N. KESRI, Service d’Endocrinologie, Centre Pierre et Marie Curie, Alger.
Résumé : L’insuffisance ovarienne prématurée (IOP) communément appelée ménopause précoce, observée chez 1-2% de la population féminine, définit l’apparition d’une aménorrhée avant l’âge de 40 ans associée à des taux élevés de FSH vérifiés à deux reprises à un mois d’intervalle. Il s’agit d’une situation pathologique relevant de multiples mécanismes pathogéniques d’origine génétique ou auto immune responsables de déplétion ou de dysfonction folliculaire entrainant une altération précoce de la fonction endocrine et de la fertilité. La symptomatologie clinique traduit la carence hormonale précoce, essentiellement estrogénique, d’installation aigue ou progressive avec une importante composante psychosomatique liée à la réduction des capacités de procréation naturelle. En absence d’un diagnostic précoce, les principales complications sont d’ordre osseux et cardio-vasculaire. Au plan évolutif, des phases de réversibilité de durée imprévisible, sont rapportées notamment dans les formes débutantes. La prise en charge comprend une enquête étiologique qui reste infructueuse dans près de deux tiers des cas, et repose sur l’institution immédiate et indiscutable d’une substitution hormonale œstro-progestative dont la seule contre-indication formelle reste actuellement le cancer mammaire. Les schémas sont personnalisés, adaptés au stade diagnostique et doivent privilégier les hormones naturelles et la voie transdermique. Dans les IOP iatrogènes le pronostic de fertilité est amélioré par le recours à la cryoconservation préalable du cortex ovarien tandis que la seule thérapeutique curatrice de l’infertilité des IOP spontanées repose actuellement sur le don d’ovules, interdit par la religion musulmane. De nouvelles techniques de thérapie cellulaire sont actuellement en cours d’expérimentation telles que la maturation de follicules primordiaux résiduels ou l’isolement de cellules souches ovariennes offrant à ces jeunes femmes dans un avenir peut être proche, des possibilités de conception naturelle à partir de leurs propres ovules.
Mots-clés : Ménopause précoce, anomalies de la folliculogénèse, syndrome climatérique précoce, traitement hormonal substitutif, hypofertilité iatrogène.
Abstract : The premature ovarian failure (POF) commonly called early menopause, observed in 1-2% of female population, defines the appearance of amenorrhea before the age of 40 years associated with high levels of FSH checked twice in one month apart. It is a pathological situation within multiple pathogenic mechanisms of autoimmune or genetic origin responsible of depletion or dysfunction follicular causing an early alteration of the endocrine function and fertility. Clinical symptomatology translated early, acute or gradual installation of hormonal deficiency mainly estrogenic with significant psychosomatic component related to the reduction of the natural reproductive capacity. In the absence of early diagnosis, the main complications are bone and cardiovascular. In evolutionary terms, phases of reversibility of unpredictable duration are reported in beginner forms. The etiological investigation remains unsuccessful in nearly two thirds of the cases, and management is based on immediate and and indisputable institution of estrogen and progestogen hormonal substitution which the only formal contraindication is currently the breast cancer. Patterns are personalized, adapted to the diagnosis stage and need to focus on the natural hormones and the transdermal route. In iatrogenic POF fertility prognosis is improved by the use of prior cryopreservation of the ovarian cortex while only infertility curative therapy is currently based on egg donation, which is forbidden by Muslim religion. New cell therapy techniques are currently being tested such as the maturing of primordial follicles or isolation of ovarian stem cells providing to these young women in future may be close opportunities for natural conception from their own eggs.
Key-words : Premature menopause, folliculogenesis anomalies, premature climateric syndrome, hormonal replacement treatment , iatrogenic hypofertility.
Introduction :
La ménopause est une étape physiologique et inéluctable de la vie génitale féminine marquée par la cessation définitive des menstruations et de la fonction de reproduction relevant d’un mécanisme unique de déplétion en follicules primordiaux potentiellement fonctionnels (1). Elle est en général diagnostiquée rétrospectivement après 12 mois d’aménorrhée et son âge de survenue est stable autour de 50 ans en moyenne (46-54 ans pour 95 % femmes, et vers 40-45 ans pour moins de 5 % d’entre elles) (1).
Une situation similaire d’aménorrhée avec son cortège climatérique classique peut être observée avant l’âge de 40 ans chez environ 1-2 % des femmes de la population générale, naturellement dénommée « ménopause précoce » (2).
