M.I. KEDIHA, S. NOUIOUA, L. ALI PACHA, M. TAZIR, Service de Neurologie, CHU Mustapha Bacha, Alger.
Résumé : Les accidents vasculaires cérébraux (AVC) sont une pathologie fréquente et grave dont la prise en charge à la phase aigüe constitue une urgence diagnostique et thérapeutique. 85 % de ces AVC sont des infarctus cérébraux qui peuvent bénéficier d’une prise en charge spécialisée et d’un traitement thrombolytique. Cette prise en charge urgente s’articule autour d’une filière de soin (filière neurovasculaire) et d’unités spécialisées « unités neurovasculaires ou UNV » avec une équipe multidisciplinaire et ce, afin d’administrer le rt-PA dans un délai maximum de 4h30 après l’installation de la paralysie. Les UNV commencent à se développer en Algérie mais pour augmenter le nombre de patients correctement traités, il faut améliorer la connaissance du grand public et du médecin sur les signes d’alerte, sur la conduite à tenir et sur la notion de temps limité « Time is Brain ».
Mots-clés :
Abstract : Stroke is a common and serious pathology whose management in the acute phase is a diagnostic and therapeutic emergency. 85% of these strokes are cerebral infarctions that can benefit from specialized management and thrombolytic therapy that has been shown to be effective. This urgent care is organized around a neurological care network and specialized neurovascular units with a multidisciplinary team, in order to administer the RT-PA within 4h30 after installation of the paralysis. These units are beginning to develop in Algeria but to increase the number of properly treated patients, it is necessary to improve the knowledge of the general public and the doctor on the warning signs, on the conduct to be held and on the notion of limited time. “ Time is Brain ”.
Key-words : Stroke, emergency, thrombolysis
Introduction :
D’après l’OMS, l’accident vasculaire cérébral (AVC) est «le développement rapide de signes cliniques localisés ou globaux de dysfonction cérébrale avec des symptômes durant plus de 24 heures, pouvant conduire à la mort, sans autre cause apparente qu’une origine vasculaire «. On parle aussi d’attaque cérébrale, d’apoplexie et le terme courant anglais est « stroke ».
Les accidents vasculaires cérébraux (AVC) posent un problème majeur de santé publique car ils représentent une pathologie fréquente avec un risque de mortalité et de handicap très élevé. Les AVC touchent plutôt les sujets âgés, avec un âge médian de 77 ans (1). Toutefois, 1/4 d’entre eux surviennent avant 65 ans (1).
L’incidence en Algérie est passée de 60 (1984) à 201/100.000 habitants (2010) (2) et elle devrait continuer à augmenter avec le vieillissement de la population.
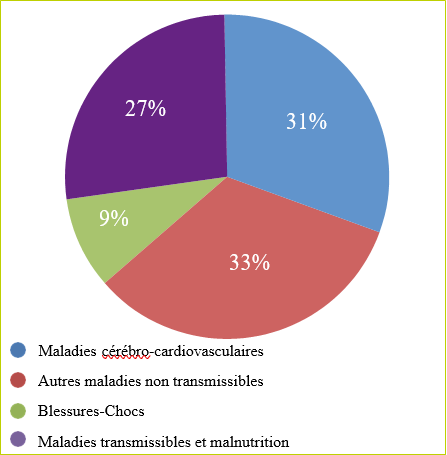
Il y a plus de 6,2 millions de décès dans le monde par an (données OMS). Les AVC constituent la première cause de décès chez la femme et la troisième chez l’homme (Figure 1) ; ils sont la première cause de handicap acquis de l’adulte, (75 % des survivants l’étant avec séquelles), et ils représentent aussi la seconde cause de démence, après la maladie d’Alzheimer (3).
Les séquelles des AVC rendent souvent la réinsertion sociale et professionnelle difficiles puisque les infrastructures familiales et environnementales n’y sont pas toujours adaptées.
