L. ALI PACHA (1), T. BENHASSINE (2), S. ASSAMI (1), S. NOUIOUA (1), M. KOENIG (3), M. TAZIR (1). Service de Neurologie, CHU Mustapha Bacha et Laboratoire de Neurosciences, Université 1, Alger Laboratoire de biologie moléculaire, Université Houari Boumediene, Bab Ezzouar, Alger Institut de génétique et de biologie moléculaire et cellulaire (IGBMC) Ill kirch, Strasbourg, France
Résumé : L’ataxie-télangiectasie est l’une des plus fréquentes ataxies autosomiques récessives survenant avant l’âge de 5 ans, liée à la mutation du gène ATM sur le chromosome11q22.3. Elle se caractérise par une hypotonie, et des troubles de l’équilibre évoluant progressivement vers l’aggravation, entrainant la perte de la marche vers l’âge de 10 ans. L’ataxie cérébelleuse est souvent associée à des télangiectasies conjonctivales, une apraxie oculomotrice, des mouvements choréiques et dystoniques et une augmentation du taux de l’alfa-fœtoprotéine. Ces patients nécessitent une prise en charge particulière en raison notamment de leur prédisposition aux cancers (lymphomes et leucémies) et à des infections récurrentes liées à leur déficit immunitaire. Onze patients appartenant à 9 familles ont été diagnostiqués selon la clinique et les résultats biologiques. 56,52% des cas étaient issus d’un mariage consanguin. La maladie s’est déclarée dans 90 % des cas avant l’âge de 4 ans. Tous les patients présentaient un phénotype classique et 10% d’entre eux étaient sur fauteuil roulant.
Mots-clés : Ataxie, télangiectasies, alfa-fœtoprotéine.
Abstract : Ataxia telangiectasia is one of the most frequent autosomal recessive cerebellar ataxia, usually develop before 5 years of age, due to ATM mutation on chromosome 11q22.3. It’s characterized by hypotonia and clumsiness that progressively worsen, leading to the loss of independent ambulation by 10 years; cerebellar ataxia is usually associated with conjunctival telangiectasias, oculomotor apraxia, chorea, dystonia, or both; sensorimotor axonal neuropathy, and high level of alpha-fœtoprotein. Patients with ataxia telangiectasia must be monitored carefully, since they are prone to malignant conditions (especially lymphomas and leukemias) and to recurrent infections beginning at an early age because of immunoglobulin deficiency. Eleven cases belonging to nine families were ascertained for clinic and mutation analysis findings, according to which they were diagnosed as ataxia-telangiectasia. Physical, neurological, biologic, ophthalmological, cardiac examinations and cerebral magnetic resonance imaging or cerebral scanner were performed. 56,52% of cases were born of consanguineous marriage. The age of onset was before 4 years in 90% of cases, all the patients had classic phenotype and almost 10 % were wheelchair bound.
Key-words : Ataxia, telangiectasia, alfa-fœtoprotein.
Introduction :
L’Ataxie télangiectasie (AT) ou syndrome de Louis Barr est une maladie héréditaire à transmission autosomique récessive liée à une mutation du chromosome 11q22.3 codant pour le gène ATM. Elle concernerait environ une naissance sur quarante milles.
C’est une affection qui se manifeste par des anomalies neurologiques et conjonctivo-cutanées.
Le tableau classique associe une ataxie cérébelleuse progressive survenant entre un et quatre ans, une apraxie oculomotrice, des infections fréquentes secondaires à un déficit immunitaire essentiellement humoral, une choréo-athétose, des télangiectasies apparaissant tardivement mais surtout évidentes au niveau de la conjonctive et une augmentation du risque de cancer, en particulier de leucémie et de lymphome. Le diagnostic de cette maladie est relativement aisé en raison d’une élévation quasi systématique de l’alpha-fœtoprotéine ainsi que d’un déficit en Ig A et Ig E.
Patients et méthodes :
Cette étude est à la fois rétrospective et prospective et a concerné un nombre de 162 patients appartenant à 100 familles qui présentent une ataxie cérébelleuse autosomique récessive (ACAR) d’évolution progressive ; les cas sporadiques ont également été inclus dans ce travail.