De nombreux auteurs considèrent cette appellation inadéquate car en plus de son âge de survenue, cette entité s’oppose à la ménopause naturelle par plusieurs aspects :
- Les mécanismes pathogéniques nombreux, de mieux en mieux cernés grâce aux progrès de génétique moléculaire réalisés au cours de ces vingt dernières années.
- Les modalités évolutives fluctuantes et imprévisibles de la fonction endocrine et même exocrine responsables d’une grande variabilité clinique et biologique.
- Le caractère indiscutable et prolongé du traitement hormonal de substitution (THS) dans cette situation, auquel ne sauraient s’appliquer les conclusions des études WHI.
Ainsi, les dénominations de « dysfonction ovarienne prématurée » ou « d’insuffisance ovarienne prématurée », sont actuellement privilégiées (2) dont l’avantage principal est d’éviter la connotation psychologique péjorative, inéluctablement associée à l’annonce d’un diagnostic aussi dévastateur dans cette tranche d’âge que celui de « ménopause précoce ».
Quelques données épidémiologiques :
Une estimation précise de la prévalence des IOP manque encore, il faut distinguer les :
- IOP spontanées dont la prévalence serait de 1 % avant l’âge de 40 ans, de 1/1.000 avant 30 ans et de 1/10.000 avant 20 ans. Leur prévalence est stable d’après les dernières études épidémiologiques.
- IOP iatrogènes dont la prévalence est en nette progression, du fait de l’amélioration de la survie liée à l’efficacité indéniable des traitements carcinologiques mais au prix d’effets gonadotoxiques inévitables.
- Certaines études épidémiologiques rapportent une association avec le tabagisme et certains facteurs ethniques vu la prévalence moindre chez les femmes chinoises ou japonaises comparées aux femmes caucasiennes ou africaines. Il n’a pas montré de lien entre la survenue d’une IOP et l’âge des premières règles, le niveau d’éducation ou la prise de contraception orale.
Aspects cliniques :
Diverses circonstances peuvent révéler une IOP, essentiellement des troubles du cycle menstruel, un bilan d’infertilité, un syndrome génétique ou une maladie auto–immune ainsi que les suites de traitements gonadotoxiques ou d’une ovariectomie bilatérale.
Le plus souvent l’IOP spontanée s’installe après une puberté normale suivie de cycles réguliers de durée variable. Quelques fois les cycles peuvent s’arrêter brusquement : absence de retour des cycles après une grossesse ou à l’arrêt d’un contraceptif oral mais le plus souvent l’aménorrhée est précédée par des prodromes sur un terrain de stress tels qu’une oligo-spanioménorrhée, polyménorrhée, ou métrorragies.
La symptomatologie est inconstante, corrélée au degré du déficit oestrogénique, variable du fait de la possibilité d’une réversibilité transitoire dans les formes débutantes, mais aussi selon l’ancienneté du déficit et de l’étiologie : en général minime dans les causes génétiques, et intense si les causes sont chirurgicales. Les conséquences des insuffisances ovariennes liées aux carences hormonales précoces sont mal connues ; elles sont décrites à partir d’observations sur des séries d’IOP secondaires essentiellement chirurgicales. Initialement ce sont des signes de déficits oestrogénique et progestatif qui apparaissent à court ou moyen terme : manifestations vasomotrices à type de bouffées de chaleur, crises sudorales surtout nocturnes, sécheresse vaginale par diminution des sécrétions vaginales source de dyspareunie gênante.
Des signes de retentissement sur l’appareil urinaire se surajoutent avec risque d’incontinence d’effort, d’infection urinaire, de dysurie et de miction impérieuse.
A long terme, le tableau se complète par l’installation de troubles osseux et cardiovasculaires.
Le tableau peut comporter également un syndrome de déficit androgénique non spécifique suspecté en absence d’amélioration sous substitution œstro-progestative, baisse de vitalité, asthénie croissante et fatigabilité, modifications corporelles comme la perte de la forticilisation des poils pubiens, et l’atrophie génitale. De même que la baisse de la masse et du tonus musculaires, des anomalies de la répartition des graisses et l’installation de troubles sexuels comme la perte du désir, des fantasmes, des rêves érotiques, et de la capacité orgasmique.
L’examen physique est capital, permet d’apprécier, en plus de la symptomatologie fonctionnelle, le degré du développement pubertaire (absent, incomplet ou normal) et de retrouver des signes d’orientation étiologique d’une IOP syndromique génétique ou auto-immune ou conclure à une IOP isolée spontanée.
