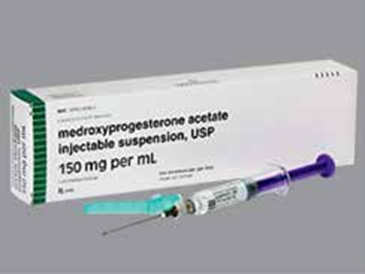
en empêchant l’œuf d’une femme de se développer complète- ment. Il entrave le cheminement du sperme vers l’ovocyte en épaississant les mucosités qui entourent le col utérin.
Pour la contraception, il existe deux présentations de ce mé- dicament. La dose intramusculaire (IM) recommandée est de 150 mg injectés à l’intérieur d’un muscle par un profession- nel de la santé tous les 3 mois. Il ne doit pas s’écouler plus de 13 semaines entre les injections. La dose sous-cutanée (SC) recommandée d’acétate de médroxyprogestérone est de 104 mg injectés sous la peau, toutes les 12 à 14 semaines. La prise de ce médicament commence habituellement au cours des 5 premiers jours qui suivent le début des menstruations normales.
A- Contre-indications
- Une allergie à la médroxyprogestérone ou à l’un des ingrédients du médicament ;
- Des antécédents d’Accident Vasculaire Cérébral ;
- Des antécédents de maladie cardiaque ;
- Des antécédents de désordres de la coagulation;
- Des antécédents de migraines précédées d’aura;
- Un cancer présent ou soupçonné sensible aux progestatifs;
- Une grossesse confirmée ou soupçonnée ;
- L’hypercholestérolémie importante, un tabagisme important chez une personne de plus de 35 ans, la présence de problèmes vasculaires secondaires au diabète ;
- Des troubles de la vue consécutifs à une affection vasculaire ;
- Pathologie du sein (par ex. un cancer) ;
- Un saignement non diagnostiqué du vagin ou des voies urinaires;
- Un trouble du foie.
B- Effets secondaires
Les troubles menstruels constituent l’effet secondaire le plus fréquent.
Ils disparaissent habituellement après quelques mois d’uti- lisation pour faire place à l’aménorrhée ou l’absence de règles. Les cycles menstruels se rétablissent dans les 6 mois suivant la dernière injection dans la majorité des cas; toute- fois, l’absence de règles peut durer jusqu’à 12 à 18 mois.
La prise de poids constitue un autre effet secondaire fré- quent, mais une diminution de poids est notée chez 20 % des femmes. Le médicament peut provoquer d’autres ef- fets secondaires comparables à ceux rencontrés avec les contraceptifs oraux :
- Maux de tête, douleur ou malaise abdominal;
- Fatigue, nausée;
- Diminution du désir sexuel, sensibilité des seins;
- Changement d’humeur, dépression.
Mais cette méthode a tendance à être de moins en moins utilisée, ceci est lié aux effets secondaires et est réservée aux femmes ne pouvant prendre en charge leur contraception, principalement en psychiatrie.
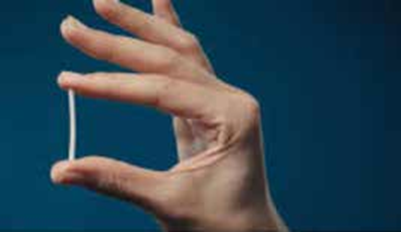
L’implant est une méthode contraceptive hormonale. Il s’agit d’un bâtonnet cylindrique de 4 cm de longueur et 2 mm de diamètre (de la taille d’une allumette). Il est inséré sous la peau du bras en quelques minutes, sous anesthésie locale. Une fois en place, l’implant ne se voit pas, ne sent pas, mais il est palpable à travers la peau.
L’implant est efficace pendant 3 ans et peut être retiré dès que la femme le désire. Inséré à la face interne du bras, l’implant libère de manière continue un progestatif : l’étonogestrel. Il agit en inhibant l’ovulation, en supprimant le pic de LH, avec épaississement de la glaire cervicale qui limite le passage des spermatozoïdes et amincissement de la muqueuse utérine, qui empêche la fixation dans l’utérus d’un éventuel œuf. Il commence à agir dès les 24 premières heures qui suivent sa pose. L’implant contraceptif est efficace à 99,9 % (efficacité théorique). Certains médicaments peuvent rendre l’implant moins efficace comme les médicaments utilisés pour traiter l’épilepsie, la tuberculose, certaines maladies infectieuses et aussi les médicaments à base d’une plante appelée millepertuis, utilisée pour traiter les états dépressifs. Le retour à la fertilité antérieure intervient dans la très grande majorité des cas dans les trois semaines suivant le retrait de l’implant.
