A. CHIALI, A. KHELIL, N.H. MAHMOUDI, Service de Dermatologie, CHU Benaouda Benzerdjeb, Oran.*
Résumé : La dermatite atopique (DA) est une dermatose chronique, inflammatoire et prurigineuse survenant sous forme de poussées d’eczéma aigu sur un fond de xérose cutanée. Elle est fréquente chez l’enfant et l’adulte jeune. La DA touche des sujets génétiquement prédisposés et elle est favorisée par des facteurs environnementaux. Elle est déterminée par l’association d’une altération fonctionnelle innée de la barrière cutanée, d’un développement d’une réaction inflammatoire cutanée impliquant l’immunité innée et adaptative et de l’action d’une anomalie de diversité des microbiomes digestif et cutané. Le diagnostic de la DA est clinique, facilité par le recours à certains critères. Les aspects cliniques varient selon l’âge. Chez le nourrisson, ils sont représentés par des poussées de lésions inflammatoires aiguës et prurigineuses débutant sur le visage. Chez l’enfant de plus de 2 ans, l’adolescent et l’adulte, les lésions cutanées sont lichénifiées et chroniques touchant les plis. La prise en charge d’un patient atteint de DA doit comporter une éducation thérapeutique et l’éviction de facteurs aggravants potentiels (allergie alimentaire, surinfection, facteurs psychologiques et eczéma de contact). L’objectif du traitement est d’améliorer la qualité de vie du patient. Les dermocorticoïdes sont utilisés en première intention en traitement d’attaque de la DA. Le tacrolimus peut être utilisé en seconde intention. La photothérapie peut être indiquée surtout chez l’adulte. Les indications des traitements systémiques (immunosuppresseurs et thérapies ciblées) sont très rares. Le seul traitement préventif est basé sur l’administration d’hydrolysats de protéines de lait de vache chez les nouveau-nés à risque d’atopie et des probiotiques et/ou des prébiotiques.
Mots-clés : Dermatite atopique, xérose, barrière cutanée, éducation thérapeutique, dermocorticoïdes
Abstract : Atopic dermatitis (AD) is a chronic, inflammatory and pruriginous dermatosis occurring in the form of acute eczema flares on a cutaneous xerosis background. It is common in children and young adults. AD affects genetically predisposed subjects and is favored by environmental factors. It is determined by the association of an innate functional impairment of the cutaneous barrier, the development of a cutaneous inflammatory reaction involving innate and adaptive immunity and the action of an anomaly of the digestive and cutaneous microbiomes diversity. The diagnosis of AD is clinical, facilitated by the use of certain criteria. Clinical aspects vary according to age. In infants, they are represented by outbreaks of acute and itchy inflammatory lesions starting on the face. In children over 2 years old, adolescents and adults, cutaneous lesions are lichenified and chronic, affecting folds. The management of a patient with AD must include therapeutic education and potential aggravating factors eviction (food allergy, superinfection, psychological factors and contact eczema). The objective of the treatment is to improve patient’s quality of life. Topical corticosteroids are used as first-line treatment for AD. Tacrolimus can be used as a second line. Photo- therapy may be indicated especially in adults. Indications of systemic treatments (immuno-suppressive agents and targeted therapies) are very rare. The only preventive treatment is based on the administration of cow’s milk protein hydrolysates to newborns at risk of atopy, and probiotics and/or prebiotics.
Key-words : Atopic dermatitis, cutaneous xerosis, cutaneous barrier, therapeutic education, topical corticosteroids.
Introduction :
La dermatite atopique (DA) (ou eczéma constitutionnel) est une dermatose inflammatoire, prurigineuse et chronique, se développant préférentiellement chez le nourrisson et le jeune enfant mais parfois présente chez l’adolescent et l’adulte. C’est une pathologie polymorphe dans son expression clinique variant en fonction de l’âge du sujet atteint. Elle constitue une des manifestations d’hyper-sensibilité et s’intègre dans le cadre de la maladie atopique au même titre que l’asthme ou la rhinite [1]. La DA débute dans les premiers mois de vie généralement vers 3 mois, mais parfois dès le premier mois [2,3]. La prise en charge de la DA de l’enfant est multidisciplinaire impliquant les pédiatres, les allergologues et les dermatologues en fonction de la gravité de la maladie et de l’âge des enfants.
Épidémiologie :
En Algérie, la prévalence de la DA est de 5 % [4]. Par ailleurs, cette affection se rencontre chez 10 à 20 % des enfants dans les pays industrialisés à niveau socio- économique élevé où il existe un doublement de sa prévalence en une vingtaine d’années [5]. Une étude chinoise a mis en évidence un gradient de prévalence de la DA urbain/ rural (10,2 % vs 4,6 %) [6]. Cette diminution du risque d’atopie en milieu rural peut s’expliquer par le fait qu’une plus grande biodiversité environnementale en milieu rural est très significativement associée à une plus grande diversité des bactéries commensales digestives et cutanées [7,8].