Profil hormonal des IOP :
Le profil est variable fonction du stade au diagnostic, de la cause et de l’ancienneté de l’affection ; l’IOP spontanée correspond à un phénomène de vieillissement ovarien précoce évoluant en deux phases :
La première, celle de la baisse de la réserve folliculaire caractérisée par des ovulations moins fréquentes : la FSH est élevée, la 17βestradiol(E2) reste élevée pendant quelques années puis la phase folliculaire et l’intervalle intercycle se raccourcissent.
La seconde est un stade d’épuisement folliculaire où les taux d’E2 baissent à moins de 50 pg/ml, les taux de FSH s’élèvent à plus de 30µU/ml ; la LH s’élève mais à des valeurs moindres ce qui différencie l’IOP d’un dosage effectué lors d’un pic ovulatoire. Par ailleurs, diminution des sécrétions androgéniques de Delta 4 androstènedione, Testostérone, DHEA et de la sécrétion des peptides ovariens (inhibines, AMH…).
Critères diagnostiques des IOP :
L’absence de standardisation des critères diagnostiques est encore cause de retard diagnostique des IOP, les critères actuellement retenus sont d’ordre clinique et biologique.
Au plan clinique, il n’y a pas encore de réel consensus sur la durée de l’aménorrhée : l’aménorrhée de plus de 4 mois chez une femme de moins de 40 ans est actuellement retenue.
Au plan biologique : les dosages d’E2, FSH et d’AMH sont proposés.
- L’intérêt du dosage d’E2 est encore discuté : du fait de sa variabilité selon l’ancienneté de l’IOP, il n’est actuellement pas retenu comme critère diagnostique.
- Le niveau d’élévation de la FSH n’est pas encore consensuel, le seuil de 30-40 µU/ml est admis, à condition de le vérifier à deux reprises, à au moins un mois d’intervalle. Certains proposent un seuil inferieur de 20-40 µU/ml en gardant présent à l’esprit que l’élévation de FSH en dessous de ce seuil, n’est pas toujours liée à une IOP (3).
- Certaines équipes proposent d’introduire parmi les paramètres diagnostiques d’IOP le dosage de l’hormone anti Mullerienne (AMH), qui reflète le nombre des follicules primitifs et l’âge ovarien. En plus du fait que l’AMH peut être dosée à tout moment durant le cycle menstruel, ce dosage semble être dans l’IOP plus utile que celui de la FSH car ses modifications apparaissent avant celles de la FSH.
Dans ces études, la précision diagnostique de l’AMH est plus élevée que celle de la FSH et à des différences significatives (4).
La dernière réunion du Groupe d’Étude des IOP a recommandé en 2016 l’utilisation des critères diagnos- tiques suivants (5) :
- Survenue d’une oligo-aménorrhée depuis au moins 4 mois ou la présence de signes de déficit E2.
- Avant l’âge de 40 ans.
- Des taux élevés de FSH supérieurs à 25UI/ml contrôlés deux fois à un mois d’intervalle.
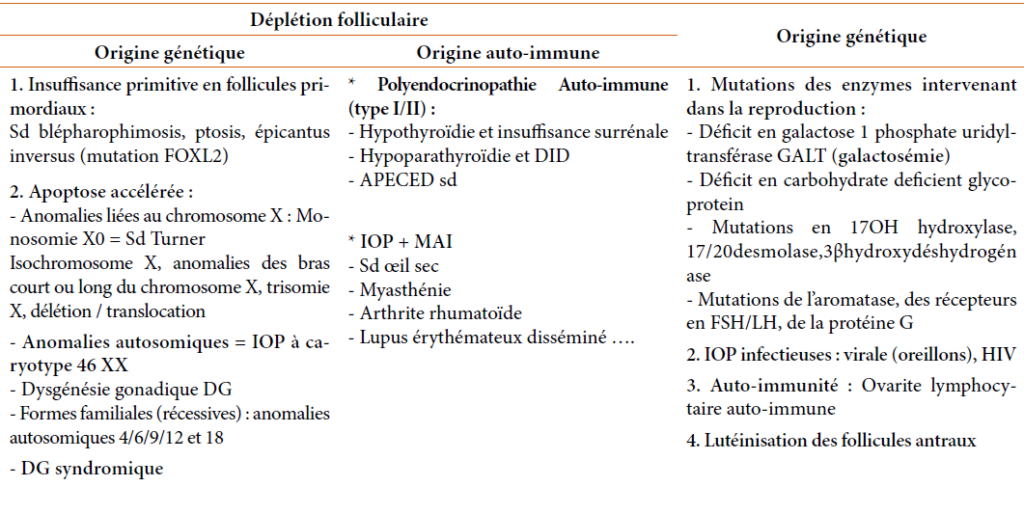
Aspects étiopathogéniques :
A l’opposé de la ménopause physiologique qui résulte d’un épuisement folliculaire, plusieurs mécanismes sont incriminés dans la survenue d’une IOP, il s’agit :
- Soit d’une déplétion folliculaire de cause génétique ou auto-immune : il n’y a pas de follicules primordiaux dans l’ovaire par échec primitif de constitution ou du maintien d’un pool suffisant de follicules primordiaux, ou atrésie des follicules, secondaire à une apoptose accélérée ou une destruction auto immune ou toxique des follicules.