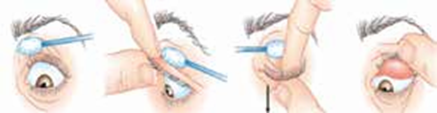
A- Pose de l’implant
Le médecin, le gynécologue ou la sage-femme insère l’implant sous la peau du bras, sous anesthésie locale. Pour renouveler ce moyen de contraception, il est possible d’insérer un nouvel implant à la place de celui qui vient d’être retiré, en une seule fois.

B- Retrait de l’implant
Le retrait est réalisé également sous anesthésie locale par un médecin, un gynécologue ou une sage-femme. Le professionnel de santé réalise une incision sous cutanée de 2 mm puis, avec une pince, il retire l’implant.
C- Avantages
Simplicité et longue durée d’action : après la pose, vous êtes protégée durant 3 ans. Aucun contrôle n’est nécessaire.
D- Inconvénients
– Modifications des cycles menstruels : certaines femmes n’au- ront pas de règles pendant 3 ans. D’autres, auront des règles moins régulières ou moins fréquentes que d’habitude et par- fois beaucoup plus courtes, parfois longues. Le profil de sai- gnement est difficile à prédire, mais il tend le plus souvent à une diminution ou une disparition des règles.
– Prise de poids chez certaines femmes (en cas de surpoids important, plus de 80 kilos, il est conseillé de changer l’implant plus tôt (au bout de 24 à 30 mois) et non trois ans comme chez une femme avec un poids normal.
Acné chez certaines femmes.
Références
- Société de médecine maternelle et fœtale (SMFM). Adresse électronique : [email protected] 1, Vricella LK 1, Gawron LM 1, Louis JM 1.
- Long-acting reversible contraception in adolescents: a systematic review and meta-analysis, Justin T. Diedrich, MD, MSCI; MAJ David A. Klein, MD, MPH; Jeffrey F. Peipert, MD, PhD.
- C. Moreau, J. Trussell, G. Rodriguez et al Contraceptive failure rates in France: results from a population-based survey. J Human Repro- duction Vol 22, n°9 p 2422-2427, 2007.
- Agence nationale d’accréditation et d’évaluation en santé et coll « stratégies de choix des méthodes contraceptives chez la femme » argu- mentaire, déc 2004 : 198p.
- Stanford JB et Mikolajczyk RT « Mechanisms of action of intraute- rine devices: update and estimation of postfertilization effects » Am J obstet Gynecol 2002; 187 (6): 1699-1708.
- Black A. « Dispositifs intra-utérins » Black A et coll Consensus cana- dien sur la contraception. J Obstet. Gynaecol Can 2004 ; 26 (3) : 289-296.
- Stratégies de choix des méthodes contraceptives chez la femme. Re- commandations de l’ANAES. Décembre 2004. 47p.
- Lyus R, Lohr P, Prager S Use of the Mirena LNG-IUS and Paragard CuT380A intrauterine devices in nulliparous women. Contraception. 2010 May; 81 (5): 367-71. Epub 2010 Feb.
- Truitt ST, Fraser AB, Grimes DA Combined hormonal versus non- hormonal versus progestin-only contraception in lactation. Cochrane Database Syst Rev. 2003;(2) : CD003988. Review.
- Farley TM, Rosenberg MJ, Rowe PJ, et al. Intrauterine devices and pelvic in- flammatory disease: An international perspective. Lancet 1992; 339: 785-88.
- Sivin I. Dose- and age-dependent ectopic pregnancy risks with in- trauterine contraception. Obstet Gynecol 1991; 78: 291-298.
- Kurz KH, Berger M. Intrauterine contraceptive devices for diabe- tics. Lancet. 1982 Sep 25; 2 (8300): 707.
- Walsh T, Grimes D, Frezieres R Randomised controlled trial of prophylactic antibiotics before insertion of intrauterine devices. IUD Study Group. Lancet. 1998 Apr 4; 351 (9108): 1005-8.
- Haute Autorité de Santé Recommandations pour la pratique cli- nique : Stratégies de choix des méthodes contraceptives chez la femme, HAS, Décembre 2004 http://www.has-sante.fr/portail/upload/docs/ application/pdf/recommandations_contraception_vvd-2006.pdf.
- Harrison-Woolrych M, Ashton J, Coulter D Uterine perforation on intrauterine device insertion: is the incidence higher than pre- viously reported? Contraception. 2003 Jan; 67 (1): 53-6.