Sur le plan thérapeutique, les infarctus cérébraux ont cessé d’être une fatalité brutale, imprévisible et incurable mais ils doivent être considérés comme l’une des plus grandes urgences médicales depuis l’avènement du rt- PA, premier traitement de reperfusion artérielle cérébrale ou thrombolyse.
Cette thrombolyse s’effectue dans des unités de soins spécialisées appelées unités neurovasculaires (UNV) qui commencent à fleurir dans notre pays.
Unité neurovasculaire : quelques définitions (4)
L’UNV est une unité fonctionnellement identifiée et reconnue, comprenant des lits de soins intensifs neurovasculaires et des lits d’hospitalisation « classiques » dédiés. L’UNV de territoire ou de référence est la structure pivot d’une filière organisée pour la prise en charge des AVC ; elle peut être dite « UNV de recours », si elle dispose de l’appui de services de neurochirurgie et de neuroradiologie interventionnelle.
L’UNV de proximité (à l’intérieur du pays) est chapeautée par une UNV de référence (intérêt de la téléthrombolyse). Ces établissements doivent accueillir les patients qui
vivent à moins de 30 minutes du service des urgences. Plusieurs UNV de référence ont vu le jour en Algérie : Blida (CHU Frantz Fanon), Oran (EHU), Tlemcen (CHU), Alger (CHU Mustapha et Bab el oued), Tizi Ouzou, Sétif et Constantine et quelques rares UNV de proximité (Médéa, Ain Defla).
L’UNV accueille et prend en charge 24h/24 et 7 jours/7 des patients présentant une pathologie neurovasculaire aigüe. Il a été démontré que les UNV réduisent le risque de décès de 18% et celui de garder un handicap invalidant de 21 % par rapport à une prise en charge non spécialisée (5).
L’UNV assure un rôle d’expertise diagnostique et thérapeutique permettant (6) :
- D’effectuer un bilan diagnostic précis et précoce,
- D’assurer la surveillance de l’état neurologique et des différents paramètres : pouls, tension artérielle, saturation en oxygène, température, glycémie,
- De débuter rapidement les traitements médicamenteux et la rééducation,
- De prévenir au mieux les complications secondaires,
- D’informer le patient et sa famille,
- De mettre en place le plus précocement possible, le projet de réadaptation et de réinsertion du patient,
- D’assurer une évaluation pluri professionnelle des patients 4 à 6 mois après l’accident,
- De proposer, si nécessaire, des programmes d’éducation thérapeutique portant sur la prévention des récidives et des complications secondaires,
- D’effectuer la formation initiale et continue des personnels médicaux et paramédicaux,
- De conduire et ou de participer à des actions de recherche.
La filière neurovasculaire :
L’UNV est insérée dans une filière organisée et dynamique. Il est recommandé de (6) :
- Préparer, en amont de l’accueil des patients, une filière bien coordonnée (phase pré-hospitalière) en informant la population générale et les médecins traitants (sensibilisation par affiches, médias…) et ce, afin d’optimiser le délai de prise en charge des AVC depuis le domicile du patient.
- Organiser l’accueil du patient AVC dans l’établissement où tout doit être mis en œuvre pour obtenir rapidement un avis spécialisé et une imagerie avec accès en urgence et prioritaire 24h/24 ainsi que la disponibilité de lits dans l’UNV pour accueillir en urgence les AVC.
Formaliser dans des protocoles, les modalités de recours au plateau technique (biologie, neuro imagerie, explorations ultra sonores cervicales, transcraniennes et cardiologiques), et à l’expertise de médecins spécialistes (cardiologues, neurochirurgiens, neuroradiologues, chirurgiens vasculaires et réanimateurs) le cas échéant.
- Mettre en place des filières d’aval pour poursuivre la prise en charge de ces patients dans les services de soins de suites et de réadaptation,
- Organiser des formations pour l’ensemble des professionnels intervenant dans la prise en charge des AVC.