Physiopathologie :
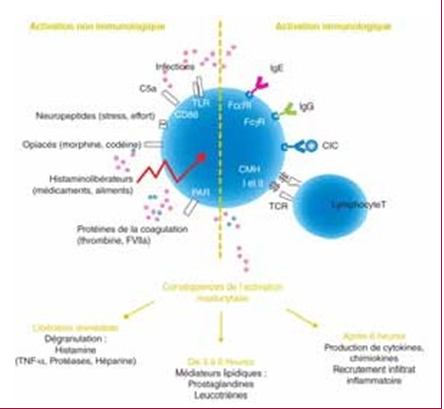
La DA est une maladie multifactorielle due à l’association de facteurs génétiques et environnementaux [9]. Sa physiopathologie fait intervenir plusieurs mécanismes dont une altération fonctionnelle innée de la barrière cutanée liée en partie à une mutation du gène de la filag plus élevée chez le sujet atteint de DA), tend à polariser l’activation lymphocytaire T vers un profil de type Th2 (associé à la production d’interleukines 4, 5, 9, 13, 31) et Th22 (avec production d’interleukine 22) [13]. À la phase chronique, une activation lymphocytaire T de type Th1 (associée à la production d’interféron gamma, de GM- CSF [Granulocyte macrophage colony-stimulating factor] et d’interleukine 12) s’associe à la réponse Th2. La sti- mulation des neurones par une cytokine proinflammatoire (thymic stromal lymphopoietin : TSLP), secrétée par les kératinocytes est responsable de la sensation de prurit [14].
Réaction inflammatoire induite
L’état défectueux de la fonction de la barrière cutanée permet la pénétration dans la peau d’allergènes ainsi que des irritants chimiques externes. Ce phénomène va stimuler l’inflammation [12]. L’immunité adaptative contribue à initier le processus inflammatoire. À la phase aiguë, l’activation des cellules de Langerhans (cellules dendritiques épidermiques dont la concentration est plus élevée chez le sujet atteint de DA), tend à polariser l’activation lymphocytaire T vers un profil de type Th2 (associé à la production d’interleukines 4, 5, 9, 13, 31) et Th22 (avec production d’interleukine 22) [13]. À la phase chronique, une activation lymphocytaire T de type Th1 (associée à la production d’interféron gamma, de GM- CSF [Granulocyte macrophage colony-stimulating factor] et d’interleukine 12) s’associe à la réponse Th2. La stimulation des neurones par une cytokine proinflammatoire (thymic stromal lymphopoietin : TSLP), secrétée par les kératinocytes est responsable de la sensation de prurit [14].
Rôle du microbiome digestif
Le microbiome digestif correspond à l’ensemble des bactéries commensales colonisant le tube digestif et constituant un écosystème complexe qui intervient dans la maturation du système immunitaire. Des anomalies de diversification précoce du microbiote intestinal ont été observées chez les enfants et les nouveau-nés à risque d’atopie [15].
Rôle du microbiome cutané
En dehors des poussées, on observe dans la DA une grande diversité de souches bactériennes commensales à la surface de la peau (microbiome cutané). Cette diversité décroît au cours des poussées de la maladie au profit des souches de staphylocoques [16]. La colonisation de la peau par le staphylocoque doré est très fréquente au cours de la DA (90 % vs 5 % des sujets sains). Un déficit de l’immunité innée cutanée semble être en partie en cause [17].
Clinique :
Le diagnostic de DA est aisé et s’effectue sur des données cliniques. Les critères diagnostiques le plus souvent utilisés sont ceux de l’UK Working Party [18] (Tableau 1).
