A.F. BOUTALEB(1), H. SAOULA(1) M. AISSAOUI(1), D. HAMIDOUCHE(1), R. OSMANE(1), H. MAHIOU(1), Y. AISSAT(1), A. MITICHE(1), Y. ZMIRI(1), F. HAMCHAOUI(2), M. NAKMOUCHE (1) Service d’Hépato-gastro-entérologie, Service d’Epidémiologie, CHU Lamine Debaghine, Bab El Oued
Résumé : La maladie cœliaque est une entéropathie auto-immune secondaire à la consommation de gluten. Les manifestations cliniques sont très variées et non spécifiques. L’objectif de ce travail est de déterminer la fréquence des formes cliniques de la maladie cœliaque (typique, atypique et silencieuse) au sein d’une population de malades cœliaques adultes et d’étudier leurs caractéristiques cliniques et biologiques ainsi que d’établir le délai diagnostique de la maladie en fonction de la forme clinique.
Le recrutement des patients cœliaques s’est effectué de Janvier 2009 à Janvier 2014. Le diagnostic de la maladie cœliaque chez nos patients tous âgés de plus de 16 ans, repose sur les critères cliniques, sérologiques, histologiques de la maladie cœliaque. Les patients sont classés selon la symptomatologie révélatrice en formes typique, atypique ou silencieuse de maladie cœliaque (classification d’Oslo). Les différentes caractéristiques cliniques et biologiques sont analysées. Le délai diagnostique est déterminé grâce à l’interrogatoire, il est défini par la période qui sépare le début des symptômes à la date du diagnostic.
Nous avons diagnostiqué 317 patients cœliaques. 78,8% ont une forme atypique et 21,2% ont une forme typique. Aucune forme silencieuse n’a été retrouvée. La forme typique est plus sévère que la forme atypique. En effet, elle est caractérisée par un retentissement important sur le plan clinique (moyenne du BMI=19,44 versus 21,15 pour la forme atypique, p=0,0005) et un syndrome carentiel plus sévère. Les manifestations extra-digestives sont observées dans les deux formes cliniques sans différence statistiquement significative. Le délai diagnostique de la plupart de nos patients (54,2%) est compris entre 1 et 10 ans. Un diagnostic tardif (au-delà de 10 ans) est posé chez 15,6% des malades.
La forme atypique de la maladie cœliaque est la forme clinique prédominante dans notre population de malades cœliaques. Il s’agit d’une forme clinique moins sévère que la forme typique. Le délai diagnostique de la maladie cœliaque est le plus souvent compris entre 1 et 10 ans. Un diagnostic tardif est fait chez 15,6% des patients.
Mots-clés : Maladie ccoeliaque, formes cliniques, delai diagnostique.
Abstract : Coeliac disease is an autoimmune reaction to gluten. The clinical manifestations are variable and not specific. The objective of this study was to determine typical, atypical and silent cases according to presenting symptoms and to evaluate their clinical and biochemical parameters. We also aimed to determine diagnosis delay of coeliac disease (interval between first symptoms and diagnosis).
317 adults patients with coeliac disease in our department between January 2009 and January 2014 were diagnosed. The coeliac diagnosis was made by serological and histological examinations. Patients were divided according to their symptoms into typical, atypical or silent form of coeliac disease.
Patients presenting atypical form of coeliac disease were predominant (78,8%). Those with typical signs were present in 21,2%. Silent form was absent in our cohort. The typical form of coeliac disease was more severe than the atypical form. In fact, patients with typical symptoms had severe clinical impact of the disease. The average of BMI (body mass index) in typical form was 19,44 versus 21,15 for the atypical form (p=0,0005). Nutrional deficiencies were also more severe. Extra-digestive clinical features were similarly observed in the two forms. In 54,2%, diagnosis delay was between 1 and 10 years. Prolonged diagnosis delay (>10 years) was seen in 15,6% of cases. Atypical form of coeliac disease was predominant in our study. It was less severe than the typical form. Diagnosis delay was between 1 and 10 years in the majority of patients. The diagnosis was considered delayed in 15,6% of cases.
Key-words : Coeliac disease, clinical forme, diagnosis delay.
Introduction :
La maladie cœliaque est une entéropathie auto-immune qui survient chez des sujets prédisposés génétiquement (HLA QQ2 ou DQ8) et consommant du gluten[1].
Sa prévalence est de 1% environ dans les pays d’Europe et d’Amérique du nord[2,3]. La prévalence dans les pays du Maghreb semble tout aussi élevée. En effet, une étude récente ayant fait l’objet d’une thèse réalisée dans une population d’écoliers de la wilaya d’Alger, a retrouvé un taux de 0,59% de cœliaques[4].
La maladie cœliaque est une maladie protéiforme. Les manifestations cliniques sont très variées. Les symptômes digestifs peuvent être frustes, voire absents[5]. Les signes extra-digestifs peuvent être prédominants ; le diagnostic n’est alors pas évoqué précocement, engendrant ainsi un retard diagnostique[7].