Il est à noter qu’en Algérie, peu de malades bénéficient d’une prise en charge en UNV avec thrombolyse car ils arrivent en majorité en ayant dépassé le délai et ce, par manque d’information et de sensibilisation, d’où l’intérêt d’optimiser cette filière en amont avec un combat quotidien (formations, informations, affiches, réseaux sociaux …) et avec la participation des médias lourds.
Diagnostic positif :
Les signes faisant suspecter un infarctus cérébral sont souvent méconnus du grand public et parfois des professionnels de santé.
Il est donc impératif d’alerter la population générale et d’améliorer sa connaissance sur la conduite à tenir en cas de suspicion d’AVC grâce notamment aux campagnes d’information répétées.
Devant toute survenue brutale de malaise ou de chute avec au moins l’un des signes suivants : déformation de la bouche et du visage (Face), chute d’un bras (Arm), difficulté pour parler (Speech) ; le patient ou toute personne témoin doit noter l’heure de survenue de ces symptômes (Time) et appeler les secours ou faire transférer le patient dans les plus brefs délais vers l’UNV la plus proche (Figure 2).

L’imagerie cérébrale :
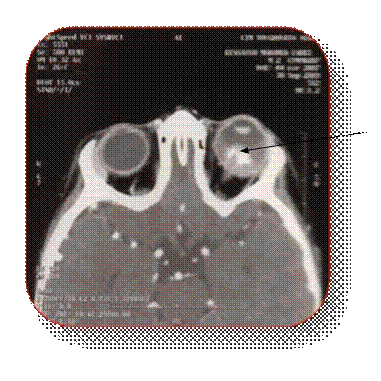
Seule l’imagerie permet d’éliminer une hémorragie cérébrale, étape fondamentale et obligatoire avant de procéder à tout traitement de phase aigüe par thrombolyse ou par traitement anti thrombotique. Cette imagerie cérébrale doit être réalisée, le plus rapidement possible, par scanner ou au mieux par
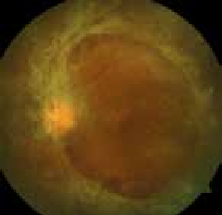
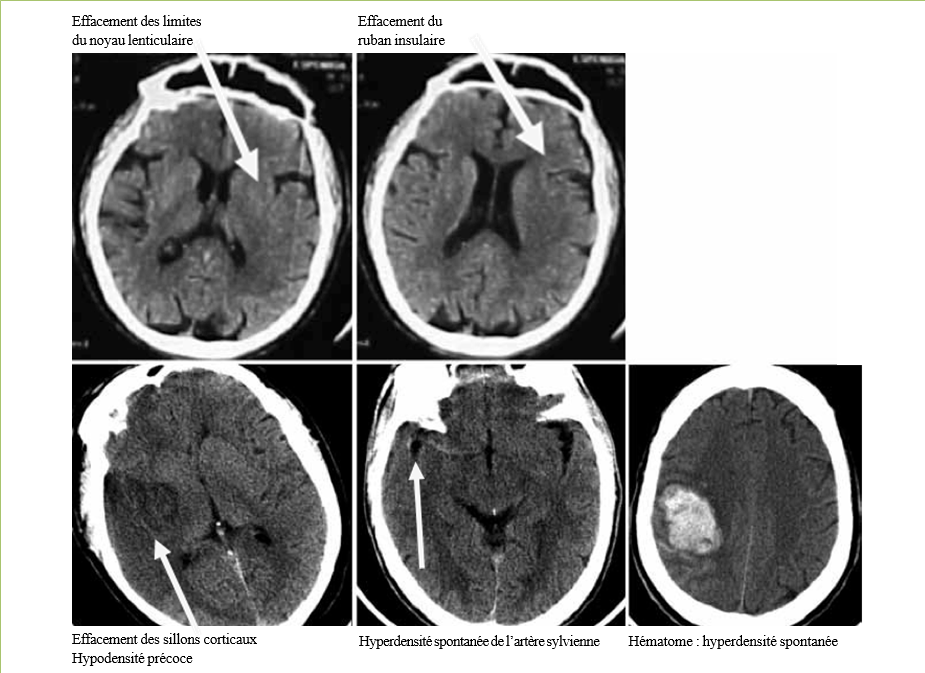
IRM. Le scanner a l’avantage de la disponibilité et de la rapidité d’examen. Il permet d’éliminer une hémorragie et d’identifier des signes précoces d’ischémie cérébrale : effacement des limites du noyau lenticulaire, effacement du ruban insulaire, effacement des sillons corticaux et l’hyperdensité spontanée de l’artère sylvienne (Figure 3)
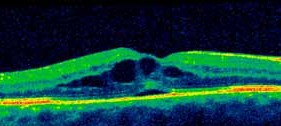
L’IRM cérébrale avec des séquences spécifiques (diffusion, FLAIR, echo de gradient T2, et ARM du polygone de Willis) permet de faire le diagnostic positif d’infarctus cérébral (même de petite taille ou situé dans la fosse cérébrale postérieure), et d’identifier d’autres lésions vasculaires d’âges différents (Figure 4).