Le registre rétrospectif s’est étalé sur la période allant de Janvier 2001 à Avril 2008, tandis que la partie prospective a concerné la période allant de Mai 2008 à Mars 2013.
Ces patients ont été adressés au service de Neurologie du CHU Mustapha Bacha, en général par des médecins généralistes, des neurologues, du secteur public ou privé, des pédiatres, des médecins rééducateurs, des médecins internistes, ainsi que des chirurgiens orthopédistes.
Pour chaque patient une anamnèse rigoureuse a été réalisée pour notamment rechercher la notion de consanguinité des parents, préciser l’âge de début de la maladie, les signes inauguraux, le mode évolutif, et l’existence de cas similaires dans la fratrie ou le reste de la famille avec établissement d’un arbre généalogique.
Chaque patient a bénéficié d’un examen neurologique détaillé, d’un bilan ophtalmologique et auditif ainsi qu’un bilan cardiaque comprenant un examen clinique, un électrocardiogramme et une échocardiographie. Nous avons également réalisé un bilan biologique standard pour chaque patient comprenant le dosage de la glycémie, du cholestérol et des triglycérides, une électrophorèse des lipides et des protéines, l’albuminémie, la créatine kinase, les lactates, l’étude des Béta hexosaminidases et de l’aryl sulfatase, ainsi que le dosage de la vitamine E et de l’alpha-fœtoprotéine. Un électromyogramme (EMG) avec étude des vitesses de conduction nerveuse, l’étude des potentiels évoqués visuels (PEV), auditifs (PEA) et somesthésiques (PES), ainsi qu’une tomodensitométrie (TDM) ou imagerie par résonnance magnétique (IRM) cérébrale ont été effectués. Le MMSE (minimental test examination) a également été réalisé. Après consentement éclairé, nous avons effectué pour tous les patients un prélèvement d’ADN du propositus, des parents, des membres atteints et sains de la fratrie en vue d’une étude génétique.
Pour chaque patient un score fonctionnel a été établi selon l’échelle SPATAX qui varie de 0 à 7 :
| • 0 : normal • 1 : discrètes anomalies à l’examen neurologique • 2 : impotence fonctionnelle légère, patient capable de marcher et de courir • 3 : capable de marcher sans aide jusqu’à 500 mètres, incapable de courir • 4 : nécessite une aide unilatérale pour marcher • 5 : nécessite une aide bilatérale pour marcher • 6 : confiné au fauteuil roulant • 7 : alité |
Résultats :
Nous avons colligé 11 patients appartenant à 9 familles atteints d’ataxie-télangiectasies, représentant 6,99% de tous nos patients ACAR.
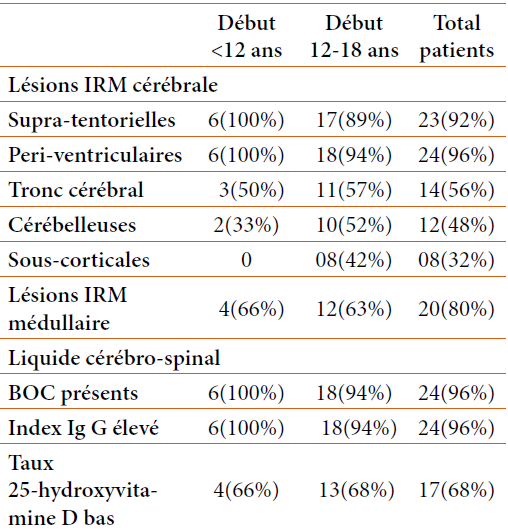
Grâce à l’étude moléculaire, le diagnostic définitif a été confirmé chez trois de ces patients. La consanguinité était retrouvée chez 6 familles. Les caractéristiques liées à l’âge de début et la durée d’évolution sont résumés dans le tableau 1.