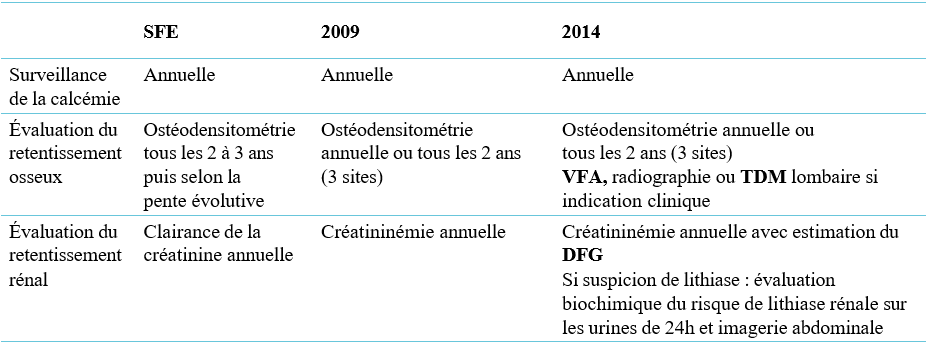
| Critère majeur : dermatose prurigineuse chronique |
| Associée à moins 3 des critères suivants : Eczéma visible des plis de flexion (ou des joues et/ou des faces d’extension des membres avant l’âge de 18 mois) Antécédent personnel d’eczéma des plis de flexion (ou des joues et/ou des faces d’extension des membres avant l’âge de 18 mois) Antécédent personnel de peau sèche au cours de la dernière année Antécédent personnel d’asthme ou de rhinite allergique (ou antécédent familial direct d’atopie chez l’enfant de moins de 4 ans) Apparition des lésions avant 2 ans (critère utilisé chez les enfants de plus de 4 ans) |
Aspects cliniques
Les aspects cliniques varient avec l’âge :
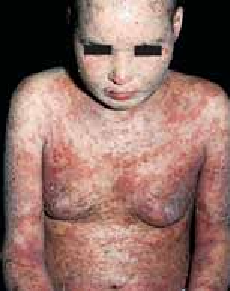
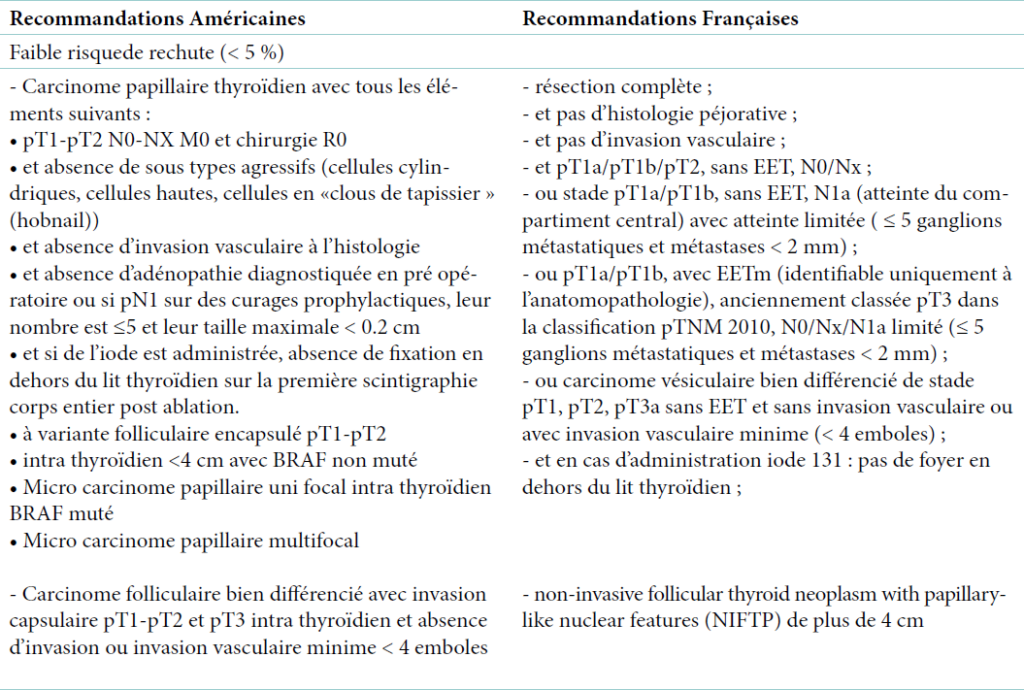
- Chez le nourrisson, les lésions débutent sur les joues, le front et le cuir chevelu. Secondairement, elles s’étendent sur les faces d’extension des membres et le tronc. Le siège et la région médiofaciale (en particulier la pointe du nez) sont épargnés (figure 1). Ce sont des lésions aiguës, érythémateuses, mal limitées, suintantes, croûteuses et prurigineuses. Les excoriations cutanées dues au grattage sont fréquentes. L’évolution est marquée par des poussées entrecoupées de rémissions.