En Algérie, il existe très peu d’études qui concernent la maladie cœliaque. La plupart ont fait l’objet de thèse mais n’ont pas encore été publiées.
L’objectif de ce travail est de déterminer la fréquence des formes cliniques de maladie cœliaque (typique, atypique et silencieuse) au sein d’une population de malades cœliaques adultes et d’étudier leurs caractéristiques cliniques et biologiques ainsi que d’établir le délai diagnostique de la maladie en fonction de la forme clinique.
Matériels et méthodes :
Notre étude est transversale, observationnelle et prospective. Le recrutement des patients cœliaques s’est effectué de Janvier 2009 à Janvier 2014. Il est essentiellement réalisé au niveau de la consultation du service de gastro-entérologie du CHU Lamine Debaghine, de Bab El Oued. Certains patients proviennent des autres services du CHU Lamine Debaghine de Bab El Oued avec lesquels nous avons collaboré ; ce sont les services de médecine interne, de rhumatologie, d’endocrinologie et de pédiatrie (individus de plus de 16 ans). De rares patients proviennent des autres CHU (Mustapha Bacha, Hôpital Central de l’Armée).
Le diagnostic de maladie cœliaque chez nos patients tous âgés de plus de 16 ans, repose sur les critères diagnostiques de l’ESPGHAN (European Society for Pediatric Gastroenterology, Hepatology and Nutrition), à savoir, des symptômes cliniques évocateurs, une sérologie positive, une histologie typique et une réponse au traitement.
Les patients sont classés en forme typique, lorsque la diarrhée chronique et le syndrome carentiel sont les manifestations révélatrices de la maladie cœliaque. Les patients qui ont des symptômes digestifs banals comme une constipation, un ballonnement, des douleurs abdominales ou un reflux gastro-œsophagien, sont considérés comme ayant une forme atypique. Des manifestations extra-digestives peuvent exister, elles peuvent dominer le tableau clinique, la maladie cœliaque est dite aussi atypique. Les patients peuvent être asymptomatiques et sont diagnostiqués grâce au dépistage sérologique. On parle alors de maladie cœliaque silencieuse.
Une fois le diagnostic posé, les renseignements concernant le patient sont reportés sur une fiche préétablie comprenant : les données démographiques, le délai diagnostique, l’examen physique avec le calcul du BMI (body mass index) selon les données du national « heart, lung and blood institute », la recherche de signes carentiels tels que la pâleur cutanéo-muqueuse, les troubles des phanères, le retard staturo-pondéral et/ou pubertaire et les résultats des examens biologiques. Ceux-ci comprennent : la formule de numération sanguine, la ferritinémie, le bilan lipidique, l’albuminémie, le bilan phosphocalcique, la vitamine D, le taux de prothrombine, la glycémie, le bilan hépatique et le bilan thyroïdien.En cas de suspicion d’une hépatopathie, un bilan étiologique est pratiqué.Une ostéodensitométrie osseuse est réalisée chez tous les patients au moment du diagnostic.
Analyse statistique :
L’analyse des données est faite sur le logiciel EPI info et le logiciel SPSS. Les tests statistiques utilisés sont le test de khi deux (variables qualitatives) pour la comparaison de pourcentages et le test de Student (variables quantitatives) pour la comparaison de deux moyennes.
Résultats :
Le nombre de patients cœliaques retenus pour cette étude est de 317 patients dont la grande majorité est féminine (258 femmes pour 59 hommes). Le sex-ratio (H/F) est de 0,23. L’âge moyen de la population est de 31,64 ans avec des extrêmes de 17 à 72 ans. Les patients qui ont 50 ans et plus sont au nombre de 22, ce qui représente 7% de la population cœliaque. Le délai diagnostique a pu être déterminé chez 153 patients soit dans 48,3% des cas. Ainsi, dans 7,2% des cas le diagnostic est précoce (moins de 6 mois), dans 23% il est compris entre 6 et 11 mois ; 54,2% ont un délai diagnostique compris entre 1 et 10 ans, enfin 15,6% ont un diagnostic tardif c’est-à-dire au-delà de 10 ans Le symptôme digestif le plus fréquent est le ballonnement abdominal (50%), les douleurs abdominales sont présentes dans 43,2%, la diarrhée chronique dans 21%, les vomissements chroniques dans 10%, la constipation chronique dans 8,8%, la dyspepsie dans 7,2%, et enfin le reflux gastro-œsophagien dans 2,5%.
4,7% des patients ont des symptômes entrant dans le cadre du syndrome de l’intestin irritable selon les critères de Rome III (dans sa forme diarrhée-constipation). Voir l’histogramme représentant les symptômes digestifs.