| P1a | P2a | P3 | P4 | P5b | P6b | P7 | P8 | P9 | P10 | P11 | |
| Sexe | F | F | F | F | F | F | F | F | F | M | M |
| Consanguinité | + | + | – | + | + | + | – | + | – | + | + |
| Cas similaires | 1 sœur | 1 sœur | – | – | 2 sœurs | 2 sœurs | 1 frère décédé | 2 sœurs | 1 frère décédé | – | 1 frère décédé |
| Âge début | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 1 | 3 | 7 |
| Âge actuel | 7 | 8 | 7 | 9 | 7 | 10 | 4 | 7 | 5 | 8 | 13 |
| Durée évolution | 6 | 7 | 6 | 8 | 5 | 8 | 2 | 5 | 4 | 5 | 6 |
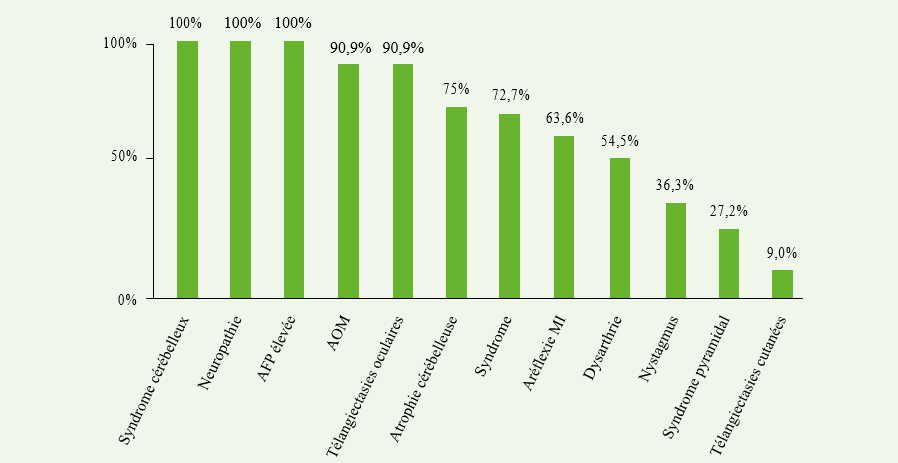
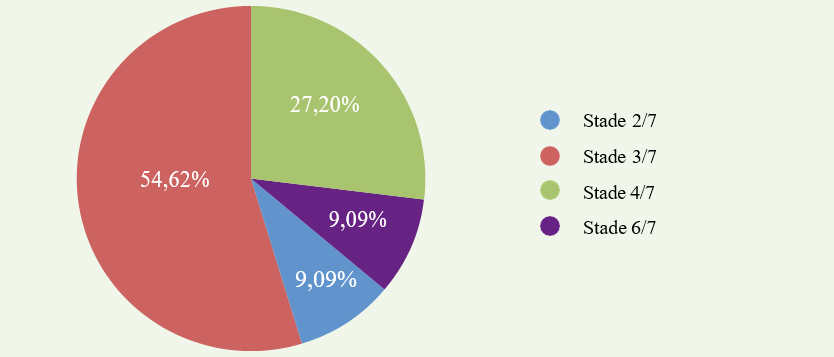
Chez un patient il a été noté une hypotonie néonatale, et 9 patients avaient des antécédents d’infections ORL ou pulmonaires récurrentes. Chez 4 patients (36%) l’affection s’est déclarée par un retard aux acquisitions psychomotrices, c’est-à-dire un retard à l’acquisition de la marche et du langage ; chez les 7 autres patients, la maladie s’est initiale- ment manifestée par des troubles de l’équilibre. Les signes cliniques retrouvés chez nos malades sont résumés dans le graphe 1 et les handicaps moteurs dans le graphe 2.
Le dosage de l’AFP, pratiqué chez 9 patients, était élevé dans 100% des cas, Il variait entre 2 et 40 fois la normale.
Le dosage des immunoglobulines, a été réalisé chez 6 patients, il était normal dans seulement un cas.
Chez 5 patients soit dans 83 %, il a montré 3 cas de déficit isolé des Ig A, un cas de déficit isolé des Ig G, et un cas de déficit combiné d’Ig A et Ig M.
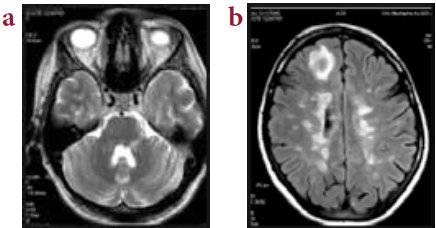
L’IRM cérébrale, réalisée chez 8 patients a montré chez 6 d’entre eux (75 %) une atrophie cérébelleuse, associée dans la moitié des cas à une atrophie corticale.