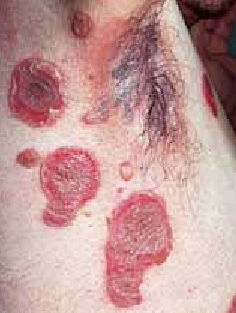
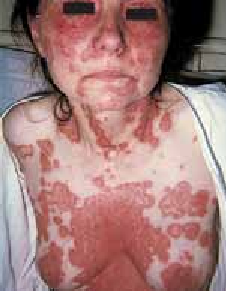
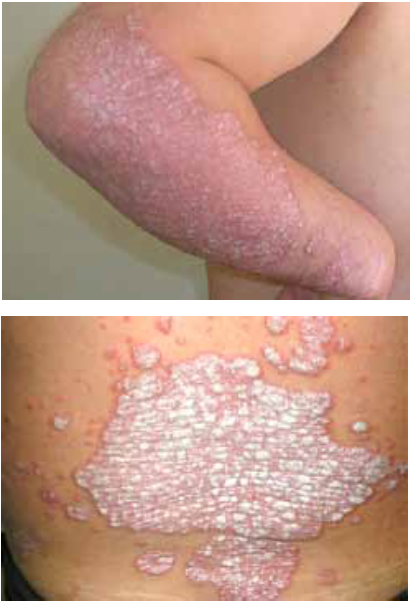
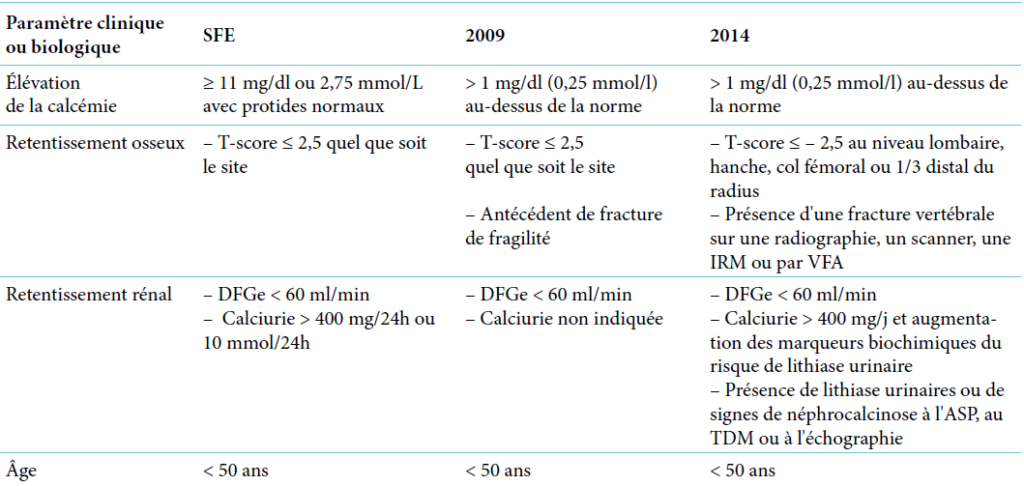
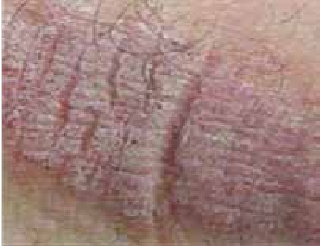
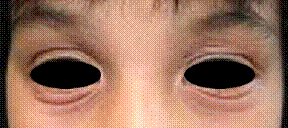
Chez l’enfant de plus de 2 ans, les lésions cutanées siègent au niveau des plis (cou, coudes, genoux, fissures sous et rétroauriculaires) chroniques. Elles se manifestent par l’aspect d’une lichénification (épaississement de la peau) (figure 2). D’autres signes s’ajoutent parfois tels qu’un prurigo, un double pli palpébral inférieur (signe de Dennie-Morgan) (figure 3) et une xérose cutanée (sécheresse).
- Chez l’adolescent et l’adulte, le tableau clinique est similaire à celui de l’enfant de plus de 2 ans avec en plus des lésions qui se localisent au visage et au cou et sont franchement lichénifiées sur les membres.
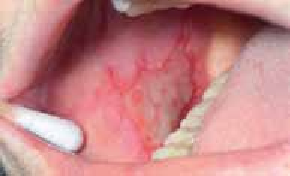
Certaines localisations sont rares mais caractéristiques : l’atteinte des mamelons, l’atteinte des lèvres (chéilite atopique) et des paupières [9].
Des formes cliniques existent [9] :
- L’eczéma nummulaire est caractérisé par des lésions rondes, inflammatoires, résistantes aux traitements,
- L’atteinte des mains peut être observée au cours de la DA. Elle se caractérise par les lésions périunguéales avec parfois une dystrophie unguéale associée
- L’atteinte pulpaire entretenue par un facteur aggravant de contact ;
- une érythrodermie [5].
D’autres manifestations d’atopie sont souvent associées (asthme, rhinite allergique, allergie alimentaire).
Examens complémentaires
Le diagnostic de la DA est clinique et anamnestique. Une hyperéosinophilie sanguine et une augmentation des IgE sériques sont fréquentes [7].
Histopathologie
L’aspect histologique comporte une atteinte épidermique prédominante avec un afflux de lymphocytes T (exocytose) qui s’accompagne d’un œdème intercellulaire (spongiose) réalisant des vésicules. En cas de lichénification, l’épiderme s’épaissit. Le derme superficiel comporte un infiltrat mononucléé périvasculaire.
La dilatation des capillaires superficiels est responsable de l’érythème et l’extravasation de protéines plasmatiques de l’œdème cutané (papules œdémateuses) [3].
Immunohistochimie
De façon caractéristique, des éosinophiles dégranulés sont objectivables (présence de major basic protein) [3].