Nos patients ont ainsi été classés : 21,2% ont une forme typique de maladie cœliaque et la grande majorité des patients, soit 78,8% ont une forme atypique. Aucun de nos patients n’a de forme silencieuse de maladie cœliaque.
Le BMI (body mass index) moyen de toute la population cœliaque est normal (20,78 +/-3,6). La moyenne du BMI des patients ayant une forme atypique (21,15+/-3,62) est supérieure à celle de la forme typique (19,44 + /-3,19) de façon statistiquement significative (p=0,0005). À l’examen physique, les signes carentiels (pâleur cutanéo-muqueuse, troubles des phanères et de la peau, retard staturo-pondéral et/ou pubertaire) ne sont présents que dans 25% des cas. Ils sont plus fréquents chez les patients ayant une forme typique mais de façon non significative (p=0,09). Le tableau 1 reporte les différents signes carentiels selon la forme clinique.
| Signes carentiels | Total | Forme atypique | Forme typique |
| Pâleur | 72 (22,7%) | 50 (20%) | 22 (32,8%) |
| Troubles de la peau et des | 59 (18,6%) | 35 (14%) | 24 (35,8%) |
| phanères | |||
| Retard staturo-pondéral | 19 (6%) | 11 (4,4%) | 8 (12%) |
| et/ou pubertaire |
Sur le plan biologique, l’anémie est la carence la plus fréquente (83%), elle est retrouvée le plus souvent dans la forme typique (p=0,03). Dans les deux formes cliniques, l’anémie est ferriprive et microcytaire hypochrome dans la quasi-totalité des cas. Le tableau 2 représente les taux d’anémie et ses caractéristiques selon la forme clinique. Les autres paramètres du syndrome carentiel sont significativement plus présents dans la forme typique, sauf pour la vitamine D (le p n’est pas significatif). Dans le tableau 3 sont rapportés les différents paramètres biologiques du syndrome carentiel selon la forme clinique.
| Anémie et ses caractères | Total | Forme atypique | Forme typique |
| Anémie absente | 54 (17%) | 49 (19,6%) | 5 (7,4%) |
| Anémie présente | 263(83%) | 201 (80,4%) | 62 (92,5%) |
| p=0,03 | |||
| Microcytaire hypochrome | 261 (99,2%) | 200 (80%) | 61 (91%) |
| Mégaloblastique | 2 (0,8%) | 1 (0,4%) | 1 (1,5%) |
| Fer sérique normal | 30 (9,6%) | 28 (11,4%) | 2 (3%) |
| Fer sérique bas | 283 (90,4%) | 218 (88,6%) | 65 (97%) |
| Ferritinémie normale | 10 (7%) | 9 (8,1%) | 1 (3%) |
| Hypoferritinémie | 134 (93%) | 102 (91,9%) | 32 (97%) |
| p=0,28 |
| Paramètres | Total | Forme atypique | Forme typique |
| Calcémie normale | 231 (74,5%) | 198 (81,2%) | 33 (50%) |
| Calcémie basse | 79 (25,5%) | 46 (18,8%) | 33 (50%) |
| p=0,000001 | |||
| Vitamine D normale | 50 (30%) | 42 (31,4%) | 8 (24,2%) |
| Vitamine D | |||
| Basse | 117 (70%) | 92 (68,6%) | 25 (75,8%) |
| p=0,42 | |||
| Lipides | |||
| Normaux | 230 (74%) | 196 (80,3%) | 34 (50,7%) |
| Lipides bas | 81(26%) | 48 (19,7%) | 33 (49,2%) |
| p=0,000001 | |||
| Albuminémie normale | 258 (82,4%) | 222 (89,9%) | 36 (54,5%) |
| Albuminémie basse | 55 (17,6%) | 25 (10,1%) | 30 (45,5%) |
| p=0,000001 | |||
| TP normal | 300 (94,6%) | 243 (97,2%) | 57 (85%) |
| TP bas corrigé | |||
| par la vit K | 10 (3,2%) | 4 (1,6%) | 6 (9%) |
| TP bas non | |||
| corrigé/ Vit K | 7 (2,2%) | 3 (1,2%) | 4 (6%) |
| p=0,006 | |||
De façon générale, les signes extra-digestifs sont présents aussi bien dans la forme atypique que dans la forme typique.
L’anémie récidivante est le symptôme extra-digestif le plus fréquemment révélateur de la maladie cœliaque. En effet, 86,7% des patients ont eu une anémie.
Le taux d’anémie chute à 83% au moment du diagnostic en raison du traitement martial administré aux patients avant le diagnostic de la maladie. Les manifestations osseuses sont présentes dans 42,3%.