L’EEG pratiqué chez 5 patients était dénué d’anomalies, sauf dans un cas où il a montré un tracé globalement ralenti. Quant aux potentiels évoqués visuels, auditifs et somesthésiques, ils étaient normaux chez tous les patients qui en avaient bénéficié.
L’EMG a été pratiqué chez 5 patients et a montré dans tous les cas des signes de neuropathie axonale sensitive ou sensitivomotrice, dont une axonomyélinique (Tableau 2). TUGTGE
| P 3 | P5b | P6b | P10 | P11 | |
| Médian moteur droit | Nf | ||||
| LD (ms) | 2,46 | 3,25 | 3,8 | 2,85 | |
| A1 (mV) | 1,13 | 1,42 | 5,61 | 3,58 | |
| A2 (mV) | 1,41 | 2,04 | 5,01 | 3,21 | |
| VCN (m/s) | 55 | 51,9 | 56,7 | 47,5 | |
| Médian moteur gauche | Nf | Nf | |||
| LD (ms) | 2,45 | 3,95 | 2,55 | ||
| A1 (mV) | 2,71 | 3,69 | 4,38 | ||
| A2 (mV) | 2,38 | 3,61 | 3,07 | ||
| VCN (m/s) | 46,7 | 63,8 | 43,2 | ||
| Cubital Nf Nf | Nf | ||||
| LD (ms) | 2,8 | 2,3 | |||
| A1 (mV) | 4,61 | 2,44 | |||
| A2 (mV) | 4,55 | 2,56 | |||
| VCN (m/s) | 59,6 | 54 | |||
| Médian sensitf | Absent | ||||
| LD (ms) | 1,18 | 1,5 | 2,4 | 2,1 | |
| A (µV) | 1,54 | 1,5 | 1,25 | 2,78 | |
| VCN (m/s) | 49 | 56,7 | 45,8 | 52,4 | |
| SPE droit LD (ms) | 4,23 | 3,3 | 2,25 | 6,75 | 4,1 |
| A1 (mV) | 1,33 | 3,65 | 1,54 | 1,85 | 2,42 |
| A2 (mV | 1,17 | 2,95 | 1,25 | 1,47 | 2,12 |
| VCN (m/s) | 49 | 46,6 | 50,6 | 45,5 | 46,4 |
| SPE gauche LD (ms) | 4,28 | Nf | 3,7 | 8 | 4,05 |
| A1 (mV) | 1,33 | 2,03 | 1,55 | 2,26 | |
| A2 (mV) | 11,7 | 1,72 | 1,25 | 2,34 | |
| VCN (m/s) | 43 | 5,53 | 56,5 | 40,7 | |
| Sural droit | Nf | Nf | Nf | Nf | |
| LD (ms) | 1,5 | ||||
| A (µV) | 3,5 | ||||
| VCN (m/s) | 46,7 | ||||
| Sural gauche | Nf | Nf | Nf | Nf | |
| LD (ms) | 1,5 | ||||
| A (µV) | 2,5 | ||||
| VCN (m/s) | 47 | ||||
Pour l’étude moléculaire, la méthode consistait en un séquençage direct sur ADN génomique en BIG Dye terminator sur ABI 3100. Les résultats sont résumés dans le tableau 3.
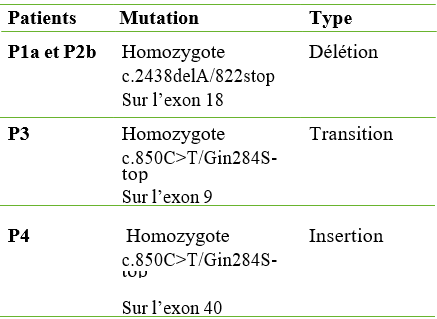
Discussion :
L’ataxie télangiectasie est une ataxie autosomique récessive dont le diagnostic doit être évoqué chez tout enfant entre 1 et 6 ans chez lequel survient une ataxie cérébelleuse associée à des télangiectasies oculaires. L’AT représente 6,99 % de notre série, occupant la quatrième place après l’ataxie de Friedreich, l’ataxie par déficit en vitamine E et l’ataxie avec apraxie oculomotrice type 2. Elle représente la forme d’ataxie la plus fréquente du nourrisson et de l’enfant avec une incidence sur la population générale estimée entre 1 et 5 cas pour 100.000 (1,2), ce qui correspond à une fréquence de porteurs hétérozygotes de 1/100 à 1/900 (3).