Explorations allergologiques
Explorations allergologiques (prick-tests cutanés, dosage des IgE sériques spécifiques, test de provocation) justifiées devant l’association d’une allergie respiratoire (asthme, rhinite ou rhinoconjonctivite), ou d’une allergie alimentaire suspectée devant sa survenue après ingestion ou contact avec un aliment, devant une DA grave ou devant une stagnation ou une cassure de la courbe pondérale [5]. Tests épicutanés ou patch tests en cas d’association avec un eczéma de contact évoqué devant l’apparition d’un eczéma de zones inhabituelles et devant une DA qui ne répond pas au traitement ou s’aggrave [5].
Évolution :
La DA comporte des formes étendues, mais son évolution est bonne avec une rémission complète dans la majorité des cas. Les formes qui persistent chez l’enfant sont plus localisées. Une recrudescence peut se produire à l’occasion de conflits psychoaffectifs.
La survenue de manifestations respiratoires (asthme ou rhinite) est d’autant plus fréquente qu’il y a des antécédents familiaux atopiques au premier degré [3].
Complications infectieuses :
- La surinfection par le staphylocoque doré (SA) : Le SA colonise constamment la peau des atopiques, même en dehors des zones cliniquement atteintes. Cette surinfection se manifeste cliniquement par la présence d’un écoulement purulent, de lésions vésiculo-bulleuses et de croûtes jaunes [9].
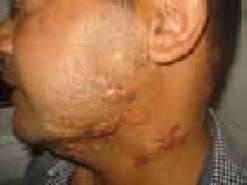
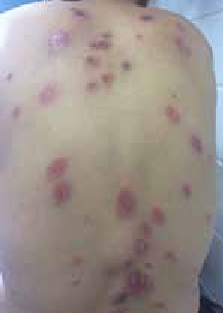
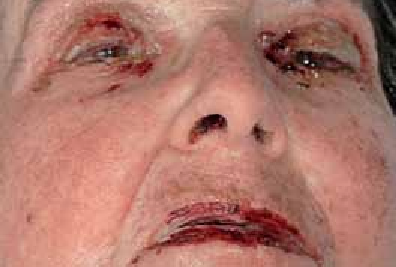
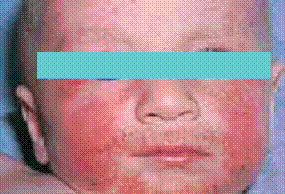
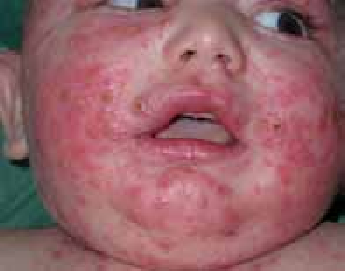
- La surinfection herpétique (HSV1) ou syndrome de Kaposi-Juliusberg : elle se traduit par une aggravation brutale de la maladie avec apparition de vésicules hémorragiques et de pustules ombiliquées rapidement extensives (figure 4) [9].
Autres complications :
- Dermatite (ou eczéma) de contact : il existe un risque important de sensibilisation chez l’enfant atteint de DA [5].
- Retard de croissance : rare mais peut être observé dans les cas de DA graves. Il se corrige habituellement quand la DA est traitée efficacement. Il doit faire rechercher une allergie alimentaire [5].
- Complications ophtalmologiques : rares à type de kératoconjonctivite, cataracte [5].
Associations morbides :
Il a été rapporté une augmentation de la fréquence de maladies auto-immunes telles que les maladies inflammatoires chroniques de l’intestin, pelade, polyarthrite rhumatoïde et lupus érythémateux, et en particulier chez les sujets jeunes [19]. Des cas de DA associant des troubles psychiatriques en particulier le déficit de l’attention et l’hyperactivité. Le prurit chronique, les troubles de sommeil et l’altération de la qualité de vie peuvent expliquer cette association [20].
Prise en charge thérapeutique de la DA :
Elle dépend de :
- L’évaluation de la gravité de la maladie. Plusieurs scores cliniques composites ont été validés (SCORAD, EASI [21], SASSAD).
- La qualité de vie : dans les formes modérées et graves, la qualité de vie des sujets atteints et de leur famille est souvent très altérée en raison du prurit, des perturbations du sommeil et du caractère affichant de la maladie. La prise en charge thérapeutique comporte :
- Les soins d’hygiène non agressifs et adaptation de l’environnement :
- Éviter le savon et utiliser un gel ou un pain sans savon doux non parfumé
- Réduire la fréquence des lavages de la peau et éviter les bains chauds prolongés
- Privilégier les textiles vestimentaires doux (éviter la laine)
- Éviter l’exposition à la chaleur (intolérance à la sueur).