- Soit d’un mécanisme de dysfonction folliculaire par blocage de la croissance folliculaire ; les follicules restent dans l’ovaire mais un processus pathologique empêche leur fonctionnement normal.
- Les principales étiologies des IOP sont résumées dans le tableau ci-dessous :
Bilan étiologique recommandé par l’ESHRE en 2016 :
Le bilan doit comporter au minimum un bilan génétique et un bilan d’auto-immunité (5) :
- Un caryotype doit être réalisé chez toute patiente présentant une IOP non-iatrogénique : une gonadectomie devrait être recommandée chez toute femme avec matériel chromosomique Y détectable complétée par la recherche systématique de la premutation de l’X fragile.
– La recherche anomalies autosomiques n’est indiquée que s’il existe des signes cliniques suggérant une mutation spécifique (ex blépharophimosis-ptosis–épicanthus inversus syndrome).
- Le dépistage des anticorps (AC) anti thyroïdiens et d’AC anti 21OHhydroxylase ou anti corticosurrénale doit être considéré chez toutes les femmes atteintes d’IOP de cause idiopathique ou si elle est associée à un désordre auto immun. En cas de positivité de ces derniers, les patientes devront être référées en endocrinologie pour évaluer la fonction surrénalienne et éliminer une maladie d’Addison.
- Il n’y a pas assez de preuves pour recommander le dépistage systématique d’un diabète sucré. et d’infections ovariennes chez les femmes porteuse d’IOP.
Cependant malgré tous les progrès réalisés, aucune cause ne peut être identifiée au terme de l’enquête étiologique chez un nombre significatif de femmes, il s’agit d’IOP inexpliquée ou idiopathique.
Risques liés à l’IOP non traitée :
ils sont la traduction de la souffrance des principaux tissus cibles secondaire à la carence hormonale :
- La première conséquence est une grande souffrance psychologique, prévisible dans une tranche d’âge aussi vulnérable qui devra nécessiter une assistance psychologique régulière et prolongée.
Elle se traduit par divers troubles de l’humeur : irritabilité, susceptibilité, hostilité, mélancolie, état anxio-dépressif menant à un épuisement psychologique, tendance à l’isolement, repli sur soi, céphalées, insomnies aggravées par les troubles vaso moteurs et réveils de plus en plus précoces aggravant l’état d’asthénie avec perte de l’estime de soi et sentiment d’infériorité (6).
- Altération de la sexualité : La fonction sexuelle chez la femme est modulée par les œstrogènes intervenant dans la congestion vaginale et la lubrification et par la testostérone dans la promotion du désir et la libido par effet central sur la dopamine. L’IOP non traitée est à l’origine de troubles sexuels tels que perte du désir, des fantasmes et des rêves érotiques, de la capacité orgasmique, et de l’activité sexuelle ainsi que de signes de carence oestrogénique, cause d’amincissement vaginal,dessèchement, perte d’élasticité et dyspareunie.
- Risque cardio-vasculaire : une augmentation significative du risque cardiovasculaire a été démontrée chez les femmes atteintes d’IOP, et de la mortalité cardiovasculaire dans 80 % par ischémie cardiaque (7,8), soit un risque plus précoce de 10 ans par rapport à une ménopause naturelle. Différents facteurs de risque ont été incriminés, essentiellement la dysfonction endothéliale, réversible sous THS (9), la baisse de l’insulino sensibilité (10), ainsi que la présence de syndrome métabolique (11) avec profil lipidique délétère (12).
- Risque osseux : Compte tenu du fait que l’IOP s’installe souvent avant l’acquisition du pic de la masse osseuse, de nombreuses études ont démontré que l’IOP est associée à une densité minérale osseuse (DMO) réduite significativement par rapport à des femmes de même âge (13) : deux tiers des IOP ont une DMO basse un an et demi après le diagnostic. La DMO des IOP idiopathiques ou malignes est beaucoup plus basse que la DMO des IOP bénignes.
Pour le groupe de l’ESHRE (5), Il est important d’évaluer la santé osseuse lors du diagnostic initial (14), par mesure de la DMO pour toutes les femmes, surtout lorsqu’il existe des facteurs de risque supplémentaires.