Les autres manifestations articulaires (9,1%), buccales (6,6%), hépatiques (6,3%) et neurologiques (4,7%) sont moins fréquentes. Le tableau 4 représente les différentes manifestations extra-digestives selon la forme clinique.
| Signes extra- digestifs | Total | Forme atypique | Forme typique |
| Anémie | 275 (86,7%) | 210 (84%) | 65 (97%) |
| Manifestations hépatiques | 20 (6,3%) | 15 (6%) | 5 (7,5%) |
| Arthralgies | 29 (9,1%) | 24 (9,6%) | 5 (7,4%) |
| Manifestations neurologiques | 15 (4,7%) | 14 (5,6%) | 1 (1,5%) |
| Aphtose buccale récidivante | 21 (6,6%) | 18 (7,2%) | 3 (4,5%) |
| Manifestations osseuses | 134 (42,3%) | 107 (42,8%) | 27 (40,3%) |
| p=0,55 | |||
Les manifestations osseuses sont représentées essentiellement par l’ostéopénie (67,4%), l’ostéoporose (26,8%) et l’ostéomalacie (5,8%).
Les hépatopathies associées à la maladie cœliaque sont représentées par les cytolyses cryptogénétiques bénignes (56%) et les hépatopathies plus sévères dysimmunitaires ou non dysimmunitaires. Dans cette étude, ces dernières sont révélatrices de la maladie cœliaque dans 100% des cas. Il s’agit de cirrhoses dysimmunitaires (15,8%), de cirrhoses cryptogénétiques (7%), d’hypertensions portales sans cirrhose (17,6%), de syndrome de Budd Chiari (1,8%) et de maladie de Wilson (1,8%).
Les manifestations neurologiques sont rares. Elles sont représentées par l’épilepsie idiopathique (5 cas), la neuropathie périphérique (4 cas dont 1 cas de névrite optique), le syndrome dépressif (5 cas) et l’ataxie au gluten (1 cas).
Étant donné la grande majorité féminine de la population cœliaque, les anomalies gynéco-obstétricales sont décrites essentiellement chez la femme. Chez les hommes, un seul cas de stérilité, un cas d’anomalie du spermogramme ayant nécessité un traitement et un autre cas de baisse de la libido chez un sujet de 30 ans, ont été retrouvés.
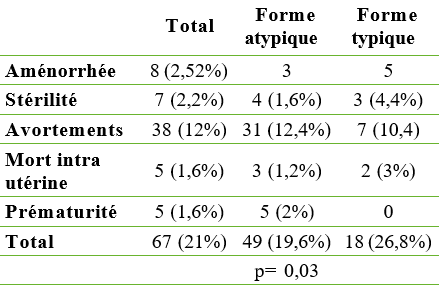
L’avortement répété représente l’anomalie gynéco-obstétricales la plus fréquente (12%). Les autres anomalies sont plus rares : stérilité dans 2,2%, mort in utero et prématurité dans 1,6%, aménorrhée primaire ou secondaire dans 0,37%. Globalement, ces anomalies gynéco-obstétricales sont plus fréquentes dans la population de patientes ayant une forme typique de maladie cœliaque, la différence étant statistiquement significative (p=0,03). Les troubles gynéco-obstétricaux selon la forme clinique sont illustrés dans le tableau 5.
Discussion :
La maladie cœliaque atteint l’adulte à n’importe quel âge avec cependant un pic de fréquence entre 30 et 40 ans [7]. Ces données concordent avec celles de notre étude, la moyenne d’âge de notre population de malades cœliaques étant de 31,6 ans.
Le diagnostic à un âge tardif est possible : 7% dans notre population, 6,5 à 9,3% dans la littérature [8,9]. Comme pour toute maladie auto-immune, la maladie cœliaque prédomine chez la femme (sex ratio de 0,50 à 0,25) [10,11]. Dans notre série, il est de 0,23.
La maladie cœliaque est une pathologie sous-diagnostiquée même dans les pays développés. En effet, aux USA pour chaque malade diagnostiqué, il en existe 7 à 8 non diagnostiqués [12]. En dépit d’une meilleure connaissance de la maladie, le délai diagnostique, défini par le temps écoulé entre le début des symptômes et le diagnostic, reste élevé. Dans l’étude de Fuchs, 32% des patients ont un délai diagnostique de plus de 10 ans [13]. Ceci est probablement dû au caractère très varié et non spécifique des symptômes liés à la maladie cœliaque. Dans notre étude, le diagnostic tardif (au-delà de 10 ans) représente 15,6% des malades et la majorité des patients (54,2%) ont un délai diagnostic entre 1 et 10 ans.