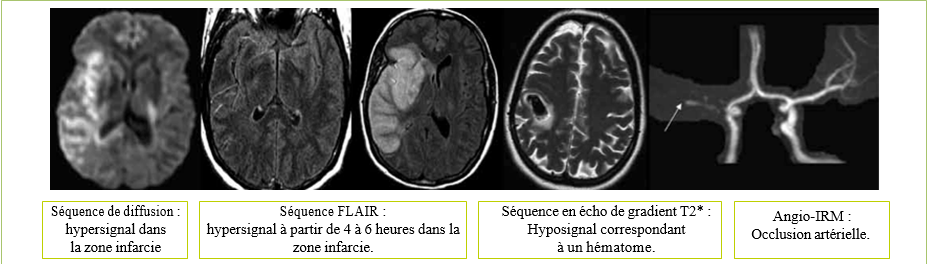
Physiopathologie : Notion de fenêtre thérapeutique étroite Après une occlusion artérielle à l’origine de l’infarctus, il se produit une souffrance neuronale qui reste réversible dans un délai étroit de 4h30 et c’est durant cette fenêtre qu’un seul traitement spécifique a démontré son efficacité la fibrinolyse par rt-PA intraveineux.
À chaque instant de la prise en charge d’un AVC, les différents intervenants doivent être guidés par le principe
« Time is Brain » et tenter de raccourcir au maximum ce délai entre le début des symptômes, l’arrivée à l’UNV (Symptom to Door) et l’administration du produit (Door To Needle).
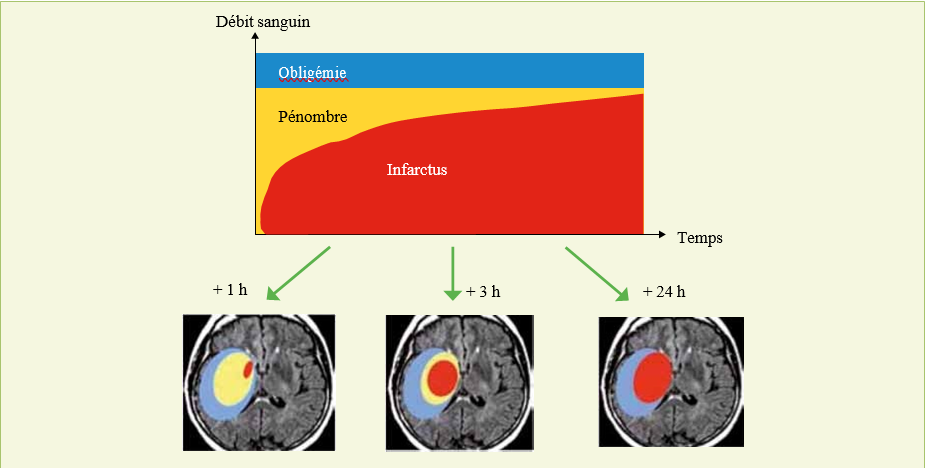
Sur le plan physiopathologique, l’objectif du traitement à la phase aigüe est de sauver « la pénombre ischémique » par la recanalisation artérielle afin d’éviter la croissance de l’infarctus au cours des premières heures (Figure 5). Cette recanalisation permet donc une meilleure récupération clinique à 3 mois et une diminution de la mortalité (7).