- Impact de l’IOP sur les fonctions cognitives : Des études d’observation ont suggéré que la carence prolongée d’E2 ovarien doublait le risque de démence et multipliait par cinq la mortalité par affections neurologiques, mais il reste encore des controverses. D’autres études ciblant les IOP ont montré un risque accru de troubles neurologiques concernant la mémoire verbale et les fonctions cognitives (15,16). Dans ces études, il est démontré que l’IOP quelle que soit sa cause, est associée au long cours avec des effets délétères sur la fonction cognitive qui ne sont pas entièrement réversibles sous THS.
Prise en charge des IOP : elle a un double objectif.
D’une part la correction du déficit oestrogénique permettant de soulager et traiter les symptômes de l’IOP et surtout d’en prévenir les effets délétères et d’autre part d’envisager les possibilités d’un traitement curatif des troubles de la fertilité. Cette prise en charge doit tenir compte de plusieurs paramètres : l’âge au diagnostic de l’IOP, son caractère induit ou spontané, l’ancienneté, l’étiologie et le terrain comportant d’éventuelles comorbidités.
- Les intérêts du THS sont multiples : de nombreuses
études ont démontré un effet de prévention primaire de la détérioration osseuse (5) : en effet, les ostrogènes naturels contenus dans le THS ont un effet protecteur indéniable sur l’os ; pris au moins 3 ans le THS diminue significativement le risque de fractures ultérieures (17). Par ailleurs, ces ostrogènes naturels auraient un effet cardio-protecteur : malgré le manque de données du devenir au long cours, l’introduction rapide du THS dès le début de l’IOP est fortement recommandée pour contrôler le risque ultérieur de maladies cardiovasculaires : il est conseillé de poursuivre le traitement au moins jusqu’à l’âge de la ménopause naturelle. Les données concernant l’effet neuroprotecteur sont encore discordantes surtout après IOP chirurgicale, cependant le THS apparait plus bénéfique s’il est initié dès la ménopause.
- Le THS est actuellement fortement recommandé par l’ensemble des sociétés savantes s’occupant de pathologie gonadique féminine (6) et ce au moins jusqu’à l’âge de la ménopause naturelle, voire au-delà si les symptômes persistent. La correction hormonale est accompagnée de mesures hygiénodiététiques permettant le contrôle des facteurs de risque cardiovasculaires (poids, TA), et de préservation du capital osseux. Ces mesures hygiéno-diététiques reposent d’une part sur des apports vitamino-calciques adéquats pour préserver le capital osseux, soit par respect des apports alimentaires recommandés pour l’âge, sinon par compléments alimentaires. Comme il n’y a pas de données spécifiques à ces jeunes femmes, il est possible de s’inspirer des recommandations de la société nord-américaine de ménopause : 1200-1500 mg/j de calcium élément et de vitamine D, sous sa forme active D3 : 800-1000 U/j, à maintenir dans les normes recommandées (25 OHD > 30 mg/ml). D’autre part sur l’exercice physique régulier : là aussi, il n’y a pas de données spécifiques à ces femmes pour la fréquence et l’intensité, les exercices conseillés sont des séances de musculation, jogging, footing, monter les escaliers à pied, et éviter les ascenseurs.
- L’arrêt du tabac et de l’alcool sont fortement recommandés chez toutes les femmes à risque d’IOP. Certaines équipes proposent le recours à l’acupuncture qui aurait un certain effet sur l’amélioration des menstruations et du syndrome climatérique, probablement lié à l’augmentation des taux d’œstrogènes (18).
Prescription du THS : L’estrogène de choix est le 17βestradiol dont les effets sont supérieurs à ceux de l’éthynil-œstradiol ou aux œstrogènes conjugués, prescrit préférentiellement par voie transdermique, percutanée du fait de ses moindres effets sur les facteurs hémostatiques, ou orale en cas de réticence. Des études contrôlées ont montré que cette voie a moins d’accidents veineux thromboemboliques que les œstrogènes oraux (19).
La dose d’initiation
doit être supérieure à la dose des œstrogènes contenus dans le THS des ménopauses naturelles pour un meilleur contrôle des symptômes (surtout vasomoteurs), elle permettrait d’atteindre une concentration plasmatique moyenne de la mi-phase folliculaire d’un cycle d’une femme normalement réglée soit environ 300-400 pg/ml assurant la protectrice osseuse. Cette dose est autour de 100 µg/j par voie transdermique (75-200 µg/j) et de 0,06 % x 2 / j de gel par voie percutanée.