La maladie cœliaque peut se manifester par des symptômes variés ; aucun signe digestif n’est prédictif de la maladie même lorsque les symptômes sont associés [14]. Dans la littérature, les manifestations digestives décrites dans la maladie cœliaque sont variées et leur fréquence diffère d’une étude à l’autre. Ainsi, dans une étude multicentrique (Italie, Iran et Roumanie) [15], les symptômes digestifs varient d’un centre à un autre. Les symptômes prédominants sont la douleur abdominale et la diarrhée. La constipation, la dyspepsie et le ballonnement sont moins fréquents [15]. Devant ce grand polymorphisme symptomatique, le clinicien a du mal à sélectionner les patients susceptibles d’avoir une maladie cœliaque. Des algorithmes décisionnels ont été proposés pour aider le praticien à déterminer les patients à dépister. Certains de ces algorithmes informatisés semblent avoir une bonne sensibilité et spécificité [16], encore faut-il qu’ils soient disponibles et faciles à utiliser pour générer une meilleure adhésion des médecins. Dans notre série, les signes digestifs les plus fréquents sont le ballonnement abdominal 49,8%, les douleurs abdominales 43,2% et la diarrhée chronique 21,2%. Moins fréquemment, sont observés les vomissements 10,1%, la dyspepsie 7,2% et le reflux gastro-œsophagien 2,5%. Dans notre population cœliaque, 4,7% ont des symptômes qui correspondent aux critères de Rome III du syndrome de l’intestin irritable dans sa forme alternance diarrhée-constipation. Pour cette catégorie de patients, le traitement à base d’antispasmodiques et de médicaments anti-gaz était inefficace. En dépit du caractère réfractaire des symptômes, aucun dépistage sérologique n’a été réalisé. La fibroscopie digestive est effectuée devant l’apparition d’une anémie ferriprive permettant ainsi de faire le diagnostic de maladie cœliaque.
Dans la littérature, le taux du syndrome de l’intestin irritable dans la maladie cœliaque est très variable d’une étude à l’autre. Cette variabilité dépend des critères utilisés pour le diagnostic du syndrome de l’intestin irritable et de la population étudiée. Ainsi, lorsque les critères de Rome II sont utilisés pour identifier les patients ayant un syndrome de l’intestin irritable, 3,23% sont cœliaques dans une étude jordanienne [17] et 12% dans une étude iranienne [18]. Tandis que 36% des cœliaques ont des symptômes du syndrome de l’intestin irritable dans l’étude de Green et al [19]. Dans une étude plus récente, les critères de Rome III ont été utilisés, le taux de 0,8% de maladie cœliaque confirmée histologiquement a été rapporté [20]. Cela est probablement dû au recours à des critères diagnostiques plus sélectifs dans la classification de Rome III pour le diagnostic du syndrome de l’intestin irritable.
Dans notre population cœliaque, la forme atypique est prédominante ; elle représente 78,8% de la population cœliaque avec un rapport forme atypique/forme typique=3,7.
Ces données sont concordantes avec celles de la littérature. En effet, les formes atypiques sont les plus fréquentes, en particulier au cours de ces dix dernières années. Elles représentent 80% dans l’étude de Farrell [21]. Dans une série Italienne ce taux est de 66% [22]. Dans les rares études du Maghreb et du Moyen Orient les résultats sont très variables selon les régions.
Notre étude ne retrouve aucune forme silencieuse, cela s’explique :
- d’une part, par notre respect strict de la définition d’Oslo [23] concernant la forme silencieuse (en effet, nous avons considéré qu’une maladie cœliaque révélée par une anémie isolée ou une ostéopénie isolée est une forme atypique et non silencieuse),
- d’autre part, par la rareté de l’utilisation du dépistage sérologique dans notre étude.
Dans notre cohorte, les symptômes cliniques sont souvent non accompagnés de signes carentiels qui orientent vers une pathologie organique, tels que la pâleur intense observée dans seulement 22,7%, les anomalies de la peau et des phanères (18,6%), le retard staturo-pondéral (6%). Ces anomalies secondaires à des carences sévères et prolongées sont peu fréquentes dans la population cœliaque témoignant de la rareté des formes sévères. Elles sont plus observées dans les formes typiques. Cependant, la différence sur le plan statistique n’est pas significative (p=0,09).
Le signe le plus objectif lors de l’examen physique qui renseigne sur le retentissement de la maladie cœliaque est le BMI (body mass index). La moyenne du BMI de notre population de malades est normale (20,78). Toutefois, celle de la population atypique est statistiquement supérieure à celle de la population typique (21,15 vs 19,44). Cela est probablement dû au fait que la maladie soit plus sévère dans la forme typique. Ces résultats nous permettent d’attester qu’un BMI diminué n’est pas un bon indicateur de maladie cœliaque, puisque la majorité des patients ont un BMI normal. Ceci est d’ailleurs rapporté dans les études internationales [24].
Dans la littérature, l’anémie est présente lors du diagnostic de maladie cœliaque dans 12 à 69% [25]. Elle représente le symptôme extra-digestif le plus fréquent [25]. L’anémie est le plus souvent ferriprive, récidivante ou réfractaire au traitement [26]. Des carences en vitamines B12 ou acide folique peuvent être associées [25, 26]. Dans notre étude, l’anémie est le signe carentiel le plus fréquent (83%) dans les deux formes cliniques. Cette fréquence élevée (dans les deux formes cliniques) est probablement en rapport avec le fait que le fer soit absorbé dans le duodénum, qui est atteint de façon quasi constante dans la maladie cœliaque. Il est à noter que le taux d’anémie est plus important de façon significative dans la forme typique. Ce taux important observé dans notre population de malades, peut être expliqué par des carences d’apport probablement associées à la malabsorption. L’anémie est le plus souvent ferriprive (90,4%) et microcytaire hypochrome (99,2%).