- L’éducation thérapeutique qui a pour objectif d’apprendre au patient à vivre de manière optimale avec une maladie chronique
- Les moyens thérapeutiques :
- Dermocorticoïdes
Les dermocorticoïdes (DC) ont une triple action : anti-inflammatoire, immunosuppressive et antimitotique. Ils sont très efficaces à court terme sur les poussées de DA. Paradoxalement, la corticothérapie locale (sans antibiothérapie associée) est un moyen efficace de réduire la colonisation cutanée par le staphylocoque doré [22,23].
Les DC sont classés selon leur puissance et leur forme galénique (crème, pommade, lotions et gels). Ils représentent le traitement topique de référence [24].
Le choix est fait en fonction de l’âge, de la sévérité de la DA, du site et de l’étendue à traiter.
Les DC d’activité très forte (classe IV) sont contre-indiqués chez le nourrisson et le jeune enfant, sur le visage, les plis, et le siège. En règle générale :
- Les formes « pommade » sont réservées aux zones cutanées lichénifiées et sèches ;
- Les formes « crème » sont réservées aux zones suintantes, aux plis et aux grandes surfaces cutanées ;
- Les formes « lotion » sont réservées aux zones pileuses et aux plis ;
- Les formes « gel » sont réservées au cuir chevelu ; Une seule application par jour est suffisante.
Le « wet wrapping »
Cette technique de double bandage (une couche de bandes humidifiées, une couche de bande sèche) associée à une corticothérapie locale semble être plus efficace sur le prurit à court terme que la corticothérapie seule [19].
La corticophobie est un phénomène fréquent, ayant pour conséquence une faible adhésion thérapeutique dans la DA. La prise en charge de la corticophobie s’impose afin d’améliorer l’adhésion au traitement et diminuer les échecs thérapeutiques.
- Émollients-hydratants
Les émollients (exemples : Dexeryl© Crème, préparations magistrales), améliorent les signes fonctionnels dus à la sécheresse cutanée et certains d’entre eux restaurent transitoirement la fonction barrière cutanée. Ils pourraient avoir un effet d’épargne des DC et un effet préventif des poussées [25].
- Inhibiteurs topiques de la calcineurine
Le tacrolimus topique sous forme de pommade est indiqué après l’âge de 2 ans chez l’enfant (tacrolimus 0,03%) et chez l’adulte (tacrolimus 0,1 %) dans la DA modérée à sévère, en cas d’échec ou de contre-indications aux DC. Prescrit en traitement d’entretien, il prévient les poussées [26]. Une application 2 fois par jour est préconisée jusqu’à disparition des lésions puis la posologie peut être diminuée à 1 fois par jour. Il est impératif d’éviter la photo-exposition après l’application afin de réduire les risques d’intolérance immédiate transitoires, de brûlures et d’exacerbation du prurit au cours des premières applications [9].
- Antihistaminiques oraux (anti-H1)
Seuls, ils sont insuffisants pour traiter une poussée de DA. Les antihistaminiques de 1ère génération peuvent être utilisés pour le confort du patient. Ils diminuent le prurit et les réveils nocturnes (du fait de leur effet sédatif).
Ils n’ont pas d’intérêt au long cours en prévention des poussées de DA [27].
- Antiseptiques et antibiotiques locaux ou systémiques utiles en cas de surinfection des lésions [27].
- Photothérapie
La photothérapie UVA-UVB et UVB à spectre étroit est un traitement de seconde ou de troisième ligne de la DA à partir de l’âge de 8-10 ans. Elle n’est indiquée que dans des DA sévères, résistantes aux autres thérapeutiques, mais pendant des périodes courtes (risques carcinogènes potentiels à long terme).
- Traitements immunosuppresseurs systémiques
Les immunosuppresseurs systémiques sont des traitements de deuxième ou troisième ligne de la DA chez l’adolescent et l’adulte, exceptionnellement chez l’enfant.
Le traitement systémique de première ligne chez l’enfant et l’adulte est la ciclosporine. Les traitements de deuxième ligne sont représentés par l’azathioprine et le méthotrexate.
- La ciclosporine est prescrite par voie orale à la dose de 3 à 5 mg/kg/j. La durée du traitement est de 6 à 9 mois. L’efficacité est souvent rapide (une semaine) mais les rechutes sont fréquentes à l’arrêt du traitement. Cette thérapeutique nécessite une surveillance de la tension artérielle et de la fonction rénale [28].
- Le délai d’action du méthotrexate (hors AMM) est de l’ordre de 4 semaines.
- Des thérapies ciblées sont en cours d’évaluation chez l’adulte.
Le rituximab (anti-CD20), les anti-TNFα et le tocilizu- mab (anti-IL6) ont montré une efficacité mais au prix d’effets indésirables sévères pour le tocilizumab [29,30].
La thérapie ciblée la plus intéressante actuellement est le dupilumab. Cet anticorps monoclonal humanisé anti- IL4/IL13 efficace dans l’asthme, a donné une réponse favorable dans la DA modérée à sévère avec une meilleure tolérance [31].