Pour certains, elle correspond probablement à la deuxième ataxie autosomique récessive la plus fréquente après la maladie de Friedreich (2,4).
Cette affection, à prédominance féminine, a débuté avant l’âge de 4 ans (91%) chez la majorité de nos patients. La plupart des auteurs ont rapporté un âge de début précoce, dans la petite enfance en général, entre 2 et 3 ans (5,6,7,8). Toutefois, des cas plus tardifs ont été décrits (9).
Principal motif de consultation, l’ataxie cérébelleuse représentait le premier symptôme de l’affection chez les 7 autres patients de notre série.
Sur le plan clinique, l’ataxie est le premier signe à apparaître, responsable d’une ataxie du tronc durant les premiers mois de la vie, s’étendant par la suite aux membres et responsable d’un retard à l’acquisition et/ou des comme un symptôme précoce et quasi constant qui précède en général la survenue des télangiectasies (1,7,14,9).
Les télangiectasies conjonctivales oculaires étaient présentes chez la grande majorité de nos patients (10 cas soit 90,90%), et les télangiectasies cutanées dans 2 cas (18,18%), siégeant au niveau des joues et des lobes des oreilles, et apparues très tôt chez la majorité des patients.
Les télangiectasies, qui représentent un élément essentiel du diagnostic (6), souvent invisibles au début de la maladie, sont présentes dans 90% des cas par la suite. Il s’agit de veinules dilatées, apparaissant vers 2 à 8 ans, d’abord au niveau des conjonctives oculaires, puis au niveau cutané.
Dans notre série, les mouvements anormaux étaient présents chez huit patients soit dans 72,72% des cas. Le pourcentage des mouvements choréiques et de la dystonie a été rapporté par divers auteurs et son taux estimé à environ 90% par certains (1,7,15). On a même décrit des cas où la dystonie était au premier plan et masquait l’ataxie (16).
Nous n’avons pas retrouvé de troubles de la sensibilité profonde, ce point distinguant l’atteinte neurologique de l’AT des autres ataxies récessives.
Dans cette affection, le pronostic fonctionnel est sévère, et les enfants se retrouvent sur fauteuil roulant vers l’âge de 10-11 ans (7,17,2), comme c’est le cas pour l’un de nos patients. Chez nos malades, le degré du handicap était corrélé souvent mais pas toujours à la durée d’évolution. Dans cette affection, la durée de vie n’est pas corrélée à la sévérité de l’affection neurologique et le décès survient avant l’âge de 20 ans en moyenne, celui-ci étant plutôt lié à des développements néoplasiques ou aux infections sino pulmonaires avec bronchectasie et insuffisance respiratoire (10). Cependant, depuis quelques années, et notamment à la faveur de l’introduction des immunoglobulines, la qualité de vie des patients a été améliorée et la durée de vie prolongée, grâce à une meilleure prise en charge des infections, des désordres immunologiques, et des néoplasies, certains patients pouvant vivre au-delà de 20 ans (certains patients ont entre 30 et 50 ans)(18,19).
Nous n’avons pas retrouvé de développement néoplasique chez nos patients, mais cet élément a été retrouvé parfois chez les frères ou sœurs plus âgés qui étaient atteints de leucémie en général ou de déficit en immunoglobulines et dont certains étaient décédés. Cette susceptibilité aux cancers, essentiellement des leucémies ou lymphomes, est dramatiquement élevée dans l’AT, et constitue l’un des principaux facteurs de mauvais pronostic de cette affection (20,2).