Les autres carences sont beaucoup moins fréquentes que l’anémie. Elles témoignent d’une malabsorption plus sévère. En effet, l’hypocalcémie est présente dans 25,5%, l’hypolipidémie dans 26%, l’hypo albuminémie dans 17,6% et la carence en vitamine K dans 3,2%. Ces carences sont, de façon très significative, plus souvent observées dans la forme typique, ce qui témoigne de la plus grande sévérité de cette forme par rapport à la forme atypique.
Dans la littérature, les carences en albumine, électrolytes et lipides sont rares [27]. Elles sont observées surtout dans les formes typiques de maladie cœliaque comme il apparaît dans notre étude.
Les manifestations extra-digestives sont présentes aussi bien dans les formes atypiques que typiques. Comme précédemment décrit, l’anémie est le signe extra-digestif le plus fréquent (86,7%). En deuxième position viennent les manifestations osseuses (42,3%). Elles sont souvent asymptomatiques et doivent être recherchées systématiquement dès que le diagnostic de maladie cœliaque est posé. Dans la littérature, près des 2/3 des patients ont une ostéodensitométrie pathologique (ostéopénie et ostéoporose) [28]. Ces anomalies osseuses sont présentes dans les deux formes de maladie cœliaque sans différence significative, elles sont dues essentiellement à la malabsorption du calcium et de la vitamine D [29]. L’absorption du calcium se fait au niveau du duodénum constamment atteint quelle que soit la forme clinique [30].
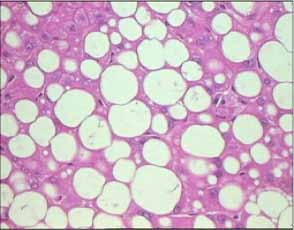
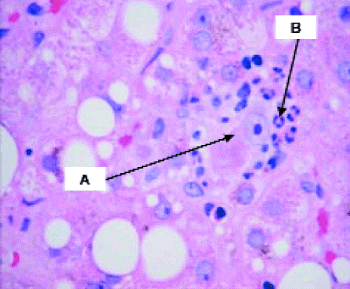
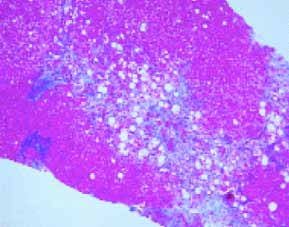
Dans la littérature, l’hypertransaminasémie idiopathique ou « hépatite » cœliaque est présente chez près de la moitié des cœliaques non traités [31]. Dans notre cohorte, elle n’est retrouvée que chez 32 patients soit dans 10% des cas. L’augmentation de la perméabilité intestinale semble jouer un rôle non négligeable dans la survenue de ces « hépatites » [32], plus fréquentes lorsque l’atteinte intestinale est sévère. Le taux faible « d’hépatite » cœliaque observé dans notre étude pourrait être expliqué par le fait que les formes sévères soient peu fréquentes dans notre cohorte.
Dans notre série, les atteintes hépatiques sévères sont retrouvées dans 8,2%. L’étiologie dysimmunitaire des cirrhoses constitue la cause la plus fréquente (9 cirrhoses dysimmunitaires sur 13 cirrhoses). Ces données sont concordantes avec celles de la littérature. En effet, la cholangite biliaire primitive et l’hépatite auto-immune sont fréquemment associées à la maladie cœliaque [33,34,35]. La même prédisposition génétique est probablement à l’origine de cette association [36]. Le dépistage de la maladie cœliaque est recommandé au cours de ces maladies auto-immunes [37].
Dans près de la moitié des cas (14 malades sur 26), le diagnostic étiologique de l’hépatopathie n’a pas pu être établi. Il s’agit des quatre cirrhoses cryptogénétiques et des dix HTP intra-hépatiques idiopathiques. Certains auteurs ont décrit l’existence de cirrhoses cryptogénétiques [38] ou d’HTP intra-hépatique [39] associées à la maladie cœliaque. La relation entre ces atteintes hépatiques (non étiquetées) et la maladie cœliaque a été évoquée devant l’absence d’étiologie et l’amélioration sous régime sans gluten.