Les progestagènes restaurent correctement un endomètre sécrétoire et protègent contre le cancer de l’endomètre, ils sont prescrits par voie orale durant 12-14 j par mois. Il s’agit d’isomères de la progestérone à la dose de 10 mg / j ou de la progestérone micronisée orale à la dose de 200 mg / j.
Il existe d’autres voies d’administration : transdermiques / gel : 12 j / mois ou la voie utérine par dispositif intra-utérin à libération continue de progestérone.
- Les schémas de prescription mimant parfaitement la sécrétion physiologique des E2/P n’existent pas et il y a nécessité d’associer la patiente dans le choix du schéma et de la voie d’administration pour une meilleure compliance au long cours, parfois même de considérer les produits contraceptifs (20).
Les schémas proposés sont :
- Le schéma séquentiel avec règles qui permet l’obtention d’hémorragie de privation à l’arrêt du progestatif, a un impact psychologique extrêmement bénéfique par prescription d’estradiol du J1 au J25 associé à la progestérone du J14 au J25.
- Le schéma combiné sans règles, comportant la prescription continue de comprimés contenant de l’E2 et de la progestérone du J1 au J25, généralement utilisé après 1 an de THS séquentiel.
- Si l’IOP comporte une activité résiduelle, le progestatif prescrit seul du J14 au J25 est suffisant pour l’obtention de règles.
Sous THS, la dose d’E2 doit être modulée selon la réponse clinique et la DMO : il n’y a pas d’intérêt à répéter les dosages d’E2 ni de FSH laquelle peut ne pas se normaliser.
- L’androgénothérapie peut être envisagée chez des femmes ovariectomisées, en cas d’absence d’amélioration sous THS bien conduit. Cependant il y a lieu d’informer ces patientes que les données concernant ce type de traitement sont encore limitées et que ses effets au long cours ne sont pas encore clairs : ils doivent être évalués après 3–6 mois et devraient être limités à 24 mois (6).
Indications du THS :
A l’opposé des recommandations actuelles concernant les ménopauses naturelles de ne traiter par THS que les femmes avec symptômes modérés à sévères, de carence oestrogénique, toutes les IOP spontanées quel que soit l’âge au diagnostic sont une indication formelle au THS sauf contre- indication absolue. Les femmes atteintes d’IOP doivent être informées qu’il n’a pas été démontré de sur-risque de cancer du sein sous THS avant l’âge de la ménopause, (21). La prévention du cancer de l’endomètre passe par la précaution d’associer un progestatif avec l’estradiol pour protéger l’endomètre des femmes avec utérus intact (22).
IOP et situations particulières :
- IOP induites : le THS reste une contre-indication formelle après un cancer du sein (23), chez les femmes porteuses de mutation du gène BRCA, cependant certaines équipes ont montré que le THS pouvait constituer une option thérapeutique bénéfique pour les femmes porteuses des mutations BRCA1/2 sans antécédent personnel de cancer du sein, institué après salpingo-ovariectomie bilatérale prophylactique (24).
- IOP et gynécologie : Dans l’endométriose, si une ovariectomie est réalisée, le THS peut être indiqué pour le traitement du syndrome climatérique et pourrait prévenir la réactivation de la maladie (25). Le fibrome ne représente pas une contre-indication au THS (26).
- IOP et migraine : la migraine ne doit pas être considérée comme une contre-indication au THS. Une éventuelle aggravation peut être réversible par le réajustement de la dose, de la voie, ou du schéma de prescription du THS. La voie transdermique présente le risque le plus faible d’une migraine avec aura (27).
- IOP et situations à haut risque cardiovasculaire : plusieurs études ont montré que ces dernières ne constituent pas de contre-indications au THS, telles que des antécédents de thrombo-embolie veineuse ou de troubles thrombophiliques, d’HTA, obésité, excès de poids ; dans ces cas la voie transdermique est fortement recommandée (28).
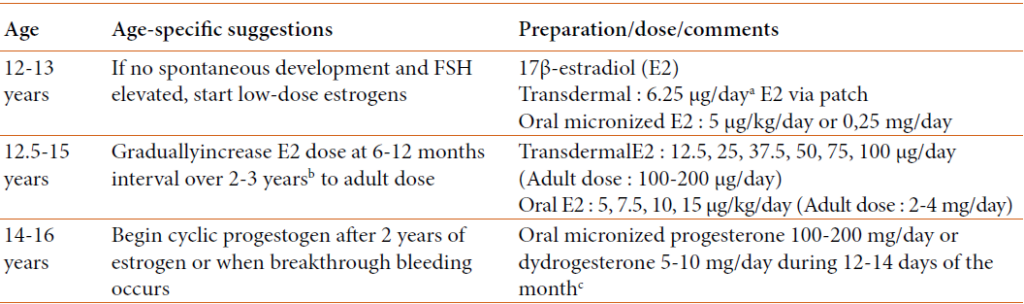
- Cas particulier de l’IOP à adolescence : Les modalités d’induction pubertaire proposées par le groupe d’étude du syndrome de Turner en 2007 sont résumées dans le tableau ci-dessous.