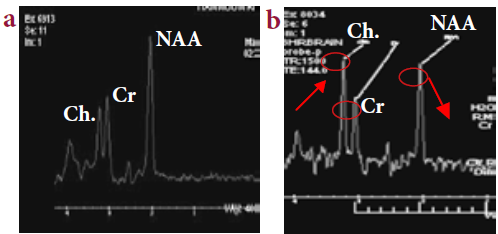
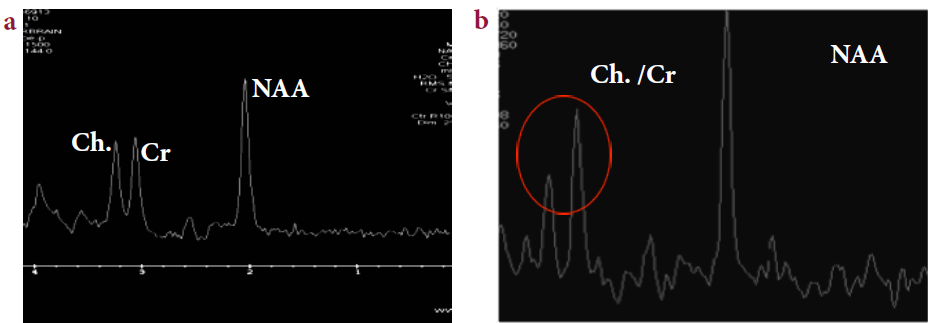
Les parents du sujet atteint sont obligatoirement hétérozygotes et ont, à un moindre degré une instabilité et une radiosensibilité chromosomique. Le risque de cancer est multiplié par 3,5 chez les hétérozygotes avec, chez la femme, un risque de cancer du sein multiplié par 5 (21). Plus de la moitié des patients testés avaient un taux d’AFP supérieur à 145u/ml, pas toujours corrélée à la sévérité du handicap ou à la durée d’évolution.
La majorité des auteurs rapporte l’augmentation du taux de l’AFP souvent au-delà de dix fois la normale. Cette anomalie constitue un bon marqueur biologique de cette affection (22,2). Le dosage des immunoglobulines, a été réalisé chez 6 patients ; il était normal dans seulement un cas.
Ces troubles immunologiques ont été estimés par les auteurs à 60 à 80% des cas (1). Cette immunodéficience, non progressive, peut ne pas se voir au début, et s’aggraver progressivement avec l’âge, d’où l’intérêt de faire des dosages répétés.
Le déficit en Ig A est le plus fréquent, les déficits en Ig G et Ig E sont plus rares.
On retrouve également dans cette affection des anomalies chromosomiques à type de translocations qui sont acquises, et dont la multiplicité serait caractéristique de la maladie. Pour beaucoup d’auteurs, la cytogénétique constitue donc un test simple et spécifique pour établir le diagnostic. Un test d’irradiation lymphocytaire, évaluant le taux de survie des lymphocytes après une irradiation de 1Gray, permet de mettre en évidence une sensibilité accrue des cellules aux radiations ionisantes (23).
La neuropathie sensitive ou sensitivomotrice est très fréquente dans l’AT, et son taux a été évalué à 71% par certains auteurs (7).
L’IRM cérébrale a montré une atrophie cérébelleuse chez les trois quarts de nos patients. Cette atrophie est fréquemment retrouvée, souvent au stade précoce. Il a même été décrit de rares cas de lésions de leuco-encéphalopathie (24).
Il est important de souligner que les tomodensitométries et les radiographies standards devraient être évitées autant que possible chez ces patients, en raison du risque accru de radiosensibilité.
Tous nos patients présentaient un phénotype classique mais il faut savoir qu’il existe des phénotypes moins sévères, présents dans 10 à 15% des cas, appelés « AT-Variant» où l’âge de début peut être plus tardif, l’évolution moins sévère, la radiosensibilité moins prononcée, et la durée de vie plus longue et/ou les signes cardinaux de la maladie peuvent manquer, rendant le diagnostic plus difficile (25,26).
Une forme particulière de l’AT variant est la forme AT Fresno combinant les signes du syndrome Nigmegen breakage (NMS) aux signes de la forme classique, caractérisée par une microcéphalie et un retard mental (25).
Génétique :
Le gène de l’ataxie télangiectasie est localisé sur le bras long du chromosome 11q22-23 et comporte 66 exons (22, 1) ; s’étalant sur environ 150kb (27). Il code pour une phosphatidyli-nositol-3 kinase, l’AT mutated protein (ATM) (28).