Les manifestations gynéco-obstétricales telles que l’aménorrhée, la stérilité ou les avortements répétés ont été décrites, associées à la maladie cœliaque dans de nombreux écrits. Elles sont dues à la malabsorption de l’acide folique, du zinc et du sélénium [40,41,42], ainsi qu’à une déficience en ghreline et leptine [43], mais aussi à une dysrégulation du système immunitaire [44]. Dans notre cohorte de patientes cœliaques, ces anomalies gynéco-obstétricales sont significativement plus fréquentes dans la forme typique de la maladie (26,8% versus 19,6% ; p=0,03), témoignant de la sévérité des carences dans cette forme clinique de maladie cœliaque.
Pour conclure : Il est important que le praticien sache évoquer la maladie cœliaque devant des symptômes digestifs banals tels que des ballonnements, des douleurs abdominales atypiques ou un syndrome de l’intestin irritable réfractaire au traitement. La connaissance des manifestations extra-digestives de la maladie cœliaque est importante. En effet, la maladie peut se manifester par une simple anémie chronique et réfractaire, une ostéopénie ou des manifestations hépatiques. De même, la forme historique avec diarrhée chronique et syndrome carentiel est certes plus sévère, mais elle ne doit plus être considérée comme la forme prédominante chez nous.
Histogramme représentant les symptômes digestifs en pourcentage
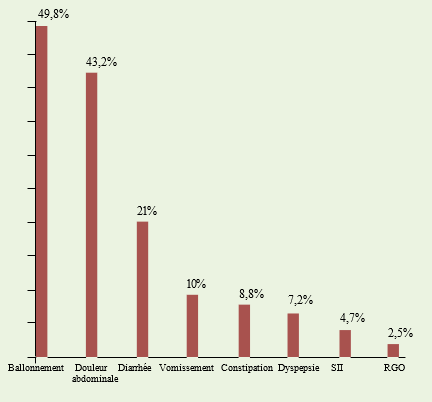
SII : syndrome de l’intestin irritable RGO : reflux gastro-œsophagien
Bibliographie :
- European Society for Pediatric, Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease (Husby S et al). J Pediatr Gastroenterol Nutr. 2012; 54(1): 136-60.
- West J, Logan RFA, Hill PG et al. Seroprevalence correlates and characteristics of undetected celiac disease in England, Gut 2003; 52:960-965.
- Cook HB, Burt MJ, Collett JA et al. Adult coeliac disease: Pre- valence and clinical significance. J Gastroenterol Hepatol. 2000; 15 :1032-36.
- Thèse de doctorat en médicine (spécialité pédiatrie) du Docteur Manoubia Bensmina soutenue en 2015 : Détermination de la Pré- valence de la maladie cœliaque chez les enfants d’âge scolaire de la wilaya d’Alger.
- Jones S, D’Souza C et Haboubi N. Patterns of clinical presentation of adults coeliac disease in a rural setting. Nutr J. 2006; 5(24): 8 pages.
- Fernandez A, Gonzalez L et De la Fuete J. Coeliac disease: Clini- cal features in adult populations. Rev Esp Enferm Dig. 2010; 202(8): 466-471.
- Snyder CL, Young DO, Green PHR, et al. Celiac disease. 2008, in Pagon RA, Adam MP, Ardinger HH, et al editors. Gene Reviews. University of Washington, Seatle; 1993-2015.
- Singh P, Sherghill S, Makharia GK, et al. Celiac disease in older adults. J Gastrointestin Liver Dis. 2013; 22: 359-60.
- Zingone F, West J, Auricchio R, et al. Incidence and distribution of coeliac disease in Campania (Italy): 2011-2013. UEGJ. 2015; 3(2): 182-89.
- Bardella MT, Fredalla C, Saladino V, et al. Gluten intolerance: Gender and age related differences in symptoms. Scand J Gastroen- terol. 2005; 40: 15-9.
- Liorente-Alonso MJ, Fernandez-Acenero MJ et Sebastien M. Glu- ten intolerance: Sex-age-related features. Can J gastroenterol. 2006 ; 20(11) : 719-722
- Mooney PD, Hadjivassiliou M, Sanders DS. Coeliac disease. BMJ. 2014; 318: g1561.
- Fuchs V, Kurppa K, Huhtala H, et al. Factors associated with long diagnostic delay in celiac disease. Scand J Gastroenterol. 2014 ; 49(11) : 1304-10.
- Nejad RM, Pourhosseingholi MA, Mojarad EN, et al. Atypical presentation is dominant and typical in celiac disease. 2009 ; 18(3) : 285-91.
- Ehsani-Ardakani MJ, Rostami-Nejad M, Villanacci V, et al. Gas- trointestinal and non-gastrointestinal presentation in patients with celiac disease. Arch Iran Med. 2013; 16(2): 78-82
- Ludvingsson JF, Pathak KJ, Murphy S, et al. Use of computerized algorithme to identify individuals in need of testing for celiac disease. J Am Med Inform Assoc. 2013; 20: e306-e310.