Alternatives au THS : Les preuves sur l’efficacité et l’innocuité de traitements alternatifs ou complémentaires dans les IOP iatrogènes sont très insuffisantes ou absentes (29).
Bilan de suivi des IOP recommande par l’ESHRE (5) :
La surveillance d’une IOP doit comporter un examen clinique annuel visant à évaluer la compliance des patientes atteintes. L’absence de signes d’appel ne justifie pas d’examen complémentaire particulier.
- Les modalités de suivi de la santé osseuse se basent sur la mesure de la DMO : Si la DMO est normale et un THS adéquat est introduit, l’intérêt de la répétition de la mesure est faible. Si une ostéoporose est diagnostiquée et le remplacement d’œstrogène ou d’un autre traitement osseux est initié, la mesure de DMO doit être renouvelée dans les 5 ans : une diminution de la DMO devrait réajuster la dose d’œstrogénothérapie et rechercher d’autres facteurs potentiels. Il y aura lieu alors de confier la patiente en milieu spécialisé.
- Les modalités de suivi des facteurs de risque cardiovasculaire comportent la surveillance annuelle du poids, de la pression artérielle, du tabagisme et le dépistage de tous autres facteurs de risque. Chez les femmes atteintes du syndrome de Turner, les facteurs de risque cardiovasculaires devraient être recherché au moment du diagnostic et réévalué chaque année (au moins PA, tabagisme, poids, profil lipidique, glycémie à jeun et l’ HbA1c)
- Les patientes avec AC TPO positifs doivent bénéficier d’une surveillance annuelle de la TSH (30,31).
Fertilité et IOP : des possibilités thérapeutiques ?
La prise en charge de l’infertilité résultant d’une IOP reste un grand challenge (32) : d’importants progrès ont été réalisés essentiellement dans les IOP iatrogènes grâce au développement des techniques de procréation médicalement assistée ayant permis chez certaines femmes une conception naturelle avec leurs propres ovules. Le préalable est l’abaissement des taux de FSH à des taux physiologiques avant d’entamer le traitement : l’éthynil œstradiol est classiquement utilisé, il permet de stimuler la glaire cervicale et le développement endométrial et n’interfère pas dans le dosage de l’estradiol. Des gonadotrophines exogènes à faibles doses sont parfois nécessaires pour booster la maturation associées à la progestérone prescrite en phase lutéale (33).
- Des techniques préventives et/ou curatrices de l’infertilité des IOP ont été développées par fertilisation in vitro utilisant le don d’ovocytes avec 50 % de succès selon l’âge du donneur, ayant permis 40-50 % de naissances vivantes par embryon transféré. Cette technique reste interdite par la religion dans les pays musulmans.
- Pour les patientes devant subir un traitement antitumoral, des techniques de cryoconservation sont proposées (34,35) utilisant soit des embryons, des ovocytes ou des fragments du cortex ovarien, suivi par la transplantation orthotopique et la maturation in vitro des oocytes. Cependant, bien que des résultats prometteurs aient été obtenus, les taux de grossesse restent très bas. Des techniques de thérapies cellulaires sont actuellement en cours d’expérimentation ciblant l’activation des follicules ovariens résiduels de la folliculogénèse ainsi que l’isolement de cellules souches ovariennes (36).
Références :
- H. Rozenbaum Ménopause -EMC d’endocrinologie nutrition [10- 035-A-10]
- A. Graff, S. Christin-Maitre : Insuffisance ovarienne prématurée EMC gynécologie [147-A-40]
- Cohen J, and al. Diminished ovarian reserve, premature ovarian fai- lure, poor ovarian responder – a plea for universal definitions. J Assist Reprod Genet. 2015
- Alipour F : Comparison of Specificity and Sensitivity of AMH and FSH in Diagnosis of Premature Ovarian Failure. Dis Markers. 2015 ;
- L. Webber and all. European Society of Human Reproduction and Embryology ESHRE 2016: « Management of women with premature ovarian insufficiency †: The ESHRE Guideline Group on POI, in Hu- man Reproduction, 2016.