Les mutations sont très variées et s’étalent tout le long de la séquence codante, compliquant singulièrement leur identification.
Plus de 400 mutations réparties dans les 66 exons du gène ont été identifiées (29,30).
Près de 70% des mutations décrites sont des mutations ponctuelles entraînant la production d’une protéine tronquée (31) et vraisemblablement une perte de fonction (mutations nulles), en accord avec le mode de transmission de la maladie.
L’analyse moléculaire du gène ATM est difficile en raison de la longueur du gène (66 exons) et de la diversité des mutations réparties tout au long de ce gène, et c’est l’une des raisons majeures pour lesquelles seuls trois de nos patients ont bénéficié de l’étude génétique.
Le conseil génétique sera celui d’une maladie génétique à pénétrance complète.
Le diagnostic prénatal est de réalisation simple dans les familles d’AT, puisqu’il consiste en un simple haplotypage par une étude des microsatellites localisés dans le gène et de part et d’autre du gène.
Conclusion :
L’ataxie télangiectasie est une ataxie autosomique récessive dont le diagnostic doit être évoqué chez tout enfant entre 1 et 6 ans chez lequel survient une ataxie cérébelleuse associée à des télangiectasies oculaires. Elle représente l’une des plus graves ataxies de l’enfant en raison notamment du déficit immunitaire et de la prédisposition aux néoplasies qui en grèvent lourdement le pronostic.
Références:
- Gatti, R.A., Boder, E., Vinters, H.V., Sparkes, R.S., Norman, A., Lange, and K.: Ataxia-telangiectasia: an interdisciplinary approach to pantho- genesis. Medicine 1991 .70: 99-117.
- Chun HH, Gatti RA, Ataxia-telangiectasia. An evolving phenotype DNA repair (Amst) 2004; 3:1187-96
- Jeret JS, Lechtenberg R, Ataxia-telangiectasia. In: Lechtenberg R, ed. Handbook of cerebellar diseases. New York: Marcel Dekker 1993: 477-90.
- Anheim M. Les ataxies cérébelleuses autosomiques récessives. Rev Neurol (Paris) 2011, 167. ; 372-384
- Louis-Bar Mme. Sur un syndrome progressif comprenant des télangiec- tasies capillaires cutanées et conjonctivales symétriques, à disposition nae- voïde et des troubles cérébelleux. Conf Neurol (1941);4: 32-42.
- Sedgwick RP, Boder E (1991). Ataxia-telangiectasia. In: Vinken PJ, Bruyn GW, Klawans HL (eds) Handbook of clinical neurology Vol 16. Elsevier Scientific , NewYork,pp347-423
- Woods.CG and Taylor AM. Ataxia telangiectasia in the British Isles: the clinical and laboratory features of 70 affected individuals. Q J Med (1992) 82: 169-79.
- Taylor. AM, Byrd. PJ. Molecular pathology of ataxia-telangiecatasia. J Clin. Pathol. 2005; 58:1009-15.
- Moin M, Aghamohamadi A, Kouh A, Tavassoli S, Rezaei N, Ghaffar SR, Gharagozlou M, Movahedi M, Purpak Z, Mirsaeid Ghazi B, Mahmoudi M, Farhoudi A. Ataxia-telangiecatasia in Iran: clinical and labo- ratory features of 104 patients. Pediatr Neurol 2007 Jul; 37(1): 21-8.
- Swift M, Heim RA, Lench NJ. Inherited ataxias. In Harding AE, Deu- fel T. (eds), Advances in Neurology 1993.Vol.61,pp115-125. Raven Press. NewYork.
- Saunders-Pullman RJ, Gatti R. Ataxia-telangiecatasia: without ataxia or telangiecatasia. Neurology 2009 Aug11; 73(6):411-5.
- Waldman TA, Broder S, Goldman CK, Frost K, Korsmeyer SJ, Medici MA. Disorders of B-cells and helper T-cells in the pathogenesis of the immunoglobulin déficiency of patients with Ataxia-telangiecatasia. J Clin Invest 1983a, 71: 282-295.