- Les mesures préventives
- Laits artificiels et allaitement maternel
L’instauration d’un lait artificiel contenant un hydrolysat de protéines de lait de vache (seul ou en complément de l’allaitement maternel) entraine une réduction modérée du risque de DA dans les premières années de vie dans la population à risque d’atopie [32]. Aujourd’hui, il n’y a pas d’argument convaincant pour suggérer que l’allaitement maternel exclusif protège du risque de DA [33].
- Probiotiques et prébiotiques
Instaurés à un âge très précoce, les probiotiques et/ou les prébiotiques per os pourraient avoir un effet préventif sur la DA. Les souches lactobacillus semblent être actuellement les plus efficaces.
Mesures contre la sensibilisation L’immunothérapie spécifique (désensibilisation). Des résultats récents ont démontré son intérêt dans la DA de l’adulte, chez des malades porteurs d’une forme sévère avec sensibilisations aux acariens et aux pollens [34].
L’utilisation de topiques potentiellement à risque (émollients ou topiques contenant des conservateurs très sensibilisants, des parfums, de la néomycine) est à limiter afin de prévenir la survenue de l’eczéma de contact.
Conclusion :
La DA s’intègre dans le cadre des maladies allergiques. C’est une maladie inflammatoire secondaire à plusieurs facteurs (altération de la fonction de la barrière épidermique et dysbiose intestinale et cutanée). Elle est plus fréquente dans les pays développés. Généralement, la DA débute dans la petite enfance Elle est caractérisée par une prédisposition génétique et une évolution récurrente avec des manifestations cliniques, en fonction de l’âge du patient. Concernant la prise en charge, le traitement local consiste toujours en l’usage des émollients et des dermocorticoïdes qui représentent toujours les al- ternatives de choix. Par contre, le traitement systémique s’est enrichi grâce aux progrès de la biotechnologie.
De nouvelles molécules de biothérapie visant à inhiber des mécanismes effecteurs clés dans la physiopathologie de la DA représentent une perspective thérapeutique très prometteuse.
Références :
- Irvine AD, Mclean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med 2011; 365: 1315-27.
- Queille-Roussel C, Raynaud F, Saurat JH: A prospective computerized study of 500 cases of atopic dermatitis in childhood. Acta Derm Venereol Suppl (Stockh) 1986; 114:87–92.
- Taïeb A, Hennino A, Bérard F, Nicolas JF. Dermatite atopique, Eczémas et dermatoses Spongiformes. In: Saurat JH, Lachapelle JM, Lipsker D, Thomas L. Dermatologie et infections sexuellement trans- missibles. Paris: Elsevier; 2009. p.67-80.
- Baghou S, Bensaad D, Taieb A, Ammar-Khodja A. Prévalence et profil clinique de la dermatite atopique en Algérie Ann Dermatol Vé- néréol 2012;139(12S):B140. Doi : 10.1016/j.annder.2012.10.200
- Auteurs et membres du Collège des enseignants en dermatolo- gie de France. Allergies cutanéo-muqueuses chez l’enfant et l’adulte : dermatite (ou eczéma) atopique. Item 114. Ann dermatol vénéréol 2008; 135S : F80—F87. Consultable sur : https://doi.org/10.1016/j. annder.2008.07.001
- Xu F, Yan S, Li F Et al. Prevalence of childhood atopic dermatitis: an urban and rural community-based study in Shanghai, China. PLoS One, 2012;7: e36174.
- Barbarot S. Physiopathologie de la dermatite atopique et perspec- tives thérapeutiques systémiques. Mises au point interactives, éd. Réalités Thérapeutiques en Dermato-Vénérologie- Performances Médicales. Cahier 1 2016 (256). p.48-49. Consultable sur : jird.info/ wp-content/uploads/2017/01/MOP4.pdf
- Barbarot S, Aubert H. Physiopathologie de la dermatite atopique. Ann Dermatol Vénéréol 2017;144 Suppl 1: S14-S20.
- Stalder JF, Barbarot S & Aubert H. Dermatite atopique. In : L. Thé- rapeutique Dermatologique. Un manuel de référence en dermatologie 27 juillet 2015. Consultable sur : http:// www.therapeutique-dermato- logique.org/spip.php?article1068
- Palmer CN, Irvine AD , Terron-Kwiatkowski A et al. Common loss- of- function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 2006; 38:441-6.
- Strachan DP. Family size, infection and atopy: the first decade of the «hygiene hypothesis». Thorax 2000;55 Suppl 1:S2-S10.
- Garn H, Neves JF, Blumberg RS et al. Effect of barrier microbes on organ-based inflammation. J Allergy Clin Immunol 2013; 131:465-78.
- Gittler JK, Shemer A, Suarez-Farinas M Et al. Progressive activa- tion of T(H)2/T(H)22 cytokines and selective epidermal proteins cha- racterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol 2012; 130:1344-54.