Prise en charge en urgence :
Cette prise en charge est organisée autour d’une filière de soins multidisciplinaires centrés autour de l’UNV et ce, notamment pour la prise en charge des patients éligibles à la thrombolyse.
Le patient doit, dans l’idéal, être évalué pendant la phase pré-hospitalière avec un transport qui doit être organisé le plus rapidement possible afin de bénéficier d’une expertise neurovasculaire et neuroradiologique d’urgence. Le transport se fera vers l’UNV la plus proche ou la structure hospitalière proche, capable de réaliser une thrombolyse (UNV de proximité), le cas échéant par télé
médecine, c’est-à-dire qu’une UNV de référence pourra guider, pas à pas, le médecin urgentiste qui pratiquera une thrombolyse et ce, en utilisant les technologies de communication et d’information modernes (via le Net). Pendant la phase hospitalière, le malade est admis dans l’UNV afin de bénéficier de la thrombolyse s’il reste toujours dans les délais requis. Une procédure d’alerte thrombolyse doit être déclenchée par l’équipe de l’UNV afin de réduire au maximum les délais entre l’arrivée du patient aux urgences et la thrombolyse. À l’arrivée, une évaluation neurologique est pratiquée à l’aide du score NIHSS (8), un bilan sanguin d’urgence est lancé, une imagerie cérébrale est faite, on recherche l’absence de contre-indications et la thrombolyse est donc initiée après réception des bilans biologiques et radiologiques.
Prise en charge thérapeutique :
La thrombolyse intraveineuse : Le seul produit ayant démontré un bénéfice est l’activateur recombinant du Plasminogene (rt-PA) en intraveineux à la dose de 0,9 mg/kg, à administrer dans les 4h30 après le début du déficit neurologique. Sa principale complication, en fait assez rare si on respecte les contre-indications, étant la transformation hémorragique qui conduit à l’aggravation de la destruction parenchymateuse (9).
La thrombectomie : consiste en l’extraction mécanique du thrombus dans une salle d’angiographie numérisée, dans un délai de 6 heures après le début de la paralysie, en introduisant une sonde dans l’artère occluse. Il s’agit d’un geste de neuroradiologie interventionnelle.
Autres traitements :
- L’aspirine à raison de 160 à 325 mg/jour est recommandée à la phase aigüe et reste bénéfique (10).
- Les anticoagulants : leur utilisation n’est pas recommandée à la phase aigüe, vu le risque hémorragique.
- Traitements généraux de la phase aigüe :
- Monitoring cardiaque, respiratoire et neurologique.
- Respect de la pression artérielle à la phase aigüe et ne la faire baisser que si PA supérieure 185/110 mmHg en cas de thrombolyse et si supérieure à 220/120 mmHg en absence de thrombolyse.
- Traitement de l’hyperthermie si supérieure à 38°.
- Traitement de l’hypo et l’hyperglycémie.
Prévention secondaire :
La prévention secondaire post-stroke est primordiale vu le risque accru de récidive (11). Cette prévention dite « vasculaire » concerne les patients ayant eu un accident ischémique transitoire (AIT) ou un infarctus cérébral (IC) après la phase aigüe. Elle comprend une prévention globale par le contrôle des facteurs de risque et un traitement spécifique en fonction de l’étiologie de l’IC ou de l’AIT. Il est donc primordial d’effectuer un certain nombre d’examens paracliniques en vue d’un diagnostic étiologique précis potentiellement traitable. Il faut pratiquer : une imagerie des artères cervicales et cérébrales (par angiographie, IRM ou Doppler), un Holter ECG (dépister une fibrillation auriculaire paroxystique) si ECG de base normal, une échographie cardiaque (dépister une cardiopathie emboligène). Un bilan de thrombophilie est également demandé en l’absence de cause cardiaque ou artérielle. De ce fait, il faut : un contrôle rigoureux des facteurs de risque vasculaire éventuellement décelés ou antérieurs à l’infarctus, à savoir : une hypertension artérielle, une dyslipidémie, un diabète, le tabac, l’alcool, l’obésité…etc.