- Border, E. Ataxia-telangiectasia: an overview. In: R.A., Gaetti, M., Swift (ends), Ataxia-telangiectasia: Genetics, Neuropathy, and Immunology of a Degenerative Disease of Childhood. Alan, R., Liss, New York, (1985). pp 1-63. 14-Lewis RF, Lederman.HM, Crawford The ocular motor abnormalities in ataxia telangiectasia. Ann Neurol (1999). 46:287-95.
- Carillo F, Schneider SA, Taylor AM, Srinivasan V, Kapoor R, Bhatra KP: Proeminent oromandibular dystonia and pharyngeal telangiectasia in atypical ataxia-telangiectasia. Cérébellum 2009 Mar;8(1):22-7.
- Bodensteiner MD, Randall M, Golblum MD, Armond S, Goldman MS. Progressive dystonia masking ataxia in Ataxia-telangiectasia. Arch Neurol 1980; 37(7):464-467.
- Perlman S, Becker-Catania S, Gatti RA. Ataxia-telangiectasia: dia- gnosis and treatment. Semin Pediatr Neurol 2003;10:173-182.
- Jabado N, Concannon P, Gatti RA. Ataxia-Telangiectasia. In handbook of ataxias édited by: Klockgether T. NewYork: Marcel Dekker, Inc;2000:164-190. 19-Nowak-Wegrzyn.A, Crawford.TO, Winkelstein JA, Carson KA, Le- derman.HM. Immunodeficiency and infections in ataxia telangiectasia. J Pediatr (2004); 144: 505-11.
- Morrel D, Cronartie E, Swift M. Mortality and cancer incidence in 263 patients with ataxia-télangiectasia. J Nat Cancer Inst 1986; 7789-9292.
- Stoppa-Lyonnet. D, Aurias, Ataxie telangiectasie. : Quel impact en cancérologie clinique. Bull Cancer (1992). 79: 645-650.
- Gatti RA, Berkel L, Boder E et al. Localization of an ataxia-telangiec- tasia gene to chromosome 11q 22-23. Nature (1988); 336: 577-80.
- Jasper NGI, Gatti RA,Baan C et al. Genetic complementation analy- sis of ataxia-télangiectasia and Nigmegen breakage syndrome: a survey of 50 patients. Cytogenet Cell Genet 1988; 49: 259-263.
- Chung EO, Bodensteiner JB, Noorani PA et al: Cerebral white-mat- ter changes suggesting leukodystrophy in ataxia telangiectasia. J Child Neurol (1994) 9:31-35.
- Gilad S, Chessa L, Khosravi R et al 1998. Genotype-phenotype relationships in ataxia-telangiectasia and variants. Am J Med Genet 1998; 62:551-561.
- Savozzi S, Saluto A, Taylor AMR et al. A late onset variant of ataxia-te- langiectasia with compound heterozygous genotype, A8030G/7481insA. J Med Genet 2002, 39:57-61.
- Savitsky K, Platzer M, Uziel T, Gilad S, Sartiel A, Rosenthal A, Elroy- Stein O, et al. Ataxia-telangiectasia: structural diversity of untranslated sequences complex posttranscriptional regulation of ATM gene expres- sion. Nucleic Acids Res1997; 25:1678-1684.
- Watters D, Khanna KK, Beamish H, Birrel G, Spring K, Kedar P, Gatei M,et al. Cellular localization of the ataxia-telangiectasia (ATM) gene product and discrimination between mutated and normal forms. Oncogène1997; 14:1911-1921.
- Li A, Swift M 2000.Mutations at the ataxia-telangiectasia locus and clinical phenotypes of A-T patients. Am J Med Genet 2000, 92 :170-177. 30-Campbell C, Mitui M, Engert. L, Coutinho G, Thorstenson Y, Gatti RA. ATM mutation on distinct SNP and STR haplotypes in ataxia-te- langiectasia patients of differing ethnicities reveals ancestral founder effects. Hum Mutat (2003) 21: 80-5.
- Telatar M, Teraoka S, Wang Z, Chun HH, Liang T, Castelvi-Bell S, Udar N, Borresen Dale AL, Chessa L, Bernatowska Matuszkiewick E, Porras O, Watanabe M, Junker A, Concannon P, Gatti RA 1998. Ataxia- telangiectasia: identification and detection of fouder effect mutations in the ATM gene in ethnic populations. Am J Hum Genet1998; 62:86-97.