- Jadallah KA et Khader YS. Celiac disease in patients with pre- sumed irritable bowel syndrome: A case-finding study. WJGE. 2009; 15(42) : 5321-5325
- Masjedijadeh R, Hajjani E, Hashemi J, et al. Celiac disease in South-West Iran. WJGE. 2006 ; 12 : 4416-19.
- Green PHR, Stavropoulos SN, Panagi SG et al. Characteristics of adult celiac disease in the USA: results of a national survey. Am J Gastroenterol 2001; 96(1) :126-31.
- Sharma H, Verma AK, Das P, Gupta SD, Ahuja V et Makharia GK. Prevalence of celiac disease in Idian patients with irritable bowel syndrome and uninvestigated dyspepsia. J Dis Dig. 2015.16(8): 443-48
- Farrell RJ, Kelly CP. Celiac sprue. NEJM. 2002 ; 17(346) : 180-8.
- Volta U, Caio G, Sanghellini V et al. The changing clinical profile of celiac disease: a 15-year experience (1998-2012) in an Italian refe- ral center. Gastroenterology. 2014 ; 14:194-8)
- Ludvingsson JF, Leffler DA, Bai JC, et al. The Oslo definitions for celiac disease and related terms. Gut. 2013 ; 62 : 43-52
- Van Der Pals M, Myleus A, Norstrom F, et al. Body mass index is not a reliable tool in predicting celiac disease in children. BMC Pediatr. 2014; 14: 165.
- Halfdanarson TR, Litzow MR, Murray JA. Hematologic manifes- tations of celiac disease. Blood. 2007; 109 : 412-21
- Hershko C et Patz J. Ironing out mechanisms of anemia in celiac disease. Haematologica. 2008; 93(12):1761—65.
- Murray JA, Van Dyke C, Plevak MF et al. Trends in the iden- tification and clinical feature of celiac disease in a North American community, 1950-2001. Clin gastroenterol Hepatol. 2003; 1(1): 19-27
- Fouda MA, Khan AA, Sultan M, et al. Evaluation and manage- ment of skeletal health in celiac disease: position statement. Can J Gastroenterol. 2012; 26(11): 819-29.
- Molteni N, Bardella MT, Vezzoli G, et al. Intestinal calcium ab- sorption as shown by stable strontium test in celiac disease before and after gluten-free diet. Am J Gastroenterol. 1995; 90: 2025-28.
- Kurpa-Kozak U. Pathologic bone alterations in celiac disease: Etiology, epidemiology, and treatment. Nutrition. 2014; 30: 16-24.
- Volta U. Liver dysfunction in celiac disease, Minerva Med 2008;99:619-29
- Volta U, De Franceshi L, Lari F, et al. Coeliac disease hidden by cryptogenetic hypertransaminasaemia. Lancet. 1998; 352: 26-9
- Floerani A et al, Prevalence of celiac disease in primary biliary cirrhosis and antimitochondrial antibodies in adults celiac patients in Italy, Dig Liv Dis 2002; 34:258-261
- Van Gerven NM, Bakker SF, DE Boer YS, et al. Seroprevalence of celiac disease in patients with autoimmune hepatitis. Eur J Gastroen- terol Hepatol. 2014 ; 26(10) : 1104-7.
- Villalta D, Girolami D, Bidoli E, et al. High prevalence of celiac disease in autoimmune hepatitis detected by anti-tissue transglutami- nases autoantibodies. J Clin Lab Anal. 2005; 19(1): 6-10.
- Ludvingsson JF, Elfstrom P, Broom U, et al. Celiac disease and risk of liver disease: a general population-based study. Clin Gastroenterol Hepatol. 2007; 5(1): 63-9 e1.
- National Institute for health and clinical excellence (NICE). Coe- liac disease: recognition and assessment of coeliac disease. 2009.
- Wakim-Fleming J, Pagadala MR, McCullough AJ, et al. Preva- lence of celiac disease in cirrhosis and outcome of cirrhosis on a glu- ten-free diet: a prospective study. J Hepatol. 2014; 61(3): 558-63.
- Maiwall R, Pulimood AB, Babji S, et al. Investigation into celiac disease in Indian population patients with portal hypertention. Ind J Gastroenterol. 2014; 33(6): 517-23.
- Hirson C. Coeliac infertility-folic acid therapy. Lancet. 1970; 1(7643): 412.
- Bedwal RSBA. Zinc, Copper, and Selenium in reproduction. Experimentia. 1994; 50: 626-40.
- Singhal N, Alam S, Sherwani R, et al. Serum zinc levels in celiac disease. Indian Pediatr. 2008; 45: 319-21.
- Martin JR, Lieber SB, McGrath J, et al. Maternal ghrelin defi- ciency compromises reproduction in femal progery through altered uterine developmental programming. Endocrinology. 2011; 152: 2060-66.
- Anjum N, Baker PN, Robinson NJ, et al. Maternel celiac disease autoantibodies from celiac patients are responsible for trophoblast damage via apoptosis in vitro. Am J Gastroenterol. 2010.