- Risque psychol Shmidt JCEM 2011)
- Jacobsen B Kand al. Age at natural menopause and total mortality and mortality from ischemic heart disease: the Adventist Health Study in J Clin Epidemiol. 1999
- Roeters van Lennep JE, and all collaborators of the Dutch Multi- disciplinary Guideline Development Group on Cardiovascular Risk Management after Reproductive Disorders : Cardiovascular disease risk in women with premature ovarian insufficiency: A systematic review and meta-analysis in Eur J Prev Cardiol. 2016.
- Kalantaridou SN, and all : Impaired endothelial function in young women with premature ovarian failure : normalization with hormone therapy J Clin Endocrinol Metab. 2004
- Corrigan EC, and all : Effects of ovarian failure and X-chromo- some deletion on body composition and insulin sensitivity in young In Menopause. 2006
- Eshtiaghi R : Menopause is an independent predictor of metabolic syndrome in Iranian women In Maturitas. 2010.
- Knauff EA, and all Lipid profile of women with premature ovarian failure. Menopause. 2008
- Popat VB and alla : Bone mineral density in estrogen-deficient young women. J Clin Endocrinol Metab. 2009
- BakhshH and all Gynecol Endocrinol. 2015 Premature ovarianinsufficiency in young girls: repercussions on uterine volume and bone mineral density. in Gynecol Endocrinol. 2015.
- Scott EL and al. Premature menopause and risk of neurological disease: basic mechanisms and clinical implications. in Mol Cell En- docrinol. 2014.
- Ryan J and all : Impact of a premature menopause on cognitive function in later life. In BJOG. 2014
- Van der Klift M and al Risk factors for incident vertebral fractures in men and women: the Rotterdam Study. In J Bone Miner Res. 2004.
- Zhongguo Zhen Jiu. 2014. [Acupuncture for premature ovarian failure : a prospective cohort study].
- Langrish JP and all : Cardiovascular effects of physiological and standard sex steroid replacement regimens in premature ovarian fai- lure. Hypertension 2009.
- Sassarini J and al : Sex hormone replacement in ovarian failure – new treatment concepts. Best Pract Res Clin Endocrinol Metab. 2015 Jan.
- Soares PM, and all Breast density in women with premature ova- rian failure or post menopausal women using hormone therapy: ana- lytical cross-sectional study. Sao Paulo in Med J 2010.
- Furness S and al. Hormone therapy in postmenopausal women and risk of endometrial hyperplasia. Cochrane Database Syst Rev 2012.
- Antoine C, and all : Safety of hormone therapy after breast cancer : a qualitative systematic review.in Hum Reprod 2007.
- Rebbeck TR et al. Effect of short-term hormone replacement therapy on breast cancer risk reduction after bilateral prophylactic oophorectomy in BRCA1 and BRCA2 mutation carriers : the PROSE Study Group. J Clin Oncol 2005.
- Dunselman GA et al. ESHRE guideline : management of women with endometriosis in Hum Reprod 2014.
- Nappi RE and all : Course of primary headaches during hormone replacement therapy in Maturitas 2001 ;
- Ciarmela P, Ciavattini A, Giannubilo SR, Lamanna P, Fiorini R, Tranquilli AL, Christman GM, Castellucci M. Management of leio- myomas in perimenopausal women in Maturitas 2014
- Langrish JP and all : Cardiovascular effects of physiological and standard sex steroid replacement regimens in premature ovarian fai- lure. In Hypertension 2009
- Rada and al. : Non-hormonal interventions for hot flushes in women with a history of breast cancer. in Cochrane Database Syst Rev 2010:
- Kim TJ, and all. Routine endocrine screening for patients with ka- ryotypically normal spontaneous premature ovarian failure in. Obstet Gynecol 1997.
- Goswami R, and all. Prevalence of thyroid autoimmunity in spo- radic idiopathic hypoparathyroidism in comparison to type 1 diabetes and premature ovarian failure. J. Clin Endocrinol Metab 2006 ;
- Check JH. : Premature ovarian insufficiency – fertility challenge. Minerva Ginecol. 2014.
- Gougeon A and all : [Present and future strategies for women at risk, or suffering from premature ovarian failure (POF)] in Gynecol Obstet Fertil. 2012.
- Blumenfeld Z and al : Gonadotropin-Releasing Hormone Agonist Cotreatment During Chemotherapy May Increase Pregnancy Rate in Survivors in Oncologist. 2015.
- Ravel C and al : Fertility preservation in pre-pubertal girls with cancer: the role of ovarian tissue cryopreservation in Gynecol Obstet Fertil. 2016
- Silvestris [Ovarian failure : New treatments in perspective ?].J Ovarian Res. 2015.