Le traitement d’une fibrillation auriculaire paroxystique ou permanente par un traitement anticoagulant oral à maintenir au long cours (anti vitamine K « AVK », ou les anticoagulants oraux non AVK), d’un infarctus du myocarde par un traitement anticoagulant surtout si fibrillation auriculaire associée ou thrombus intracardiaque. Le traitement antithrombotique en cas d’athérosclérose, ou si cause indéterminée, par un antiagrégant plaquettaire type aspirine (75 à 325 mg/jour) ou clopidogrel (75 mg/jour). Leur association n’est plus recommandée en prévention secondaire.
Conclusion :
La prise en charge des AVC dans les unités neurovasculaires devrait constituer une règle absolue car son efficacité a été scientifiquement établie. En Algérie, malgré les efforts de certaines structures sanitaires qui se sont dotées d’UNV en dépit de difficultés, elles demeurent en nombre insuffisant devant le nombre grandissant d’AVC vu l’amélioration de l’espérance de vie et le vieillissement de la population. Par ailleurs, un effort particulier, concernant l’information et la formation du grand public et des professionnels de la santé, devrait être effectué afin de raccourcir les délais de prise en charge de l’AVC au sein de l’UNV.
Un plan national de prise en charge des AVC, avec la création d’UNV de référence et de proximité couvrant le territoire national, devra être établi par les autorités sanitaires avec l’aide des professionnels de santé concernés dans des délais appropriés, étant donné la fréquence élevée de cette pathologie et son impact sur la santé publique.
Références :
- A. Léger (urgences cérébrovasculaires, hôpital Pitié Salpetrière-Paris) AVC ischémiques : prise en charge à la phase aiguë. Revue générale vasculaire mai/juin 2014
- S. Kesraoui, N. Boutarene, Z. Yahiaoui, M. Arezki, Epidémiologie des AVC ischémiques dans la région de Blida Revue neurologique 167S(2011)
- HAS : recommandations mai 2009 AVC : Prise en charge précoce (Alerte, phase pré hospitalière, hospitalisation initiale, indications de la thrombolyse)
- Agence régionale de santé Ile-de-France. Cahier des charges des unités neurovasculaires (version 2013) Revue neurologique 2001 (Paris) ; 157 :11, 1447-56
- Stroke unit Trialist’s Collaboration. Organised inpatient (stroke unit) care for stroke. Cochrane Database Syst Rev 2007;(4):CD000197
- Société Française neurovasculaire. Recommandations pour la création d’unités neurovasculaires. Revue neurologique vol157 N°11-novembre 2001
- Rha JH, Saver JL. The impact of recanalization on ischemic stroke out- come : a metaanalysis. Stroke, 2007; 38:967-973
- Josephson SA, Hills NK, Johnston SC. NIH Stroke Scale Reliability in ratings from a large sample of clinicians. Cerebrovascular disord, 2006; 22:389-395
- Derex L, Nighoghossian N. Intracerebral hemorrhage after thrombolysis for acute ischemic stroke: an update. J. Neurol Neurosurg Psychiatry, 2008; 79: 1093-9
- Jaugh EG et coll. American heart association Stroke Council. Guide- lines for the early management of patients with acute ischemic stroke. Stroke, 2013; 44:870-947.
- HAS: Prévention vasculaire après un infarctus cérébral ou un accident ischémique transitoire Juillet 2014 (mise à jour février 2015)