- Wilson SR, The L, Batia LM et al. The Epithelial Cell-Derived Ato- pic Dermatitis Cytokine TSLP Activates Neurons to Induce Itch. Cell 2013; 155:285-95.
- Ismail IH, Oppedisano F, Joseph SJ et al. Reduced gut microbial diver- sity in early life is associated with later development of eczema but not atopy in high-risk infants. Pediatr Allergy Immunol 2012; 23:674-81.
- Kong HH, Oh J, Deming C Et al. Temporal shifts in the skin micro- biome associated with disease flares and treatment in children with atopic dermatitis. Genome research 2012; 22:850-9.
- Kuo IH, Yoshida T, De Benedetto A et al. The cutaneous innate immune response in patients with atopic dermatitis. J Allergy Clin Immunol 2013; 131:266-78.
- Williams HC, Burney PG, Hay RJ et al. The U. K. Working Party’s Diagnostic Criteria for Atopic Dermatitis. I. Derivation of a minimum set of discriminators for atopic dermatitis. Br J Dermatol 1994; 131:383-96.
- Chiavreini C. Quoi de neuf en dermatologie pédiatrique. Ann Dermatol Vénéréol 2017;144 suppl 4:IVS29-IVS39.
- Strom MA, Fishbein AB, Paller AS, Silverberg JI. Association between atopic dermatitis and attention deficit hyperactivity disorder in U.S. children and adults. Br J Dermatol 2016; 175:920.
- Barbier N, Paul C, Luger T Et al. Validation of the Eczema Area and Severity Index for atopic dermatitis in a cohort of 1550 patients from the pimecrolimus cream 1% randomized controlled clinical trials program. Br J Dermatology 2004; 150:96-102.
- Stalder JF, Fleury M, Sourisse M Et al. Local steroid therapy and bacte- rial skin flora in atopic dermatitis. Br J Dermatol 1994; 131:536-40.
- Bath-Hextall FJ, Birnie AJ, Ravenscroft JC et al. Interventions to reduce Staphylococcus 23. aureus in the management of atopic ecze- ma: an updated Cochrane review. Br J Dermatol 2011; 164:228.
- Leloup P, Stalder JF, Barbarot S. Outpatient home-based wet wrap dressings with topical steroids with children with severe recal- citrant atopic dermatitis: A Feasibility Pilot Study. Pediatr Dermatol 2015;32:e177-8.
- Lucky AW, Leach AD, Laskarzewski P, Wenck H. Use of an emol- lient as a steroid-sparing agent in the treatment of mild to moderate atopic dermatitis in children. Pediatr Dermatol 1997;14:321-4.
- Schmitt J, Von Kobyletzki L, Svensson A, Apfelbacher C. Efficacy and tolerability of proactive treatment with topical corticosteroids and calci- neurin inhibitors for atopic eczema: systematic review and meta-analysis of randomized controlled trials. Br J Dermatol 2011;164:415-28.
- Barbarot S, Aubert H, Stalder JF, Bernier C. Dermatite atopique. Ency- cl Med Chir, Dermatologie 2016;11(3):1-20 [Article 98-150-A-10].
- Schmitt J, Schmitt N, Meurer M. Cyclosporin in the treatment of patients with atopic eczema – a systematic review and meta-analysis. J Eur Acad Dermatol Venereol 2007;21:606-19.
- Simon D, Hösli S, Kostylina G Et al. Anti-CD20 (rituximab) treatment improves atopic eczema. J Allergy Clin Immunol 2008; 121:122-8.
- Jacobi A, Antoni C, Manger B Et al. Infliximab in the treatment of mo- derate to severe atopic dermatitis. J Am Acad Dermatol 2005; 52:522-6.
- Simpson EL, Bieber T, Guttman-Yassky E et al. SOLO 1 and SOLO 2 Investigators. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N Engl J Med 2016; 375(24):2335-2348. ISSN 0028-4793 Consultable sur: https://doi.org/10.1056/NEJMoa1610020.
- *Von Berg A, Filipiak-Pittroff B, Kramer U et al. Allergies in high- risk schoolchildren after early intervention with cow’s milk protein hydrolysates: 10-year results from the German Infant Nutritional Intervention (GINI) study. J Allergy Clin Immunol 2013;131:1565-73.
- Flohr C, Nagel G, Weinmayr G Et al. Lack of evidence for a pro- tective effect of prolonged breastfeeding on childhood eczema: lessons from the International Study of Asthma and Allergies in Childhood (ISAAC) Phase Two. Br J Dermatol 2011;165:1280-9.
- Bae JM, Choi YY, Park CO et al. Efficacy of allergen-specific immunotherapy for atopic dermatitis: a systematic review and me- ta-analysis of randomized controlled trials. J Allergy Clin Immunol 2013;132:110-7.